

Abstract
Purpose
Cardiac troponin I (cTnI) is a well-established biomarker for stroke prediction, especially in patients with heart diseases. However, the causal effect of circulating cTnI on stroke remains unclear.
Methods
We employed Mendelian Randomization (MR) analysis to determine the associations between genetically predicted circulating cTnI levels and stroke and its subtypes. Summary-level data for exposure and outcomes were generated from different genome-wide association studies. Single-nucleotide polymorphisms (SNPs) associated with circulating cTnI at genome-wide significance level (P < 5 × 10−8) were employed as instrumental variables (IVs). We used fixed-effect inverse-variance weighted (IVW) as the main method for pooling MR estimates. Sensitivity analyses and multivariable MR analyses were carried out to assess the robustness of the results.
Results
Using the fixed-effects IVW method, we found that genetically elevated plasma cTnI levels may have a causal effect on the risk of cardioembolic stroke (CES) (odds ratio (OR), 1.58; 95% confidence interval (CI), 1.17–2.13; P = 0.003). Additional analyses including multiplicative random-effects (mre) IVW, weighted median, MR-Egger and MR-PRESSO yielded similar results, but with broader CIs that span 1.0. The total effect of cTnI on CES was attenuated by adjusting for the effect of atrial fibrillation (OR,1.26; 95% CI, 0.76–2.11; P = 0.371) and smoking (OR,1.53; 95% CI, 0.87–2.66; P = 0.137). In addition, we found no causal effect of cTnI on the risk of any stroke and other stroke subtypes, including any ischemic stroke, large artery stroke, cardioembolic stroke, small vessel stroke, and intracerebral hemorrhage. These results were consistent across sensitivity analyses.
Conclusion
This study provides little evidence that increased serum cTnI levels lead to a higher risk of stroke.
Keywords: Mendelian randomization, cardiac troponin I, stroke, causation, genome-wide association studies
Introduction
Stroke remains one of the leading causes of death worldwide. About 1 in 4 people will suffer from a stroke from the age of 25 onwards,1 and the age-standardized death rate of stroke in 2016 reaches 86.5 per 100,000 people.2 Atrial fibrillation, obesity, smoking, etc., are well-recognized risk factors for stroke.3–5
Accumulating evidence from population-based studies has demonstrated that biomarkers reflecting myocardial injury help improve risk stratification of stroke.6 Cardiac troponin I (cTnI) is one of these biomarkers and has been intensively investigated in the preclinical settings. It is a protein found in the cardiomyocytes and released into the bloodstream following myocardial injury. Elevated circulating cTnI level has been associated with a high risk of ischemic stroke, with hazard ratios ranging from 1.14 to 2.99 per 1-SD increase in cTnI7–9 in the general population. Furthermore, circulating cTnI has also been included in the ABC (age, biomarkers, clinical history) stroke score for predicting stroke in patients with atrial fibrillation.10 However, the causal association between cTnI and stroke remains largely unclear, and such an assessment will be very challenging due to the confounding or reverse causation bias inherent in the observational studies.
Mendelian Randomization (MR) is a widely used method for assessing the causal effect of clinical exposures on explicit outcomes.11 Genetic variants randomly assigned to offspring are employed as instrumental variables (IVs). Since such IVs remain constant after conception and should be free from potential confounders, the MR approach overcomes some limitations of traditional epidemiological studies.12,13 Hence, to explore the causal relationship between lifelong elevated circulating cTnI and stroke risk, we carried out a two-sample MR analysis (the data for MR was extracted from two independent datasets). Single nucleotide polymorphisms (SNPs) associated with cTnI levels were selected as IVs.
Materials and Methods
Study Design
An overview of this two-sample MR approach is displayed in Figure 1. Briefly, single nucleotide polymorphisms (SNPs) were used as instrumental variables (IVs) to assess the causal association between cTnI and stroke. The following three assumptions are recognized as the basis for this analysis: (1) IVs are robustly associated with the exposure; (2) IVs must not be associated with potential confounders; (3) IVs should not directly affect the outcome other than through the exposure.
Figure 1.

Schematic diagram showing the assumptions of Mendelian randomization analysis.
Abbreviations: SNPs, single nucleotide polymorphisms; AS, any stroke; AIS, any ischemic stroke; LAS, large artery stroke; CES, cardioembolic stroke; SVS, small vessel stroke; ICH, intracerebral hemorrhage; AF, atrial fibrillation; BMI, body mass index.
Data Sources and SNP Selection
The information on all the summary statistics used in this MR analysis is shown in Supplementary Table 1. Ethical approval and written informed consent of participants were obtained for all original studies included in the public genome-wide association meta-analysis studies (GWASs) datasets.
Summary statistics for the stroke and ischemic stroke subtypes were extracted from a publicly available database from the MEGASTROKE consortium, which included 40,585 cases and 406,111 controls of European ancestry.14 Here, stroke is defined as a rapidly developing sign of focal (or global) disturbance of cerebral function lasting more than 24 hours or resulting in death, with no apparent cause other than a vascular origin.14 In this GWAS meta-analysis, strokes were classified as any ischemic stroke (AIS; 34,217 cases and 406,111 controls), intracerebral hemorrhage (ICH), and stroke of uncertain type. Based on the Trial of Org 10172 in Acute Stroke Treatment criteria, AIS was further divided into large artery stroke (LAS; 4,373 cases and 146,392 controls), cardioembolic stroke (CES; 7193 cases and 204,570 controls), and small vessel stroke (SVS; 5386 cases and 192,662 controls). In addition, summary-level data for ICH was obtained from a GWAS released by the International Stroke Genetics Consortium, which incorporated 1545 cases and 1481 controls of European ancestry.15
Genetic variants associated with circulating cTnI levels were derived from the hitherto largest GWAS meta-analysis of 48,115 individuals.16 Twelve single-nucleotide polymorphisms (SNPs) with genome-wide significance (P < 5 × 10−8) were collected. The F-statistics were estimated to identify any weak instrument bias using the formula 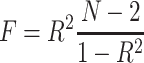 , where R2 indicates the proportion of the variance of circulating cTnI explained by the genetic instruments and N refers to the sample size.17 We used the formula R2 = 2 × EAF × (1-EAF) × Beta2 to calculate R2 of each SNP.18 Two SNPs (rs7481951 and rs151313792) were excluded from this MR study since they were in linkage disequilibrium (LD) (r2 < 0.001; region size, 10000kb) with other SNPs. Two SNPs (rs8024538 and rs9391746) were not available in the GWAS datasets from the MEGASTROKE consortium and four SNPs (rs8024538, rs9391746, rs2731672, rs4283165) were absent from the summary statistics for ICH; therefore, we found proxies using an online tool (http://snipa.helmholtz-muenchen.de/snipa3/) to replace them if available (r2 > 0.8).
, where R2 indicates the proportion of the variance of circulating cTnI explained by the genetic instruments and N refers to the sample size.17 We used the formula R2 = 2 × EAF × (1-EAF) × Beta2 to calculate R2 of each SNP.18 Two SNPs (rs7481951 and rs151313792) were excluded from this MR study since they were in linkage disequilibrium (LD) (r2 < 0.001; region size, 10000kb) with other SNPs. Two SNPs (rs8024538 and rs9391746) were not available in the GWAS datasets from the MEGASTROKE consortium and four SNPs (rs8024538, rs9391746, rs2731672, rs4283165) were absent from the summary statistics for ICH; therefore, we found proxies using an online tool (http://snipa.helmholtz-muenchen.de/snipa3/) to replace them if available (r2 > 0.8).
Statistical Analysis
We conducted a two-sample MR to assess the causal association between circulating cTnI and risk of stroke. SNPs were harmonized across the data sources via the effect alleles to ensure that the estimates are directionally matched. Then, we generated effect estimates using fixed-effect inverse variance-weighted (fe-IVW) meta-analysis. Complementary analyses were carried out using the weighted median,19 MR-Egger regression,20 and MR-pleiotropy residual sum and outlier (MR-PRESSO) methods.21 The weighted median method can provide valid estimates even when up to 50% of the information in the analysis comes from invalid IVs.19 The MR-Egger method provides more conservative causal estimates in the presence of pleiotropic variants and is less likely to generate inflated test statistics.20 The MR-PRESSO method was used to detect the presence of outliers that could bias the results.21
A series of sensitivity analyses were performed to assess the robustness of the results. Quantitative heterogeneity among the SNPs was assessed by calculating Cochran’s Q statistic and I2. Provided Cochran Q-derived P < 0.05 or I2 > 25%, heterogeneity was considered to exist,22 and a random-effect IVW (mre-IVW) model would be adopted to avoid the bias of weak IVs-exposure associations.23 Pleiotropy effects were assessed by MR-Egger analysis.24 To determine whether a single SNP drove the causal relationship, we applied leave-one-out analyses in which we excluded one SNP in turn and then re-evaluated the causal effect. The mRnd power calculator (available at http://cnsgenomics.com/shiny/mRnd/) was employed to calculate statistical power.25
In addition, multivariable MR (MVMR) was conducted to generate estimates independent of the effects of atrial fibrillation, smoking, and body mass index (BMI). We used publicly available summary statistics for atrial fibrillation from the Atrial Fibrillation Haplotype Reference Consortium for atrial fibrillation (n = 588,190),26 smoking from GWAS & Sequencing Consortium of Alcohol and Nicotine (n=1,232,091),27 and BMI from the Genetic Investigation of Anthropometric Traits (n = 322,154).28
A Bonferroni-corrected significance threshold of P < 0.083 (P < 0.05/6 outcomes) was set for all the statistical tests. All analyses were conducted using R software (version 4.1.0) with R packages including TwoSampleMR,29 MendelianRandomization,30 and MR-PRESSO.21
Results
Details of SNPs for cTnI were displayed in Supplementary Table 2. These instrumental variables were estimated to account for 1.2% of the observed phenotypic variation of cTnI. The F-statistics for all SNPs (ranging from 25.9 to 75.5) were greater than the recommended threshold of 10, suggesting no weak instrument bias in the present study.
The overall IVW meta-analysis suggested a causal effect of genetically predicted cTnI levels on the risk of CES (odds ratio (OR), 1.58; 95% confidence interval (CI), 1.17–2.13; P = 0.003) (Figure 2). Sensitivity analysis indicated moderate heterogeneity (I2 = 71.5%, P for Cochran’s Q = 0.001); therefore, we employed the multiplicative random-effects (mre) IVW method23 for more reliable estimate. The estimate was unchanged using mre-IVW methods, but with a broader CI (OR, 1.58; 95% CI, 0.93–2.67; P = 0.087). The association was directionally consistent with the weighted median and MR-Egger methods but with wider CIs that span 1.0. In addition, we used MR-PRESSO method to detect any outliers in the IVW method and generate a corrected MR estimate robust to heterogeneity. One potential outlier was detected among the 9 SNPs (rs12331618, P = 0.027), and analysis after excluding this outlier yields a similar result (Figure 2). In the leave-one-out analysis, we found the causal association was modestly driven by 2 SNPs (rs2731672 and rs8039472) (Supplementary Figure 1). Furthermore, multivariable MR analysis revealed no causal association between cTnI and CES after adjustment for atrial fibrillation and smoking, suggesting that the causal relationship suggested by IVW methods may be mediated by common risk factors (Table 1).
Figure 2.

Associations of genetic predicted cTnI levels with stroke and its subtypes. A proxy SNP (rs8039472) was used to replace SNP (rs8024538) (pairwise r2 = 0.92) in all the datasets. *No outliers detected. †MR-PRESSO outlier detected: rs12331618.
Abbreviations: IVW (fe), fixed-effects inverse-variance weighted; IVW (mre), multiplicative random-effects inverse-variance weighted; MR-PRESSO, Mendelian randomization pleiotropy residual sum and outlier method; NA, not available.
Table 1.
Multivariable MR Associations of Cardiac Troponin I with Stroke and Its Subtypes Adjusted for Atrial Fibrillation and BMI
| Outcome | Model | OR (95% CI) | P-value |
|---|---|---|---|
| AS | IVW adjusted for atrial fibrillation | 1.00 (0.85, 1.17) | 0.996 |
| IVW adjusted for smoking | 1.02 (0.88, 1.18) | 0.764 | |
| IVW adjusted for BMI | 1.03 (0.88, 1.20) | 0.747 | |
| Fully adjusted model | 0.99 (0.83, 1.19) | 0.918 | |
| AIS | IVW adjusted for atrial fibrillation | 1.08 (0.86, 1.36) | 0.503 |
| IVW adjusted for smoking | 1.08 (0.88, 1.33) | 0.47 | |
| IVW adjusted for BMI | 1.09 (0.87, 1.36) | 0.466 | |
| Fully adjusted model | 1.09 (0.80, 1.48) | 0.583 | |
| LAS | IVW adjusted for atrial fibrillation | 1.08 (0.86, 1.36) | 0.503 |
| IVW adjusted for smoking | 1.10 (0.74, 1.63) | 0.64 | |
| IVW adjusted for BMI | 1.04 (0.67, 1.59) | 0.87 | |
| Fully adjusted model | 1.23 (0.75, 2.03) | 0.409 | |
| SVS | IVW adjusted for atrial fibrillation | 0.93 (0.60, 1.44) | 0.734 |
| IVW adjusted for smoking | 0.78 (0.48, 1.28) | 0.324 | |
| IVW adjusted for BMI | 0.68 (0.39, 1.17) | 0.162 | |
| Fully adjusted model | 0.88 (0.49, 1.59) | 0.668 | |
| CES | IVW adjusted for atrial fibrillation | 1.26 (0.76, 2.11) | 0.371 |
| IVW adjusted for smoking | 1.53 (0.87, 2.66) | 0.137 | |
| IVW adjusted for BMI | 1.93 (1.14, 3.24) | 0.014 | |
| Fully adjusted model | 1.53 (0.84, 2.78) | 0.168 | |
| ICH | IVW adjusted for atrial fibrillation | 0.74 (0.27, 2.01) | 0.554 |
| IVW adjusted for smoking | 0.99 (0.40, 2.44) | 0.981 | |
| IVW adjusted for BMI | 1.10 (0.42, 2.85) | 0.852 | |
| Fully adjusted model | 0.73 (0.24, 2.23) | 0.576 |
Abbreviations: OR, odds ratio; CI, confidence interval; IVW, inverse variance-weighted; AS, any stroke; AIS, any ischemic stroke; LAS, large artery stroke; CES, cardioembolic stroke; SVS, small vessel stroke; ICH, intracerebral hemorrhage.
In addition, genetically predicted cTnI was not causally associated with the risk of other stroke subtypes. The results were consistent across complementary analyses including the weighted median and MR-Egger methods. No outliers were found in the MR-PRESSO test. No single genetic variant was found to bias the causal associations potentially in the leave-one-out analyses (Supplementary Figure 1).
Forest plots of the effect of every single SNP on the outcomes are reported in Supplementary Figure 2. No evidence of directional pleiotropy was found as measured by MR-Egger regression (P for intercept >0.176, Supplementary Table 3).
Discussion
In the present study, we carried out a two-sample MR analysis to determine the causal association between the circulating cTnI levels and the risk of stroke and its subtypes. Although fe-IVW indicated a causal relationship between cTnI and CES, complementary analyses yield estimates with broader CIs that span 1.0, and MVMR analyses suggested that this causal relationship, if any, may be mediated by atrial fibrillation and smoking. For cTnI and other types of strokes, our analysis provided little evidence of a causal association. Altogether, our study suggested that cTnI is a bystander rather than a risk factor in stroke.
cTnI has been recognized as a pragmatic biomarker for the diagnosis of myocardial damage. It may predict all-cause mortality in patients with heart failure (HF) and patients with acute ischemic stroke.31,32 Recently, several lines of evidence demonstrated that elevated cTnI levels are associated with a higher risk of stroke. In a retrospective study including 2.8 million individuals, Jacob VanHouten et al. reported that circulating cTnI levels were elevated in 20.6% of participants who suffered from an acute ischemic stroke but were free of any known heart diseases.33 On the other hand, circulating cTnI can serve as a predictor of stroke, as observational studies have shown.7,8 A novel biomarker-based stroke risk score with the introduction of cTnI was well calibrated and performed better than the extensively used CHA2DS2-VASc score.10 Nowadays, developed biosensors for cTnI detection are being promoted to aid early management decisions in acute strokes.34
However, assessing the causal effects of cTnI on stroke is challenging due to the residual confounding and reverse causality inherent in the observational studies.35 In addition, whether cTnI plays a role of driver or passenger in the process of stroke remains unknown. This MR analysis was carried out to answer these questions. Although the fixed-effects IVW method yield a result that genetically predicted cTnI may be causally linked to the risk of CES, this causal association no longer existed after adjustment for atrial fibrillation and smoking. In addition, our analysis also suggested that cTnI was not associated with any stroke, LAS, SVS, CES, and ICH. These results are different from those of previous observational studies. We think there are several explanations. First, although some confounders have been adjusted in these studies, there remain some unmeasured risk factors that remain uncontrolled due to the nature of the observational study. Circulating cTnI levels are usually higher in males, older, hypertensive, and diabetic; smoking, CVDs, and higher BMI lead to elevated cTnI levels. The existence of all these common risk factors may contribute to the observations in traditional studies. Second, the reverse causation bias was difficult to avoid in the observational studies. In addition, elevated cTnI may be just a manifest of other cardiovascular diseases (CVDs).
It was reported in animal studies that circulating cTnI induced immune responses causing HF,36,37 and it helped improve risk stratification of acute myocardial infarction (AMI) in cohort studies.38,39 Hence, to explore the causal effects of circulating cTnI on HF and AMI, Marta R. Moksnes et al performed a two-sample MR analysis, but no causal association was found.16 This conclusion corroborated with the findings in the present study. Consistently, another MR study suggested that GDF-15, a stress responsive cytokine, was not causally associated with stroke risk, except a suggested relationship with CES.40 The results of no causality did not weaken the value of the previous studies identifying the prognostic role of cTnI for CVDs (AMI, HF, stroke, and so on) but provided a greater insight into the role of this biomarker as well as the mechanisms of these diseases.
A similar MR analysis for the same subject was recently reported by Dandan Liu et al.41 Some of their conclusions shared with ours, such as no causal effect of cTnI on the risk of AS, AIS, LAS, and SVS. However, they concluded that cTnI is causally associated with CES. We think this conclusion is somewhat unconvincing because they neither considered SNPs in linkage disequilibrium nor performed MVMR to find potential confounders in their study. Since atrial fibrillation, smoking and BMI have been associated with stroke risk,4,42–44 we believe these confounders may bias the association between cTnI and CES.
Notable strengths of this MR analysis include the large sample size derived from several GWAS datasets and the consistency of the results across different complementary analyses. However, this study has several limitations. First, potential directional pleiotropy cannot be completely ruled out, although MR-egger interception tests and multivariable MR analyses did not find sufficient evidence to support this. Second, this study lacked patient-level data such as age and sex; thereby, the potential association between circulating cTnI and stroke in different sexes and ages could not be evaluated. Third, the GWAS data in this analysis is restricted to European, which might limit the scope of our findings. In addition, it is possible that there may be a non-linear association for cTnI with stroke outcomes, which might be missed by two-sample MR. Finally, given that the genetic variants were estimated to explain a very small proportion of the phenotypic variation of cTnI, our results did not reach sufficient statistical power; hence, there remains a possibility of false negatives in our analysis, and larger GWAS for cTnI is warranted in the future.
Conclusion
Our two-sample MR analysis found little evidence of a causal association between increased serum cTnI levels and risk of stroke in European populations, suggesting that efforts to decrease serum cTnI levels will probably not result in a decreased risk of stroke. The positive associations reported in observational studies may be attributable to residual confounding and reverse causation. Further studies are likely needed to confirm our results.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 81873484); the Youth Program of the National Natural Science Foundation of China (grant number 82000316); and the Nature Science Foundation of Zhejiang Province (grant number LZ16H020001). We extend our sincere thanks to Dr. Moksnes, Dr. Rosjo, Dr. Richmond et al, MEGASTROKE Consortium, International Stroke Genetics Consortium, Atrial Fibrillation Haplotype Reference Consortium, GSCAN Consortium, GIANT Consortium for sharing GWAS summary statistics.
Data Sharing Statement
The data that support the findings of this study are available in the Supplementary Material of this article.
Ethics Approval and Consent to Participate
Approval for this study was granted by the Ethics Committee of The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China.
Disclosure
The authors declare that they have no conflict of interest.
References
- 1.Feigin VL, Nguyen G, Cercy K, et al. Global, regional, and country-specific lifetime risks of stroke, 1990 and 2016. N Engl J Med. 2018;379:2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naghavi M, Abajobir AA, Abbafati Cet al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–1210. doi: 10.1016/S0140-6736(17)32152-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamel H, Healey JS. Cardioembolic stroke. Circul Res. 2017;120:514–526. doi: 10.1161/CIRCRESAHA.116.308407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsson SC, Burgess S, Michaëlsson K. Smoking and stroke: a Mendelian randomization study. Ann Neurol. 2019;86:468–471. doi: 10.1002/ana.25534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horn JW, Feng T, Mørkedal B, et al. Obesity and risk for first ischemic stroke depends on metabolic syndrome: the HUNT study. Stroke. 2021;52(11):3555–3561. doi: 10.1161/STROKEAHA.120.033016 [DOI] [PubMed] [Google Scholar]
- 6.Ng GJL, Quek AML, Cheung C, Arumugam TV, Seet RCS. Stroke biomarkers in clinical practice: a critical appraisal. Neurochem Int. 2017;107:11–22. doi: 10.1016/j.neuint.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 7.Jia X, Sun W, Hoogeveen RC, et al. High-sensitivity troponin I and incident coronary events, stroke, heart failure hospitalization, and mortality in the ARIC study. Circulation. 2019;139:2642–2653. doi: 10.1161/CIRCULATIONAHA.118.038772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camen S, Palosaari T, Reinikainen J, et al. Cardiac troponin I and incident stroke in European cohorts: insights from the BiomarCaRE project. Stroke. 2020;51:2770–2777. doi: 10.1161/STROKEAHA.120.029452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welsh P, Preiss D, Hayward C, et al. Cardiac troponin T and troponin I in the general population. Circulation. 2019;139:2754–2764. doi: 10.1161/CIRCULATIONAHA.118.038529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hijazi Z, Lindbäck J, Alexander JH, et al. The ABC (age, biomarkers, clinical history) stroke risk score: a biomarker-based risk score for predicting stroke in atrial fibrillation. Eur Heart J. 2016;37:1582–1590. doi: 10.1093/eurheartj/ehw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi: 10.1002/sim.3034 [DOI] [PubMed] [Google Scholar]
- 12.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–R98. doi: 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14:577–590. doi: 10.1038/nrcardio.2017.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik R, Chauhan G, Traylor Met al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo D, Falcone G, Devan W, et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet. 2014;94:511–521. doi: 10.1016/j.ajhg.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moksnes MR, Røsjø H, Richmond A, et al. Genome-wide association study of cardiac troponin I in the general population. Hum Mol Genet. 2021;30(21):2027–2039. doi: 10.1093/hmg/ddab124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–764. doi: 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 18.Shim H, Chasman DI, Smith JD, et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One. 2015;10:e0120758. doi: 10.1371/journal.pone.0120758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowden J, Davey smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377–389. doi: 10.1007/s10654-017-0255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greco FD, Minelli MC, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926–2940. doi: 10.1002/sim.6522 [DOI] [PubMed] [Google Scholar]
- 23.Bowden J, Del Greco M F, Minelli C, et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36:1783–1802. doi: 10.1002/sim.7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–1501. doi: 10.1093/ije/dyt179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roselli C, Chaffin MD, Weng L-C, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu M, Jiang Y, Wedow R, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann TJ, Choquet H, Yin J, et al. A large multiethnic genome-wide association study of adult body mass index identifies novel loci. Genetics. 2018;210:499–515. doi: 10.1534/genetics.118.301479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. doi: 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundström J. Myocardial biomarkers for prediction of cardiovascular disease. Dis Markers. 2009;26(5–6):235–246. doi: 10.1155/2009/436740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Y, Jiang M, Gong D, Man C, Chen Y. Cardiac troponin for predicting all-cause mortality in patients with acute ischemic stroke: a meta-analysis. Biosci Rep. 2018;38. doi: 10.1042/BSR20171178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.VanHouten J, Fricker G, Collins B, et al. Circulating troponin I level in patients with acute ischemic stroke. Curr Neurol Neurosci Rep. 2018;18:32. doi: 10.1007/s11910-018-0842-6 [DOI] [PubMed] [Google Scholar]
- 34.Dolati S, Soleymani J, Kazem Shakouri S, Mobed A. The trends in nanomaterial-based biosensors for detecting critical biomarkers in stroke. Clin Chim Acta. 2021;514:107–121. doi: 10.1016/j.cca.2020.12.034 [DOI] [PubMed] [Google Scholar]
- 35.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- 36.Göser S, Andrassy M, Buss SJ, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114:1693–1702. doi: 10.1161/CIRCULATIONAHA.106.635664 [DOI] [PubMed] [Google Scholar]
- 37.Okazaki T, Tanaka Y, Nishio R, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–1483. doi: 10.1038/nm955 [DOI] [PubMed] [Google Scholar]
- 38.Kozinski M, Krintus M, Kubica J, Sypniewska G. High-sensitivity cardiac troponin assays: from improved analytical performance to enhanced risk stratification. Crit Rev Clin Lab Sci. 2017;54:143–172. doi: 10.1080/10408363.2017.1285268 [DOI] [PubMed] [Google Scholar]
- 39.Sandoval Y, Nowak R, deFilippi CR, et al. Myocardial infarction risk stratification with a single measurement of high-sensitivity troponin I. J Am Coll Cardiol. 2019;74:271–282. doi: 10.1016/j.jacc.2019.05.058 [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Yang F, Ma M, et al. The impact of growth differentiation factor 15 on the risk of cardiovascular diseases: two-sample Mendelian randomization study. BMC Cardiovasc Disord. 2020;20:462. doi: 10.1186/s12872-020-01744-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu D, Deng Y, Wang J, et al. Genetically predicted cardiac troponin I concentrations and risk of stroke and atrial fibrillation. J Stroke Cerebrovasc Dis. 2021;31:106267. doi: 10.1016/j.jstrokecerebrovasdis.2021.106267 [DOI] [PubMed] [Google Scholar]
- 42.Hou L, Xu M, Yu Y, et al. Exploring the causal pathway from ischemic stroke to atrial fibrillation: a network Mendelian randomization study. Mol Med. 2020;26:7. doi: 10.1186/s10020-019-0133-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 STUDy. Circulation. 2014;129:837–847. doi: 10.1161/CIRCULATIONAHA.113.005119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jood K, Jern C, Wilhelmsen L, Rosengren A. Body mass index in mid-life is associated with a first stroke in men: a prospective population study over 28 years. Stroke. 2004;35:2764–2769. doi: 10.1161/01.STR.0000147715.58886.ad [DOI] [PubMed] [Google Scholar]