

Abstract
Question:
Cystic fibrosis (CF) is characterized by the accumulation of viscous, adherent mucus in the lungs. While several hypotheses invoke a direct relationship with CFTR dysfunction (i.e., acidic airway surface liquid (ASL) pH, low [HCO3−], airway dehydration), the dominant biochemical alteration of CF mucus remains unknown.
Materials/Methods:
We characterized a novel cell line (CFTR-KO Calu3 cells) and the responses of human bronchial epithelial (HBE) cells from subjects with G551D or F508del mutations to Ivacaftor and Elexacaftor-Tezacaftor-Ivacaftor (ETI). A spectrum of assays such as short-circuit currents (Isc), qPCR, ASL pH, western blotting (WB), light scattering/refractometry (SEC-MALS), scanning electron microscopy (SEM), % solids, and particle tracking were performed to determine the impact of CFTR function on mucus properties.
Results:
Loss of CFTR function in Calu3 cells resulted in ASL pH acidification and mucus hyperconcentration (dehydration). Modulation of CFTR in CF HBE cells did not affect ASL pH or mucin mRNA expression, but decreased mucus concentration, relaxed mucus network ultrastructure, and improved mucus transport. In contrast with modulator-treated cells, a large fraction of airway mucins remained attached to naïve CF cells following short apical washes, as revealed by the use of reducing agents to remove residual mucus from the cell surfaces. Extended hydration, but not buffers alkalized with NaOH or HCO3−, normalized mucus recovery to modulator-treated cell levels.
Conclusion:
These results indicate that airway dehydration, not acidic pH and/or low [HCO3−], is responsible for abnormal mucus properties in CF airways and CFTR modulation predominantly restores normal mucin entanglement.
Introduction:
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene affect mucus-producing organs, notably the lungs and gastrointestinal tract [1]. Defective CFTR protein prevents Cl− and HCO3− secretion, which alters mucus viscoelastic properties and affects organ function. In the gastrointestinal tract, accumulation of a thick, adherent mucus blocks pancreatic ducts and slows intestinal transit, hindering nutrient absorption and bodily growth [2]. In the lungs, viscous mucus causes obstruction of submucosal gland ducts and small airways, providing a favorable environment for chronic bacterial infection, which is the leading cause of mortality for CF patients [3, 4].
Mucins, the main polymeric component of mucus, are large, heavily-glycosylated proteins that organize in complex multimers to produce the viscoelastic properties of mucus. MUC5AC and MUC5B, the dominant airway mucins, are tightly packed in goblet and secretory cells via a low pH/high Ca2+ environment inside the granules [5]. Upon exocytosis, Ca2+ is proposed to be chelated by HCO3− in the extracellular milieu, a step considered critical for mucin protein unfolding and the formation of a loose mucus gel [5]. One of the hypotheses proposed for the pathophysiology of CF disease is improper anion (HCO3−) secretion, reducing extracellular HCO3− concentrations and compromising mucin expansion [6]. Another consequence of low [HCO3−] is acidic ASL pH that is proposed to affect electrostatic interactions within the mucus network and plays a role in aberrant mucus properties in CF [7, 8]. In addition to HCO3− and pH abnormalities, another prominent hypothesis is that reduced Cl− secretion leads to airway dehydration, mucin hyperconcentration, and mucus stasis [9]. In brief, under this hypothesis, defective CFTR Cl− transport results in osmotic gradients across the epithelium that favor water hyperabsorption and mucin hyperconcentration [10]. In this dehydrated environment, mucin polymer organization becomes more entangled with high osmotic pressure, causing the collapse of the periciliary layer (PCL) and cessation of mucus transport [8, 11–13].
For decades, treatment of CF was reduced to symptom management and infection control with the primary goal of slowing disease progression. In 2012, Ivacaftor (VX-770), a CFTR potentiator (a small molecule improving CFTR gating), became the first therapy to address the underlying defect but benefited only 5% of the CF population who exhibited mild mutations, e.g., the G551D gating mutation [14, 15]. Recently, Elexacaftor-Tezacaftor-Ivacaftor (ETI), a triple combination therapy consisting of two corrector compounds (VX-445 and VX-661, small molecules improving CFTR folding) with potentiator VX-770, was shown to efficiently restore function of F508del CFTR, which affects almost 90% of CF patients [16]. Robust clinical benefits from highly effective modulator therapies like ETI demonstrated that CFTR rescue restored airway clearance within days following the initiation of therapy, but the biochemical mode of action of these compounds on pathological CF mucus remains unknown.
To elucidate these biochemical changes, we used wild-type (WT) and CFTR-knockout (CFTR-KO) Calu3 cells to demonstrate that lack of CFTR function alone was sufficient to reduce ASL pH and increase the concentrations of both MUC5AC and MUC5B. Using the opposite approach, CFTR function was rescued in CF human bronchial epithelial (HBE) cells with VX-770 and ETI to study the impact of highly effective modulators on ASL pH, mucin concentration, polymeric network organization, and mucociliary transport.
Materials and Methods:
For full materials and methods see supplementary materials. Calu3 cells were genetically modified using CRISPR-Cas9 to knock-out CFTR [17]. Primary CF HBE cells were obtained from lung transplants from patients heterozygous for G551D or homozygous for F508del mutations, and CFTR function was restored using VX-770 and ETI, respectively [18]. Lack and rescue of CFTR function was confirmed by short-circuit current (Isc) measurements. Gene expression was assessed by qRT-PCR [19]. Cell washings were analyzed by western blotting (WB) to determine the relative changes in MUC5AC and MUC5B content and by light scattering/refractometry (SEC-MALS) for total mucin concentration as described previously [20, 21]. ASL pH was measured using a microprobe in a temperature/CO2 controlled chamber. Mucociliary transport was measured by video tracking, and mucus biophysical properties were analyzed using particle tracking microrheology (PTM) as previously described [12, 22]. Mucus network ultrastructure was analyzed via SEM.
Results:
Loss of CFTR function Affects ASL pH and Mucin Concentration
CFTR pharmacologic inhibition by adding CFTRinh172 or GlyH-101 to the apical side of the cells is commonly utilized for bioelectrical measurements. However, this approach is known to hydrate and crosslink the extracellular mucus due to the presence of fluid and/or DMSO, leading to misinterpretation of findings [23]. Hence, the use of a CFTR-KO cell line allowed for direct comparison of cells with functioning and defective CFTR in the absence of pharmacologic treatments. We utilized Calu3 cells, a lung-derived adenocarcinoma cell line normally expressing CFTR and producing MUC5AC and MUC5B, in which the CFTR gene was knocked out using CRISPR-Cas9 to analyze the effects of CFTR loss of function on mucus properties [17].
Measurements of mRNA levels showed a significant knockdown of the CFTR gene in CFTR-KO cells, while MUC5B and MUC5AC gene expression remained unchanged (Figure S1A). Complete knockout of CFTR function was confirmed by absence of forskolin or CFTRinh172 responses in transepithelial Isc measurements (Figure 1A). One of the characteristics of CF airways is lower ASL pH that has been shown to impair bacterial killing and affect mucus biophysical properties [7, 22, 24]. An ASL pH measurement assay using a pH microprobe in a temperature- and CO2-controlled chamber showed that CFTR-KO Calu3 cells exhibited a lower ASL pH than WT cells (~0.25 pH unit) and confirmed that CF HBE secretions were significantly more acidic than non-CF controls (~0.2 pH unit) (Figures 1B and S2).
Figure 1. Loss of CFTR function affects ASL pH and mucin concentration in CFTR-KO Calu3 cells.
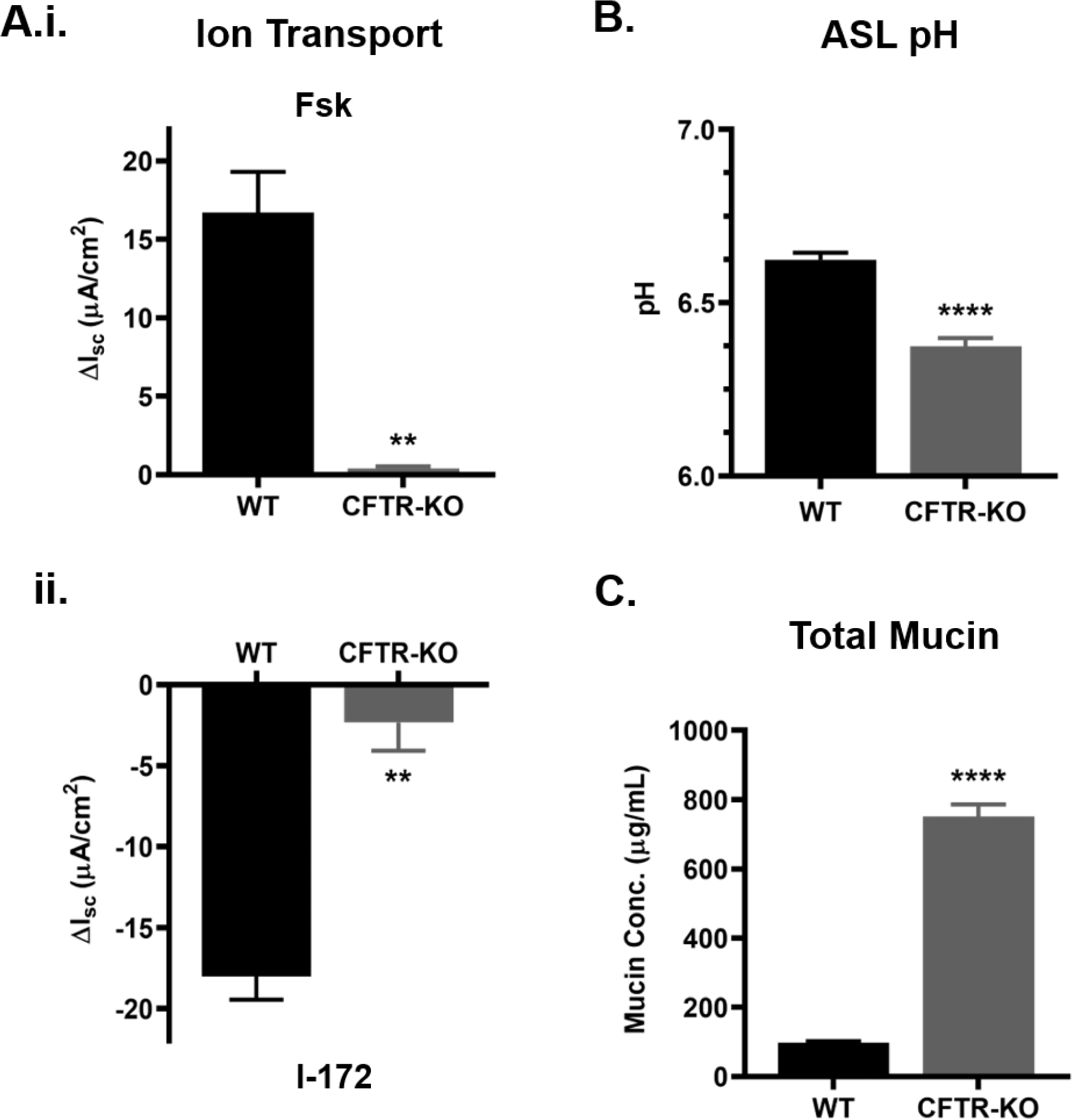
CFTR gene was knocked out in Calu3 cells via CRISPR-Cas9 to generate the CFTR-KO cell line. Wild-type (WT) and CFTR-KO Calu3 cells were analyzed via different assays. A.) Bioelectrical measurements of CFTR-mediated Isc for WT and CFTR-KO Calu3 cells showing cell response to forskolin (Fsk) (i.) and CFTR inhibitor (I-172) (ii.). n=3 per group **P<0.01, Student’s t-test B) ASL pH of WT and CFTR-KO cells measured by microprobe under CO2 and temperature-controlled conditions, n=5 per group. ****P<0.0001, Student’s t-test C) Absolute extracellular mucin concentration from WT and CFTR-KO apical washings quantified by SEC-MALS. n=4 per group. ****P<0.0001, Student’s t-test. Error bars indicate SEM.
To investigate the effect of CFTR function on mucin concentration, 30 min PBS washings from Calu3 cells were analyzed by SEC-MALS, CFTR-KO cells showed a 7.7-fold increase in total mucin concentration compared to WT cells (Figure 1C). In parallel, western blotting was used to quantify the relative change in MUC5AC and MUC5B content, and CFTR-KO cells showed a 2.4-fold increase in MUC5AC and a 4-fold increase in MUC5B signal compared to WT (Figure 2A–B). When observed by SEM, the mucin network ultrastructure was altered in CFTR-KO cultures compared to the WT meshwork (Figure 2C). The CFTR-KO polymeric network appeared more compact and exhibited numerous entanglements. Histological sections revealed the presence of a thicker mucus layer in the CFTR-KO compared to WT Calu3 cells, which correlated with a reduced intracellular signal, suggesting intense goblet cell degranulation in response to CFTR dysfunction (Figure S3).
Figure 2. Elevated mucin concentration in CFTR-KO Calu3 cell supernatant correlates with a tighter mucin network.
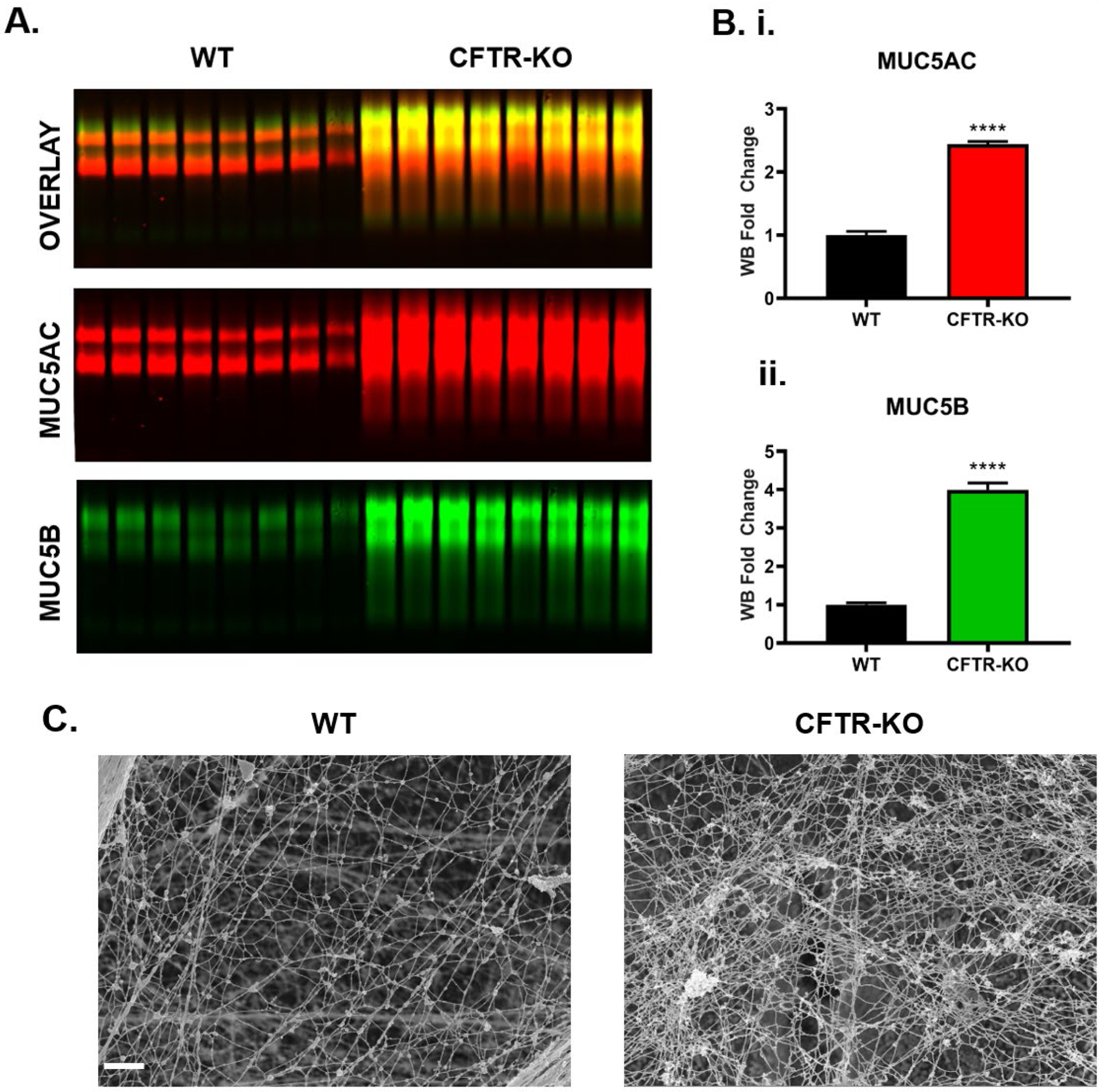
A.) Mucin western blot of apical washings reveal differences in MUC5AC (red) and MUC5B (green) signals in CFTR-KO compared to WT Calu3 cells. B.) Graphs showing average signal intensities of immunoblots of supernatants from CFTR-KO signal intensity normalized to WT for i) MUC5AC and ii) MUC5B. n=8 per group. ****P<0.0001, Student’s t-test. C.) Representative SEM images of a thin layer of extracellular mucus, revealing the network ultrastructure in WT and CFTR-KO Calu3 cultures, Scale bar = 1 μm. Error bars indicate SEM.
CFTR rescue in G551D HBE cells reverses mucus properties
Ivacaftor (VX-770) has been reported to restore CFTR function in cells with a G551D gating mutation [14]. To assess the effects of CFTR rescue on airway cells in vitro, we utilized primary CF HBE cells heterozygous for the G551D gating mutation to study the effects of the potentiator VX-770 on airway mucus. Bioelectrical measurements confirmed that CFTR function was efficiently restored by VX-770 (Figure 3A). Similar to CF Calu3 cells, mucin gene expression was not affected by CFTR rescue (Figure S1B). As shown in Figure S2, ASL pH from CF cultures was slightly more acidic than non-CF cells (6.9 vs 7.1, respectively). Remarkably, CFTR potentiation did not cause significant alkalization of the ASL pH (6.89 vs 6.92 for DMSO- and VX-770-treated cultures, respectively) but reduced significantly (~1.8-fold) the absolute concentration of airway mucins in apical washings (Figure 3B–C), which correlated with a relaxation of the mucin network (Figure 3D). Compared to vehicle-treated cells, the mucus coating VX-770-treated cells displayed a looser appearance with decreased branching and increased mesh size (0.1 vs. 0.5 μm for the largest pores).
Figure 3. Treatment of G551D HBE cells with VX-770 reverses aberrant mucus properties.
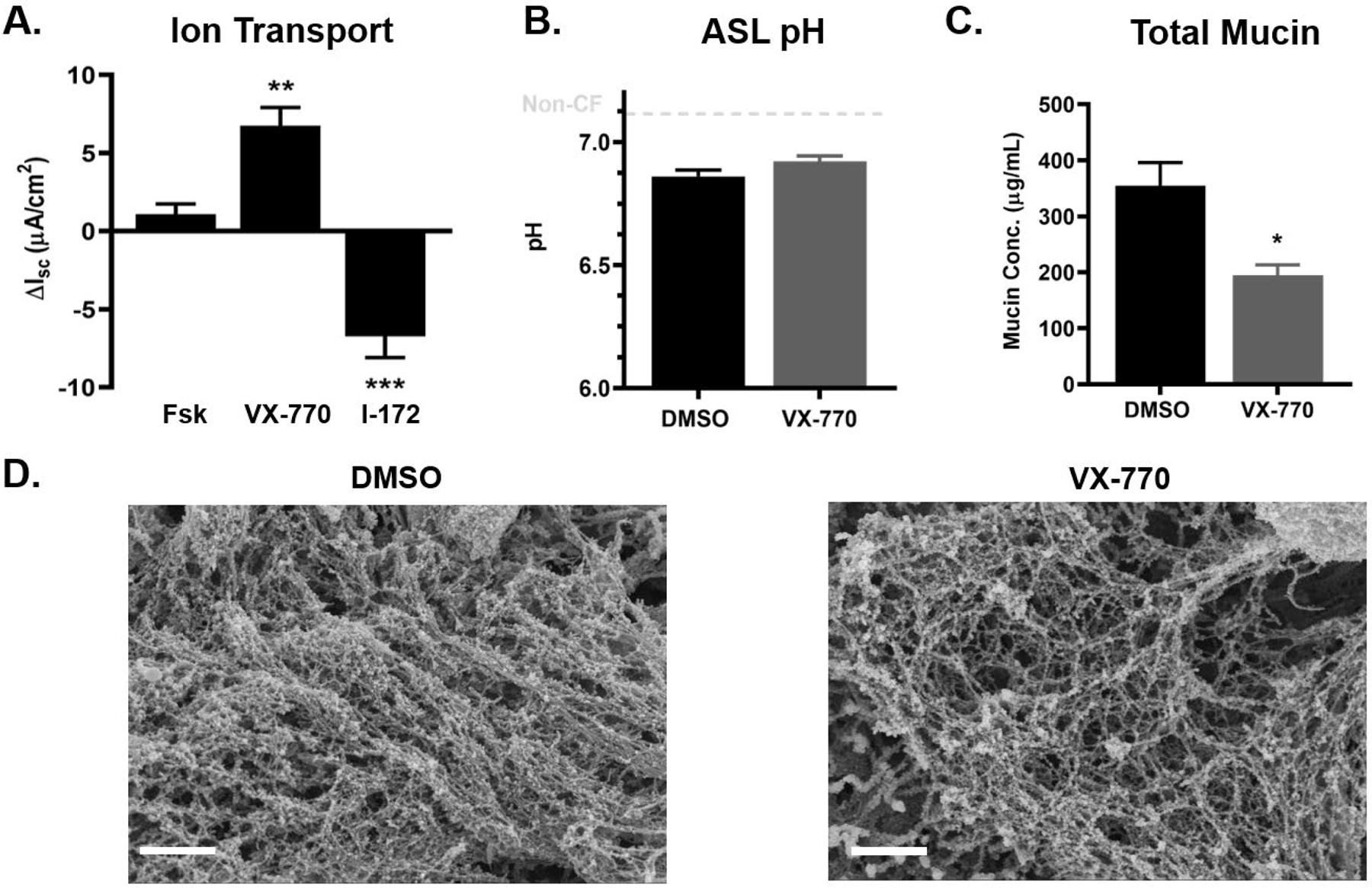
Primary HBE cells with one copy of the G551D mutation were treated with 0.05% DMSO or 5μM VX-770 acutely at the time of experimentation. A.) Bioelectrical measurements of CFTR-mediated Isc for G551D HBE cells treated acutely with VX-770 and CFTR inhibitor (I-172). n=3 inserts per group **P<0.01, ***P<0.001, Student’s t-test. B.) ASL pH of DMSO and VX-770-treated cells measured by microprobe under CO2 and temperature-controlled conditions. n=6 per group. C.) Absolute mucin concentration in apical washings as measured via SEC-MALS. n=6 per group. *P<0.05, Student’s t-test. D.) Representative SEM micrographs of the apical mucin network in DMSO- (left) and VX-770-treated (right) G551D cells. Scale bar = 1μm. Error bars indicate SEM.
ETI corrects F508del CFTR in vitro and affects mucus:
Due to limited access to G551D cells and the recent availability of ETI for research, we used F508del HBE cells for further experimentation. In Ussing chamber assays, we confirmed that ETI treatment of F508del HBE cells restored CFTR-mediated short-circuit currents (Figure 4A). Consistent with G551D cells, CFTR modulation in F508del cells did not alter ASL pH (6.85 vs 6.84 in DMSO- and ETI-treated cultures, respectively) but significantly reduced total mucin concentration in apical washings (Figure 4B–C). Mucin mRNA transcript levels remained unchanged following ETI treatment (Figure S1C).
Figure 4. ETI treatment of F508del cells restores CFTR function and affects mucin concentration but not ASL pH.
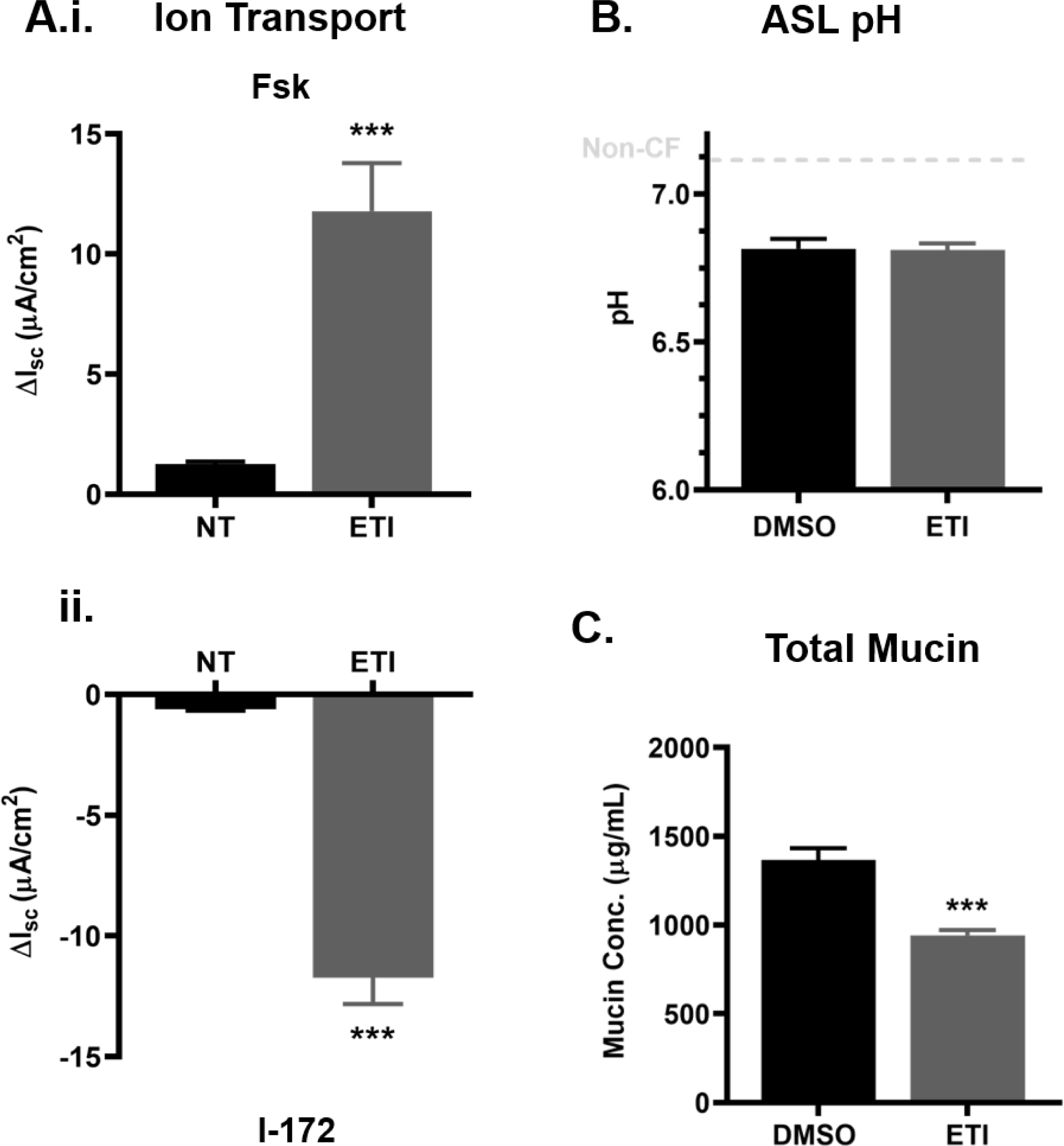
Primary HBE cells homozygous for F508del were treated for 3 days with 0.06% DMSO or 3μM VX-661, 2μM VX-445 and 1μM VX-770 (ETI) before experimentation. A.) Bioelectrical measurements of CFTR-mediated Isc for non-treated (NT) and ETI-treated F508del cells showing cell response to forskolin (Fsk) (i.) and CFTR inhibitor (I-172) (ii.). n=3 per group **P<0.01, Student’s t-test. B.) ASL pH of DMSO and VX-770-treated cells measured by microprobe under CO2 and temperature-controlled conditions. n=6 per group. C.) Absolute mucin concentration in 30 min apical washings as measured via SEC-MALS. n=6 per group. ***P<0.001, Student’s t-test. Error bars indicate SEM.
In previous work, it was shown that increases in mucin concentration correlated with increased mucus % solids content and reduced mucociliary clearance in CF lungs [11, 26]. Following 3 days of ETI treatment, mucus % solids decreased from ~9% to ~3% total solids (or 8 to 2% organic content) (Figure 5A). Mucus stasis was observed in non-treated F508del HBE cells, but following ETI treatment the average mucus velocity increased from 5 to 143 μm/sec (Figure 5B, videos 1–8). Increased mucus transport correlated with a slight but significant increase in ciliary beat frequency (11.7 vs 14.3 Hz in NT and ETI groups, respectively) (Figure 5C). When examined by SEM, DMSO- and ETI-treated cells revealed striking differences (Figure 5D and Figures S4–5). In DMSO-treated cells, large sheets of mucus (>200 μm2) covered the cell surfaces and compressed the cilia. At high magnification, cilia appeared disorganized, flattened, and mucus was seen compressing cilia shafts. In contrast, in ETI-treated cells post-fixative wash, the apical surfaces were nearly devoid of mucus and the cilia were erect and well-organized.
Figure 5. Effects of ETI treatment on mucus % solids, MCT velocity, and CBF in F508del HBE cells.
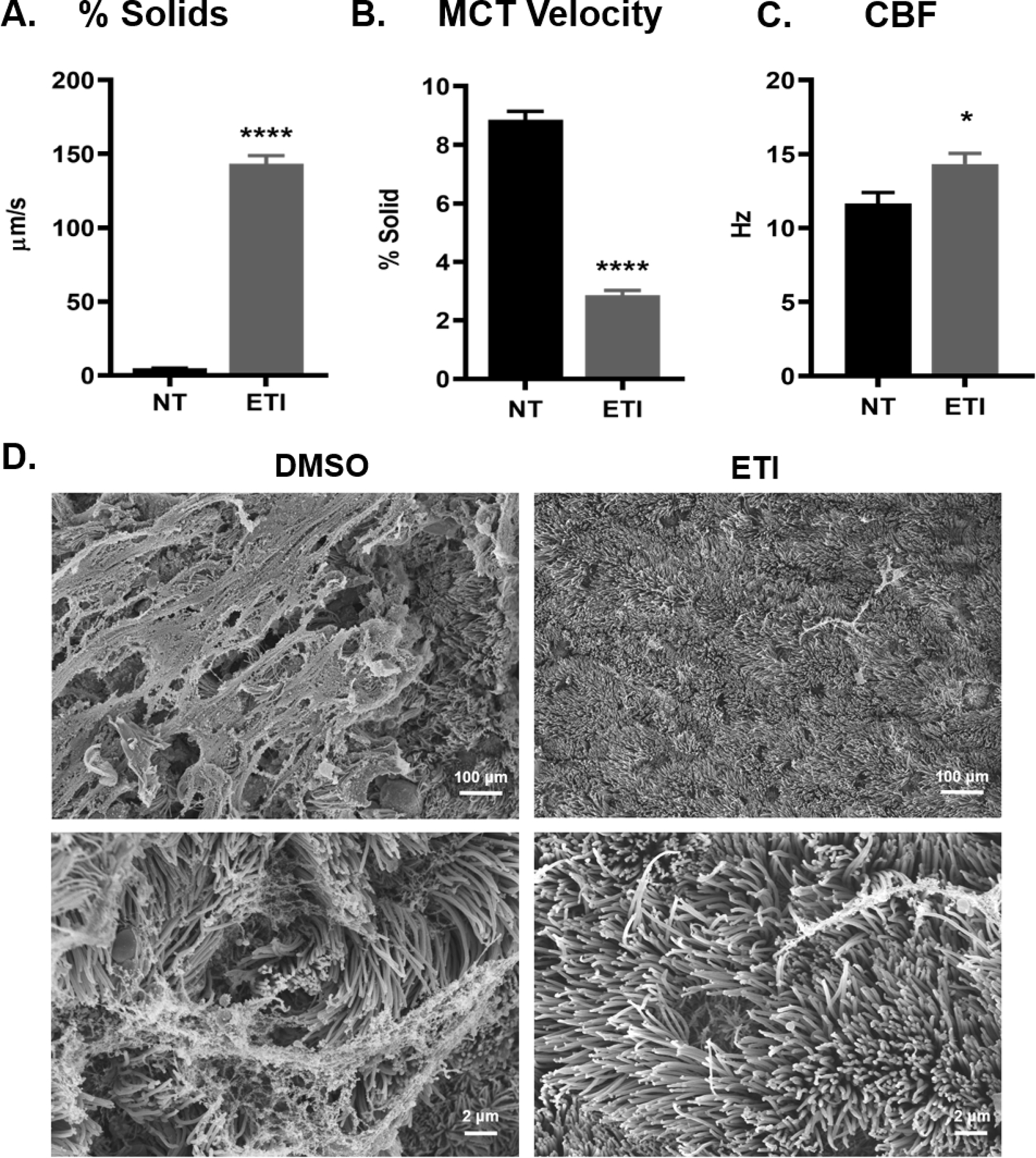
A.) Percent solids (% solids) of apical mucus was measured as wet/dry ratio before and after 3-day ETI treatment, n=3–6 per group. **** P<0.0001, Student’s t-test. B.) Mucus velocity rates were measured by video tracking before and after a 3-day ETI treatment, n=4 inserts per group. **** P<0.0001, Student’s t-test. C.) CBF was measured by video tracking for non-treated and ETI treated cultures, n=9 inserts per group. D.) Representative SEM micrographs of DMSO- or ETI-treated F508del HBE cells showing an apical view of the inserts at low (above) and high (below) magnification power. Error bars indicate SEM.
Mucus removal is facilitated by ETI treatment in F508del cells:
Apical washings from DMSO- or ETI-treated F508del cells were collected via a short (15 min) wash with PBS followed by additional 15 min wash with 1mM TCEP, a reducing agent, to completely remove mucus from the cell surfaces and establish the proportion of residual mucus post-washings [27]. Washings were then subjected to western blotting (Figure 6A). Lane intensity measurements indicated that the short PBS wash removed 33% and 60% more MUC5AC and MUC5B from ETI- vs DMSO (vehicle)-treated cells, respectively (Figure 6B.i.).
Figure 6. Western blot analysis of MUC5AC and MUC5B collection by short washings in DMSO- and ETI-treated cells, followed by washings with a reducing agent.
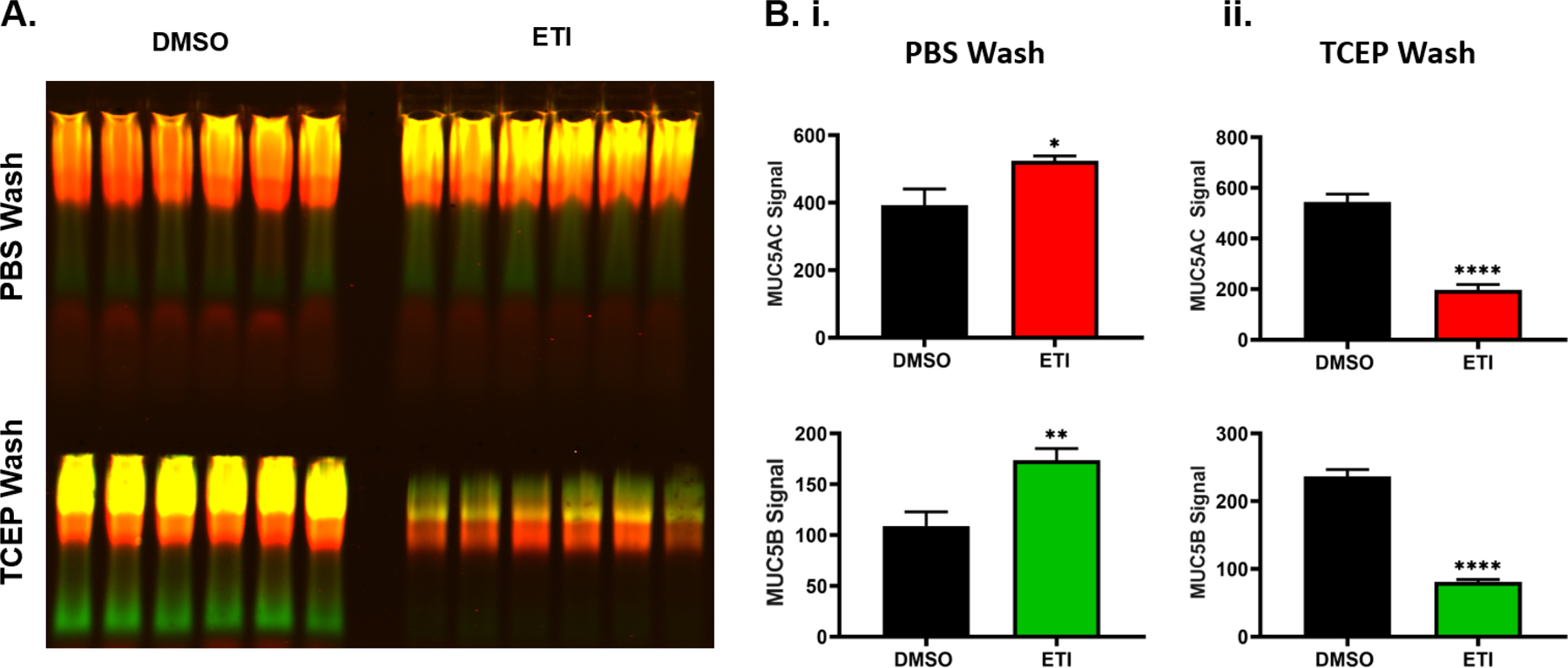
Following treatment for 3 days with 0.06% DMSO or 1μM VX-770, 2μM VX-445, and 3μM VX-661 (ETI), the apical surface of F508del HBE cells were washed with PBS for 15 min followed by an additional 1mM TCEP wash for 15 min. A.) Mucin western blot showing harvested MUC5AC (red) and MUC5B (green) by PBS (top) and additional 1mM TCEP (bottom) washings in DMSO- or ETI-treated cells. To stop the reaction, reduced samples were quenched with 1 mM iodoacetamide after 15min, n=6. B.) Graphs showing the intensity analysis of MUC5AC (top) and MUC5B (bottom) signals for PBS wash (i.) and additional TCEP wash (ii.). n=10. * P<0.05, ** P<0.01, **** P<0.0001, Student’s t-test. Error bars indicate SEM.
To test whether mucus remained on cell surfaces post-short washing, western blots were performed on the TCEP washes. Intensity analysis revealed that the majority of the mucus (~62%) remained adhered to the cells in the DMSO group post-PBS wash vs ~30% in the ETI group (Figure 6B.ii.). These results are consistent with the SEM images in Figure 5.
Hydration, but not pH or HCO3−, affects mucin recovery in CF cells:
To assess the major biochemical change(s) that mediated the efficient wash-induced mucus removal in modulator-treated cells, we washed non-treated F508del HBE cells with different solutions to determine which, if any, solution would result in removal of mucus from cell surfaces similar to ETI-treated cultures. Three of the dominant CF-related hypotheses were tested: acidic airway pH causing new/aberrant mucin interactions; lack of HCO3− secretion resulting in mucin compaction; and airway dehydration causing mucin polymer entanglement [1]. To test for the effects of volume, pH and/or [HCO3−], PBS wash buffers were delivered neat or alkalized with NaOH or NaHCO3 by 0.3 pH units to non-CF levels (see Figures 1 and S2) and administered for 15 min. To test for mucin disentanglement kinetics, normal PBS and Krebs-Ringers (containing 25 mM NaHCO3) were administered for 1h and 3h washings, respectively. Mucin western blot analysis revealed that washing with PBS alone or PBS alkalized with NaOH or NaHCO3 did not normalize mucus removal to ETI levels (Figure 7). However, long (1h) and extended (3h) washings of CF cultures with PBS alone or buffered with KBR normalized both MUC5AC and MUC5B mucin recovery to ETI levels. Extended washes removed 94% of MUC5AC and 83% of MUC5B of the total mucins present on CF cell cultures (Figure S7). Together these results revealed that hydration is sufficient to release mucus in modulator-treated cultures.
Figure 7. Hydration therapy, but not pH correction or bicarbonate addition, normalizes mucus to modulator-treated levels in F508del HBE cells.
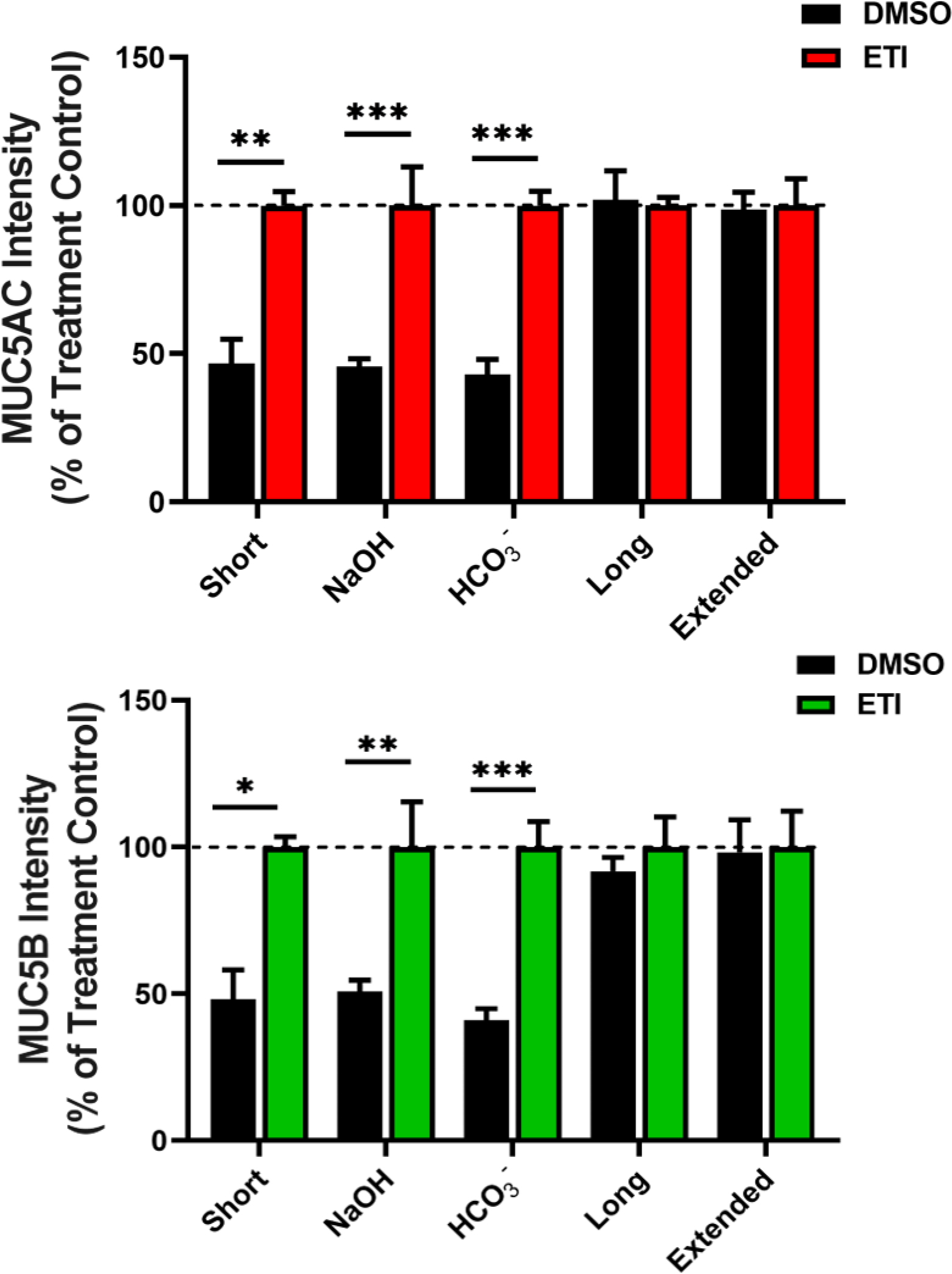
Following treatment for 3 days with 0.06% DMSO or 1μM VX-770, 2μM VX-445, and 3μM VX-661 (ETI), the apical surface was washed with adjusted buffers. Western blot quantification of MUC5AC (top graph) and MUC5B (bottom graph) from DMSO- and ETI-treated cultures washed for 15 min with PBS (Short), PBS alkalized by 0.3 pH unit with NaOH (OH−) or with NaHCO3 (HCO3−) or for 1h with PBS (Long) or 4h with Krebs-Ringers (Extended). n=4–6 per group. * P<0.05, ** P<0.01, *** P<0.001, Student’s t-test. Error bars indicate SEM.
To further test whether hydration normalizes the biophysical properties of CF mucus, DMSO- and ETI-treated cell washings were analyzed by particle tracking microrheology (PTM). From Figure 6 results, we hypothesized that washes from untreated F508del CF cells harvested only a small fraction of the total mucus mostly comprised of readily-soluble, non-adherent mucins. In contrast, washes from ETI-treated cultures contained a larger soluble fraction of total mucus (Figure 8A–B). PTM measurements of the complex viscosity (η*) of particles in washes revealed bimodal profiles with low and high viscosity peaks (Figure 8C). Using a Gaussian mixture model (see [28]), a hierarchical clustering of bead/particle movements was established for the “thin” (i.e., fluid) and “thick” components of mucus. In short-wash DMSO samples, both peaks were in sol-like phases (G’ < G”), while in ETI samples, the thick peak displayed a gel-like profile (G’ ≥ G”) (Figure 8 and S8) [12]. In long-wash samples, the rheological profiles in both control and ETI-treated groups were qualitatively similar and exclusively gel-like, confirming that sustained hydration effected the biophysical properties of control mucus similarly to modulator treatment. In addition, we showed that extended (3h in KRB) washes showed a similar rheological profile than long (1h in PBS) washes, revealing two peaks slightly shifted towards the gel-like phase, and that TCEP treatment homogenized and markedly shifted the samples towards a sol-like phase (Figure S9).
Figure 8. Short and long cell washings from DMSO- and ETI-treated F508del HBE cells analyzed by particle tracking microrheology (PTM).
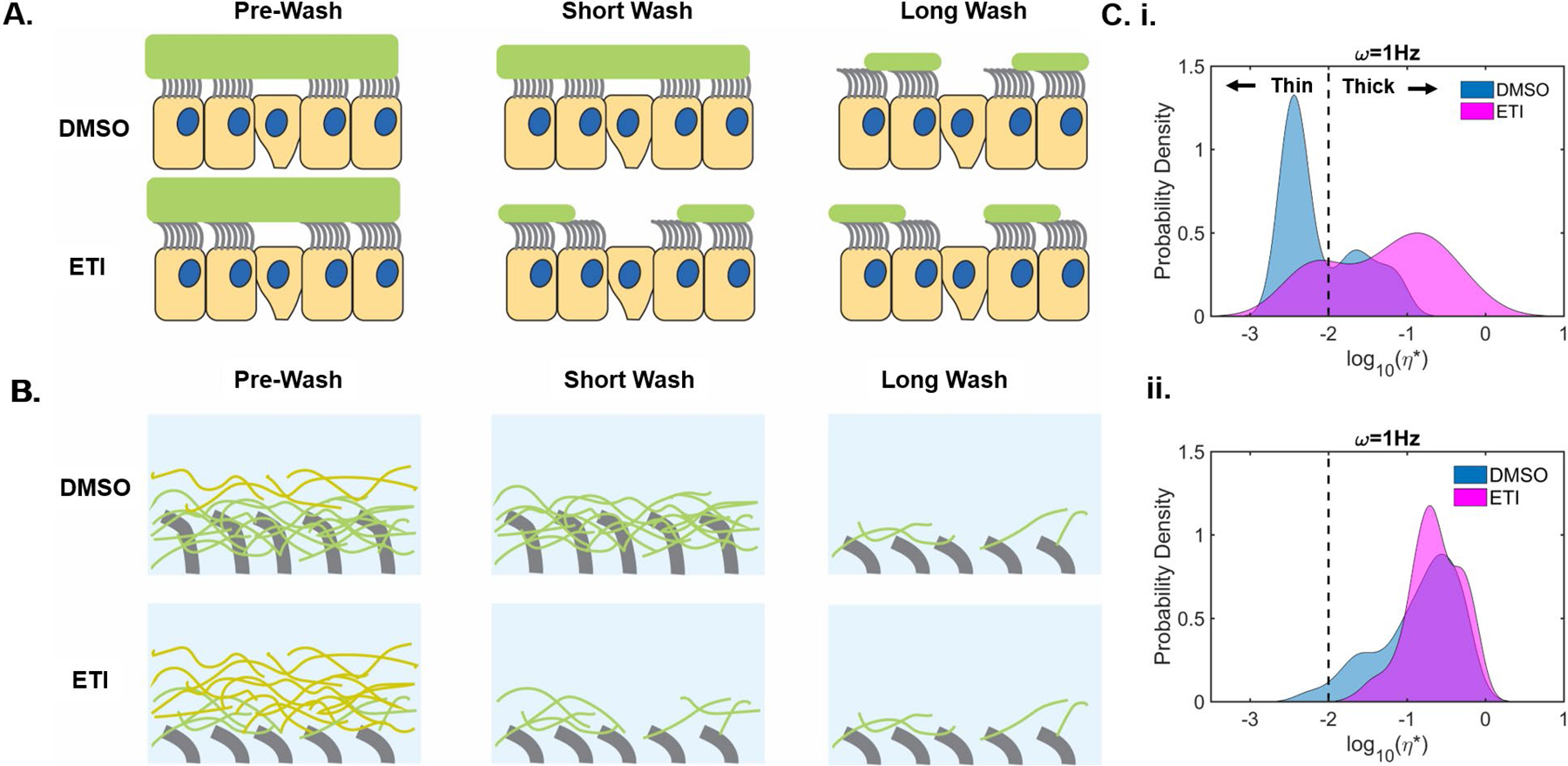
Following a 3-day treatment with 0.06% DMSO or 1μM VX-770, 2μM VX-445, and 3μM VX-661 (ETI), F508del CF HBE cells were washed with PBS for 15 min (short) or 1h (long) before PTM analysis. A.) Large-scale schematic view summarizing the effects of short and long washes on the mucus layer in DMSO- and ETI-treated cell cultures. B.) Small-scale schematic view of the periciliary and mucus layers in DMSO- and ETI-treated cultures following short and long washes. C.) Histograms of complex viscosity (ƞ*) analyzed via PTM, revealing bead probability density of short (i.) and long apical washings (ii.). Quantitative rheological behavior of short cell washings were clustered into two distinct modes of viscoelastic behavior (“thin” and “thick”) separated by a dotted line. n=6 inserts/group.
Discussion:
The recent development of effective modulator therapies has highlighted the role of corrected CFTR in the improvement of airway mucus clearance, but the underlying mechanism by which modulator compounds “reverse” mucus defects have remained unclear. We examined the three prevailing hypotheses (i.e., low pH, low [HCO3−], and dehydration). Our data indicate that dehydration dominates the biochemical changes occurring within the mucin network in response to CFTR malfunction.
Genetically modified cell lines and primary airway cells were used to investigate the effects of CFTR activation/inactivation on mucus in a milieu devoid of bacteria and inflammation. In vitro pH measurements on WT and CFTR-KO Calu3 cells, as well as non-CF and CF HBE cells, confirmed that in the absence of CFTR function ASL pH decreased by ~0.3 pH units (Figures 1 and S2). Previous studies have shown that environmental acidification affects mucus viscoelastic properties, but these changes occurred at 2–3 pH units lower than observed in airway mucus [8, 22]. Small changes (~0.5) in ASL pH, as measured in CF piglet samples, correlated with an increased mucus viscosity, but abnormal mucus clearance persisted in adult animals despite ASL pH normalization with age [7, 29]. Using G551D and F508del CF HBE cells, we restored CFTR function via modulator treatment, but ASL pH remained unchanged in treated cultures (Figures 3 and 4). The absence of pH change may reflect the fact that partial CFTR function does not fully restore HCO3− secretion or, more likely, that mucus can buffer small changes in pH as suggested by recent in vivo pH measurements in CF infants [30].
In contrast to pH regulation, we consistently showed that mucin concentration was affected by CFTR function. Mucin concentrations were high in association with CFTR dysfunction while CFTR rescue decreased mucin concentration (Figures 1, 3 and 4). Of note, neither CFTR dysfunction nor rescue affected mucin gene expression (Figure S1). Western blotting showed that both MUC5AC and MUC5B gel-forming mucin concentrations were affected by CFTR function (Figures 2 and 6). Increased mucin concentration may arise from an accelerated release rate of mucin granules but was not investigated in the study herein. In the absence of CFTR function, both mucins adhered to the cell surfaces, as evidenced by the compression of the periciliary layer by mucus and the need for a reducing agent to completely remove the mucus (Figures 5 and 6).
Increased mucin concentration in CF cultures correlated with a tightening of the mucin network ultrastructure, consistent with hyperconcentrated mucin (Figure 1 and 2). Inversely, CFTR rescue produced a relaxation of the mucin network in G551D cells treated with VX-770 (Figure 3). Compared to vehicle-treated cells, the mucus network in ETI-treated cells displayed a larger mesh size and was efficiently removed by short PBS washes or fixative submersion (Figures 5, 6, S4, S5 and S6). These physical changes were accompanied with a change in mucus solid contents from 8 to 2% organic solids before and after treatment (Figure 5). At >2% organic solids, the osmotic pressure of the mucus layer is greater than the osmotic pressure of the PCL, which causes the collapse of the cilia and abolishes mucus transport [11]. As shown in previous studies [27, 31], we observed mucus stasis in F508del cell cultures before treatment (Figure 5, videos 1–4). Following ETI treatment of the same cultures, mucus velocity was effectively restored at roughly normal rate (Figure 5 and videos 5–8) [32].
One of the most profound changes produced by modulator treatment was the facilitation of removal of adherent mucus from cell surfaces by short PBS washes. To explore genotype-agnostic therapies aimed at “reversing” abnormal mucus properties in CF, mucus collected by cell washing on ETI-treated cells was the reference standard for successful removal of mucus, and we tested in comparison various washing approaches to remove mucus from the surfaces of vehicle-treated F508del cells (Figure 7). We showed that short washes of PBS removed ~50% of mucus compared to the ETI group. Alkalization with or without HCO3− failed to normalize PBS-recovered mucus to ETI-treated levels. In contrast, sustained 1h hydration with PBS and extended 3h washings in Krebs-Ringers enabled mucus recovery at the same levels as the ETI-treated group. Hence, prolonged washes with Krebs-Ringer containing HCO3− had no additional effect compared to 1h PBS washes.
PTM assays confirmed that ETI and TCEP treatments affected the biophysical properties of mucus (Figure 8 and S8–9). Short washes on control CF cells showed that the majority of tracked particles signaled a very low viscosity solution, consistent with less mucus recovered in the lavage solution. By contrast, particles tracking in short ETI washes identified more beads in a higher viscosity environment, consistent with a higher mucin concentration. Long washes of both vehicle- and ETI-treated cultures produced bead profiles consistent with higher mucin concentrations that had sufficient time to fully swell. These data indicate that ETI treatment decreased the adherent interactions of mucins with the underlying epithelium, allowing mucins to be rapidly harvested by lavage.
In terms of hydration therapy, our data correlate with the failure of isotonic saline to restore MCC in CF patients and elucidates the limitations of short-acting osmotic therapies due to an absence of sustained hydration [33, 34]. In agreement with previous studies [27], treatment with reducing agent facilitated mucus removal (Figure 6) and may provide an additional clinical benefit option for patients who do not qualify for modulator therapy.
Although we showed that hydration significantly alters the mucin network in vitro, the presence of bacteria, inflammatory cells, and extracellular DNA in vivo may further impact mucus viscoelastic properties, which may affect the efficacy of modulator therapies. Despite their limitations, cellular models provide a controlled environment to study the direct effects of modulator treatment on mucus. Using cell models, we showed that CFTR dysfunction affected mucin concentration and ASL pH. Conversely, CFTR rescue facilitated mucus removal from the cell surfaces, and these biochemical and biophysical changes correlated with a change in mucin concentration but not ASL pH. Assessing novel therapies aimed to “reverse” abnormal mucus properties complement CFTR modulators, as mucin-based therapies can add to modulator therapy and be universal (i.e., independent of genotype and/or inflammation). Indeed, mucin-based therapies may also benefit other muco-obstructive diseases such as COPD and asthma.
Supplementary Material
Take-home message:
CFTR rescue failed to normalize acidic pH but reduced mucin concentration. Extended rehydration, not pH adjustment, facilitated mucus removal from cell surfaces, suggesting that mucus hydration is the dominant biochemical change in CF airways.
Acknowledgments:
Work in part supported by grants from Vertex Pharmaceuticals (Ehre RIA Award), the cystic fibrosis foundation (EHRE16XX1, MARKOV18F0, HILL19G0, and HILL20Y2-OUT, BOUCHE19R0), the NIDDK (2 P30 DK 065988–16) and NHLBI (5 P01 HL 108808–08). Special thanks to Victoria Madden from the Microscopy Services Laboratory at UNC, the Histology Core Department of Pathology and Laboratory Medicine at UNC, Scott Randell for providing cells through the UNC Tissue Procurement Core.
References
- 1.Morrison CB, Markovetz MR, Ehre C. Mucus, mucins, and cystic fibrosis. Pediatr Pulmonol 2019: 54 Suppl 3: S84–S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med 2013: 3(9): a009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Widdicombe JH, Wine JJ. Airway Gland Structure and Function. Physiol Rev 2015: 95(4): 1241–1319. [DOI] [PubMed] [Google Scholar]
- 4.Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol 2014: 52: 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdugo P Supramolecular dynamics of mucus. Cold Spring Harb Perspect Med 2012: 2(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 2008: 372(9636): 415–417. [DOI] [PubMed] [Google Scholar]
- 7.Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, Choudhury B, Varki A, Stoltz DA, Welsh MJ. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest 2016: 126(3): 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner CE, Turner BS, Rubinstein M, McKinley GH, Ribbeck K. A Rheological Study of the Association and Dynamics of MUC5AC Gels. Biomacromolecules 2017: 18(11): 3654–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med 2007: 13(6): 231–240. [DOI] [PubMed] [Google Scholar]
- 10.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, Sheehan JK, Boucher RC, Kesimer M. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest 2014: 124(7): 3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012: 337(6097): 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, Forest MG. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS One 2014: 9(2): e87681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson WH, Coakley RD, Button B, Henderson AG, Zeman KL, Alexis NE, Peden DB, Lazarowski ER, Davis CW, Bailey S, Fuller F, Almond M, Qaqish B, Bordonali E, Rubinstein M, Bennett WD, Kesimer M, Boucher RC. The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am J Respir Crit Care Med 2015: 192(2): 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009: 106(44): 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS, Group VXS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011: 365(18): 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, Plant BJ, Prais D, Ramsey BW, Taylor-Cousar JL, Tullis E, Uluer A, McKee CM, Robertson S, Shilling RA, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Rowe SM, Group VXS. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018: 379(17): 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hao S, Roesch EA, Perez A, Weiner RL, Henderson LC, Cummings L, Consiglio P, Pajka J, Eisenberg A, Yeh L, Cotton CU, Drumm ML. Inactivation of CFTR by CRISPR/Cas9 alters transcriptional regulation of inflammatory pathways and other networks. J Cyst Fibros 2020: 19(1): 34–39. [DOI] [PubMed] [Google Scholar]
- 18.Randell SH, Fulcher ML, O’Neal W, Olsen JC. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol 2011: 742: 285–310. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Ribeiro CMP, Sun L, Okuda K, Kato T, Gilmore RC, Martino MB, Dang H, Abzhanova A, Lin JM, Hull-Ryde EA, Volmer AS, Randell SH, Livraghi-Butrico A, Deng Y, Scherer PE, Stripp BR, O’Neal WK, Boucher RC. XBP1S Regulates MUC5B in a Promoter Variant-Dependent Pathway in IPF Airway Epithelia. Am J Respir Crit Care Med 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, Doerschuk CM, Alexis NE, Anderson WH, Henderson AG, Barr RG, Bleecker ER, Christenson SA, Cooper CB, Han MK, Hansel NN, Hastie AT, Hoffman EA, Kanner RE, Martinez F, Paine R 3rd, Woodruff PG, O’Neal WK, Boucher RC. Airway Mucin Concentration as a Marker of Chronic Bronchitis. N Engl J Med 2017: 377(10): 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsey KA, Rushton ZL, Ehre C. Mucin Agarose Gel Electrophoresis: Western Blotting for High-molecular-weight Glycoproteins. J Vis Exp 2016(112). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill DB, Long RF, Kissner WJ, Atieh E, Garbarine IC, Markovetz MR, Fontana NC, Christy M, Habibpour M, Tarran R, Forest MG, Boucher RC, Button B. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J 2018: 52(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan S, Hollinger M, Lachowicz-Scroggins ME, Kerr SC, Dunican EM, Daniel BM, Ghosh S, Erzurum SC, Willard B, Hazen SL, Huang X, Carrington SD, Oscarson S, Fahy JV. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med 2015: 7(276): 276ra227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB Jr., Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012: 487(7405): 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med 2005: 107: 183–206. [DOI] [PubMed] [Google Scholar]
- 26.Donaldson SH, Laube BL, Corcoran TE, Bhambhvani P, Zeman K, Ceppe A, Zeitlin PL, Mogayzel PJ Jr., Boyle M, Locke LW, Myerburg MM, Pilewski JM, Flanagan B, Rowe SM, Bennett WD. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight 2018: 3(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, Markovetz MR, Delion MF, Kato T, Villalon D, Thelin WR, Esther CR Jr., Hill DB, Grubb BR, Livraghi-Butrico A, Donaldson SH, Boucher RC. An Improved Inhaled Mucolytic to Treat Airway Muco-obstructive Diseases. Am J Respir Crit Care Med 2019: 199(2): 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esther CR Jr., Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, Ramsey KA, Markovetz MR, Garbarine IC, Forest MG, Seim I, Zorn B, Morrison CB, Delion MF, Thelin WR, Villalon D, Sabater JR, Turkovic L, Ranganathan S, Stick SM, Boucher RC. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med 2019: 11(486): eaav3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, Stoltz DA, Welsh MJ. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014: 345(6198): 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schultz A, Puvvadi R, Borisov SM, Shaw NC, Klimant I, Berry LJ, Montgomery ST, Nguyen T, Kreda SM, Kicic A, Noble PB, Button B, Stick SM. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat Commun 2017: 8(1): 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998: 95(7): 1005–1015. [DOI] [PubMed] [Google Scholar]
- 32.Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, Dierksen G, Mazur M, Shastry S, Li Y, Watson JD, Smith AT, Schuster BS, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. A functional anatomic defect of the cystic fibrosis airway. Am J Respir Crit Care Med 2014: 190(4): 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye PT, National Hypertonic Saline in Cystic Fibrosis Study G. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006: 354(3): 229–240. [DOI] [PubMed] [Google Scholar]
- 34.Goralski JL, Wu D, Thelin WR, Boucher RC, Button B. The in vitro effect of nebulised hypertonic saline on human bronchial epithelium. Eur Respir J 2018: 51(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.