

SUMMARY
Nuclear actin has been elusive due to the lack of knowledge about molecular mechanisms. From actin-containing chromatin remodeling complexes, we discovered an arginine mono-methylation mark on an evolutionarily conserved R256 residue of actin (R256me1). Actin R256 mutations in yeast affect nuclear functions and cause diseases in human. Interestingly, we show that an antibody specific for actin R256me1 preferentially stains nuclear actin over cytoplasmic actin in yeast, mouse, and human cells. We also show that actin R256me1 is regulated by protein arginine methyl transferase-5 (PRMT5) in HEK293 cells. A genome-wide survey of actin R256me1 mark provides a landscape for nuclear actin correlated with transcription. Further, gene expression and protein interaction studies uncover extensive correlations between actin R256me1 and active transcription. The discovery of actin R256me1 mark suggests a fundamental mechanism to distinguish nuclear actin from cytoplasmic actin through post-translational modification (PTM) and potentially implicates an actin PTM mark in transcription and human diseases.
Graphical Abstract
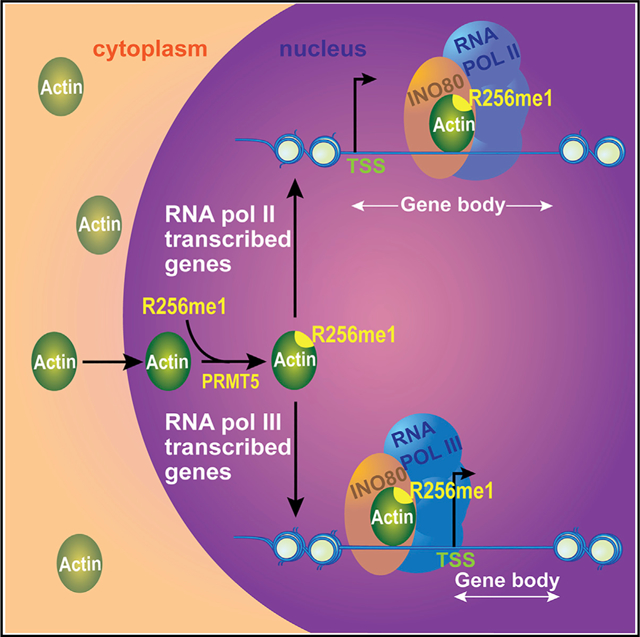
In Brief
Nuclear actin and actin PTMs are poorly understood. Kumar et al. discover a system of actin PTMs similar to histone PTMs, including a conserved mark on nuclear actin (R256me1) with potential implications for transcription and human diseases.
INTRODUCTION
Compared with the established cytoplasmic actin biology, nuclear actin remains poorly defined. Given that the same actin gene encodes both the cytoplasmic and nuclear actin, a fundamental question arises as to how nuclear actin distinguishes itself from cytoplasmic actin. For example, compounds that are commonly used to stain actin filaments in the cytoplasm, such as phalloidin, do not stain nuclear actin, despite the fact that the concentration of nuclear actin is sufficient to form stable filaments. These differences hinted that nuclear actin might have distinct biochemical and/or structural features compared with its cytoplasmic counterpart. One possibility is that nuclear actin may be specifically modified post-translationally, leading to changes in its biochemical and/or structural properties. Indeed, sumoylation of human β-actin has been suggested to regulate the transport of nuclear actin (Hofmann et al., 2009).
Actins are similar to histones in terms of their high evolutionary conservation and fundamental importance. However, compared to the wealth of knowledge on the mechanisms of histone post-translational modifications (PTMs), the mechanisms of actin PTMs are poorly understood despite the discovery of several actin PTMs (Terman and Kashina, 2013). To date, the prevailing paradigm in actin biology has been the regulation of diverse actin functions through specific actin-binding proteins (ABPs). As such, actin PTMs have not been widely considered as a major mode of actin regulation, and there have been only a few studies of actin PTMs since 1970s, such as N-terminal acetylation that occurs in many proteins, N-terminal arginylation, and a developmentally regulated tyrosine phosphorylation (pY53) in Dictyostelium (Abe et al., 2000; Baek et al., 2008; Berger et al., 1981; Howard et al., 1993; Karakozova et al., 2006; Kishi et al., 1998; Liu et al., 2006; Schweiger et al., 1992). We reasoned that actin PTMs might occur locally and in specific contexts. When bulk actin is examined, low levels of individual actin PTMs may not be easily detected. Interestingly, the actin-containing chromatin remodeling complexes such as INO80 allowed us to overcome this limitation by examining the specific pool of actin present in the nucleus. This defined system allowed us to reveal a distinct monomeric actin mechanism in the INO80 chromatin remodeling complex, which contrasts the established mechanism of cytoplasmic actin based on polymerization (Kapoor et al., 2013).
Excitingly, through mass spectrometry using specific actin pools, such as from INO80, we have identified a set of actin PTMs, similar to the histone PTMs. This finding led us to hypothesize that actin PTMs may also carry out context-specific functions, similar to the histone PTMs. As an initial proof-of-principle study for the actin PTM hypothesis, we focused our studies on actin R256 methylation, given the importance of R256 for nuclear functions in yeast, as well as the links between actin R256 (R258 in human α-actin isoforms) mutations and human diseases (Rubenstein and Wen, 2014). We show that the R256 residue of actin (R256me1) mark is present in all the three actin-containing chromatin-modifying complexes (INO80, SWR1, and NuA4) in yeast. Consistent with the nuclear role of actin R256me1, mutation of R256 in yeast actin lead to defective nuclear actin functions, without affecting the transport of actin into the nucleus. We show that the actin R256me1 mark is evolutionarily conserved in actins from different organisms and in different human actin isoforms. Strikingly, the actin R256me1 antibody stains mostly nuclear actin in various cell lines, providing a tool to track nuclear actin. Moreover, we identified that protein arginine methyl transferase-5 (PRMT5) is required for R256me1 in HEK293 cells, implicating R256me1 in transcription. A genome-wide analysis of actin R256me1 mark in yeast by using high-resolution chromatin immunoprecipitation (ChIP)-exo mapping showed that actin R256me1 mark is localized to the gene body compared to transcription start site (TSS) for the highly expressed genes transcribed by RNA polymerase II (RNA Pol II). However, for tRNA genes transcribed by RNA Pol III, actin R256me1 mark is localized directly upstream of the TSS. IP of actin using R256me1 antibodies followed by mass spectrometry analyses showed an association of actin with subunits of RNA polymerases. RNA sequencing (RNA-seq) analyses of R256 mutants of actin showed significant changes in transcription programming. Together, these studies suggest strong correlations of the actin R256me1 mark with active transcription.
Strikingly, R256 mutations (R258 in human α-actin isoforms) cause several human diseases. For example, in human smooth muscle (SM) α-actin encoded by ACTA2, R258 mutations cause thoracic aortic aneurysms and dissections (TAAD), along with an early onset of ischemic strokes due to a Moyamoya-like cerebrovascular disease (Guo et al., 2007, 2009). Analyses of cell lines derived from ACTA2 R258C patients showed a reduction in the R258me1 mark of SM α-actin, accompanied by distinct changes in gene expression profiles, raising possible connections between vascular diseases and nuclear actin PTMs. In addition to R256 methylation, analyses of other actin PTM sites also suggest a striking correlation of actin PTM sites with other causal mutations of ACTA2 in human vascular diseases.
The discovery of actin R256me1 mark suggests a fundamental mechanism of distinguishing nuclear actin through PTMs and provides a tool to track nuclear actin. Given the wide spectrum of actin PTMs similar to histone PTMs, as well as the evolutionary conservation of these actin PTM sites, the study of R256me1 may provide the foundation to establish actin biology through PTM regulations.
RESULTS AND DISCUSSION
Mass Spectrometry Studies of Distinct Actin Pools Reveal a System of Actin PTMs
To uncover the potential biochemical differences between nuclear actin and cytoplasmic actin, we analyzed nuclear-actin-containing yeast INO80 chromatin-remodeling complex using 2D gel electrophoresis. The 2D gel analysis revealed multiple spots of actin at different isoelectric points, similar to the Ies4 subunit of the INO80 complex (Figure 1A). Given that our previous studies of Ies4 2D gel spots led to the discovery that Ies4 is phosphorylated in response to DNA damage (Morrison et al., 2007), these results suggest that actin in the INO80 complex might also be post-translationally modified. To address this possibility, we isolated the actin band from purified INO80 complex separated on an SDS-PAGE gel and subjected it to mass spectrometry analyses (Figure 1B, step 1). Moreover, to test if actin PTMs are also present in cytoplasmic actin, we analyzed the actin pool purified from the cytoplasmic fraction and further expanded the spectrum of actin PTMs (Table S1).
Figure 1. Identification of Actin R256me1 from Yeast INO80 Complex.
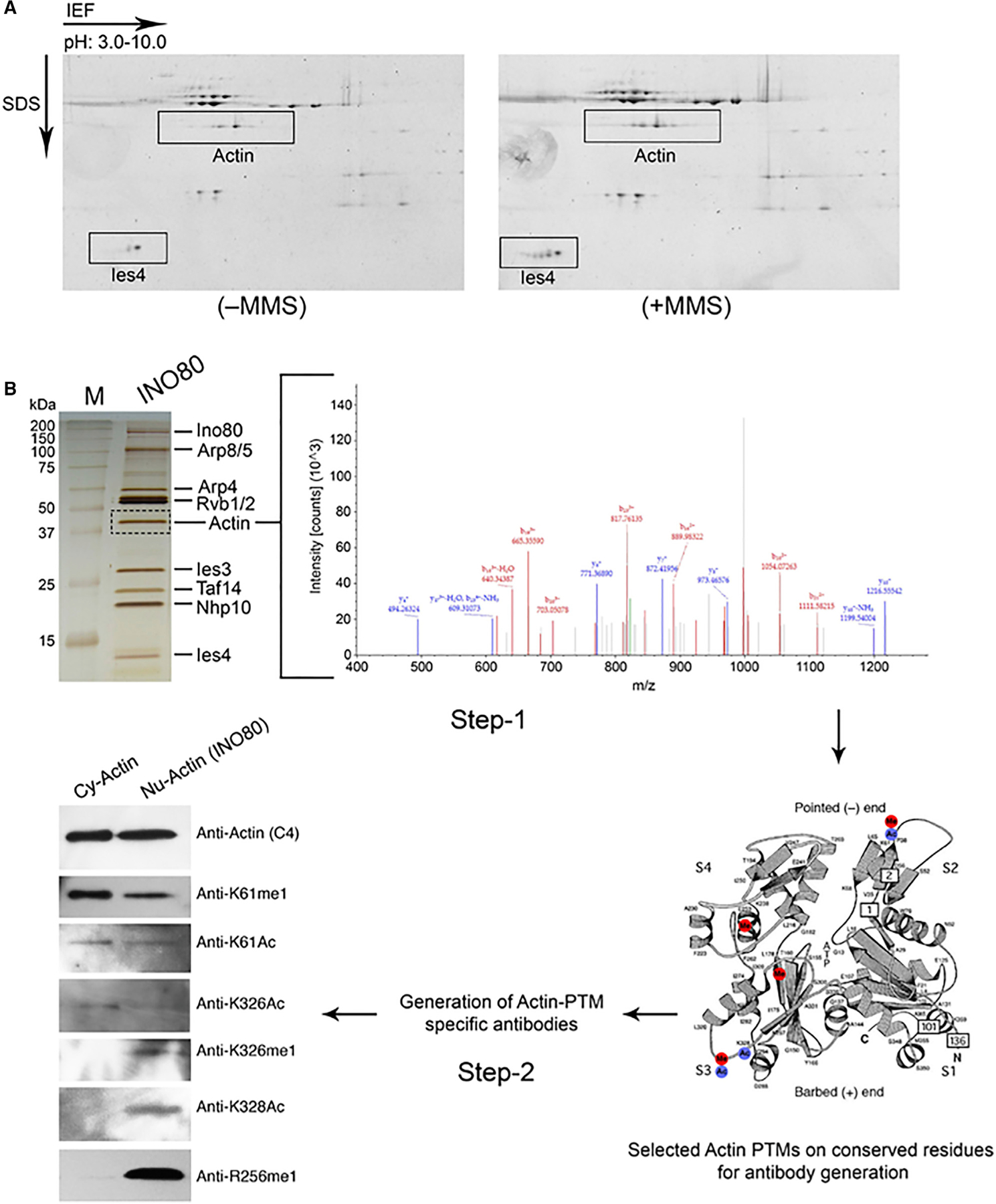
(A) 2D gel analysis using 3–10 pH immobilized pH gradient (IPG) strips of the INO80 complexes that were purified with 0.5 M KCl buffer from untreated (left panel) or MMS-treated (right panel) BY4733 strains. The actin subunits of the INO80 complex showing multiple spots are indicated in gels stained with Deep Purple.
(B) Step 1, SDS-PAGE analysis of INO80 complex after silver staining (left panel) indicating actin band that was gel sliced and subjected to mass spectrometry analysis (right panel) by using LC-MS/MS. Step 2, antibodies raised against specific actin PTMs identified through mass spectrometry analyses, followed by the western blot analyses of purified cytoplasmic actin (Cy-actin) and nuclear actin in the INO80 complex (Nu-actin), as indicated at the top, by using individual actin PTM antibodies separately, as indicated at right. Data presented are the representative of three independent biological repeats with similar results. Please also see Table S1.
Together, these analyses revealed 35 actin PTMs with high confidence (Table S1). These PTMs are located throughout the actin molecule and cover a wide spectrum similar to histone PTMs. These actin PTMs include lysine acetylation, methylation, ubiquitination or sumoylation, and arginine methylation. It should be noted that most methylation events detected are mono-methylation, possibly due to the dynamic nature of methylation. Overall, actin PTMs appear to fall into two categories. First, a set of actin PTMs shared between cytoplasmic and nuclear actins, such as K61me1, K61Ac, K326Ac, suggesting broad functions for these PTMs. Second, a subset of actin PTMs, which appear to be context specific, such as the nuclear-specific K326me1, K328Ac, and R256me1. Together, our results reveal that actin is subjected to a system of PTMs similar to histones and that some of these PTMs might be context specific. Strikingly, given the high evolutionary conservation of actin, most of the actin PTM sites are also conserved from yeast to human, suggesting that actin PTMs are likely conserved through eukaryotes, similar to histone PTMs.
Initial Survey of Actin PTMs through Specific Antibodies Implicates R256me1 as a Predominantly Nuclear Actin Mark
To begin testing the actin PTM hypothesis, we generated rabbit polyclonal antibodies against actin PTMs on several residues that are evolutionarily conserved by using individual post-translationally modified actin peptides, including those for K61me1, K61Ac, R256me1, K326me1, K326Ac, and K328Ac. To test the specificity of actin PTM antibodies, we first performed western blot analyses of actin in the INO80 complex (as a source of nuclear actin) and actin purified from the cytoplasmic fraction of yeast cells (as a source of cytoplasmic actin). As shown in Figure 1B (step 2), all actin PTM antibodies detected nuclear actin in the INO80 complex, consistent with our mass spectrometry analyses (Table S1). Excitingly, a subset of actin PTM antibodies including those for K326me1, K328Ac, and R256me1 detected nuclear actin from the INO80 complex only and showed no signal in the purified actin from the cytoplasmic fraction on the same blot (Figure 1B, step 2, bottom three western blot panels). These results were also consistent with our mass spectrometry analyses (Table S1) and confirm that nuclear actin has a set of actin PTMs distinct from cytoplasmic actin.
To test the actin PTM hypothesis, we selected the R256 methylation for further analyses as a proof-of-principle study. Two features make R256 methylation an ideal initial target: first, R256me1 appears to be context specific, as the R256me1 antibody primarily detects nuclear actin (Figure 2A); second, R256 is evolutionarily conserved and is implicated in several human diseases (Rubenstein and Wen, 2014). We first validated the specificity of the actin R256me1 antibody by using a peptide competition assay. Actin R256me1 antibody pre-incubated with an unmethylated (R256) actin peptide detected actin in the INO80 complex, whereas the signal was abolished when an actin R256me1 antibody was pre-incubated with the mono-methylated actin peptide (R256me1) (Figure 2B). These results confirm that the actin R256me1 antibody specifically recognized the actin R256me1 mark. Furthermore, when we mutated the actin R256 residue (R256C) in yeast cells, the actin R256me1 antibody no longer recognized the mutated actin in the cell extracts (Figure S1B). These results indicate that the actin R256me1 antibody is specific for the R256me1 mark.
Figure 2. Validation of Actin R256me1 Antibodies and Functional Studies of Actin R256 Residue.
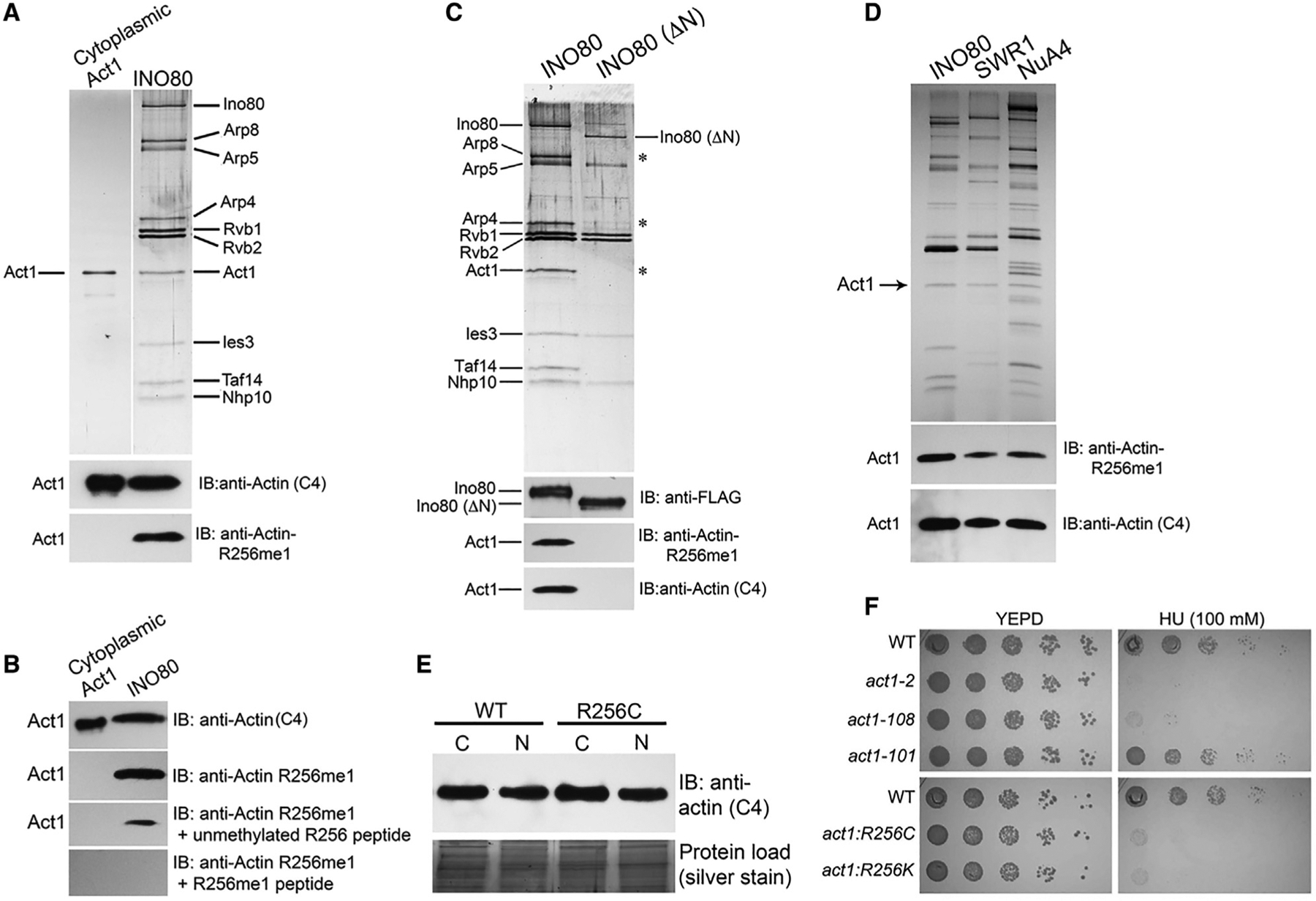
(A) Top panel, SDS-PAGE analysis of purified yeast cytoplasmic actin (Act1) and the INO80 complex after silver staining, as indicated at the top; and bottom two panels, the western blot analysis of a similar gel using antibodies as indicated..
(B) Western blot analysis of the purified cytoplasmic actin and nuclear actin containing the INO80 complex after peptide competition assay using actin (C4), and actin R256me1 antibodies as indicated..
(C and D) Top panels show the SDS-PAGE analysis after silver staining of the purified yeast INO80, ΔN INO80, SWR1, and NuA4 complexes, as indicated at the top of the gels; and bottom panels, the western blot analysis of a similar gel using antibodies as indicated..
(E) Top panel, western blot analyses of equal protein content of nuclear and cytosolic fractions of WT and R256C yeast strains using actin (C4) antibodies; bottom panel, silver-stained gel of the total protein content of the extract as loading control..
(F) Phenotypic analyses of actin mutants act1-2 (A58T), act1-108 (R256A E259A), act1-101 (D363A E364A), act1:R256C, and act1:R256K on YEPD, and HU (100 mM) plates at 30°C. Data presented are the representative of three biological repeats with similar results.
To further rule out potential artifacts, we purified the INO80 (ΔN) complex that lacks actin and actin-related proteins (Shen et al., 2003; Figure 2C, top panel). The western blot analyses of the INO80 (ΔN) complex using an actin R256me1 antibody did not detect actin in the INO80 (ΔN) complex (Figure 2C, bottom panel). These results further confirm the specificity of the actin R256me1 antibody. Moreover, the actin R256me1 antibody also detected nuclear actin in other actin-containing chromatin-modifying complexes such as SWR1 and NuA4 (Figure 2D), consistent with mass spectrometry results detecting the actin R256me1 mark in other chromatin-modifying complexes such as NuA4 (data not shown). Together, these results indicate that the actin R256me1 antibody is specific for the R256me1 mark in actin and suggest that the actin R256me1 mark is present in all known nuclear actin-containing chromatin-modifying complexes.
Actin R256 Mutation Affects Nuclear Actin Functions without Affecting the Transport of Actin into the Nucleus
We further analyzed the role of actin R256 residue in the nuclear transport of actin. As shown in Figure 2E, the western blot analyses of actin content in the nuclear and cytosolic fractions of R256C yeast strains compared to the wild-type (WT) strains showed no significant differences, suggesting that actin R256 methylation is not required for the transport of nuclear actin. We reasoned that if the actin R256me1 mark is functional in the nucleus, an actin R256 point mutation might disrupt nuclear functions. Curiously, we have previously performed genetic screens searching for actin mutants defective in nuclear functions (Kapoor et al., 2013). In this screen, we used the hypersensitivities of actin mutants to hydroxyurea (HU) and methyl methanesulfonate (MMS) as indicators of defective nuclear functions. Among a small set of actin mutants, including act1-2 (A58T) (Kapoor et al., 2013), we identified act1-108 (R256A E259A) that also showed similar nuclear defects as act1-2 (A58T), such as hypersensitivity to hydroxyurea at permissive temperature (30°C) (Figure 2F, top panel). Given that in the act1-108 mutant both R256 and E259 residues were mutated and that the effects of E259A in the nuclear functions of actin could not be ruled out, we generated actin R256K and R256C point mutants to analyze the effects that can be attributed solely to the R256 residue. Interestingly, both R256C and R256K mutants showed similar nuclear defects to those observed in act1-2 and act1-108 mutants (Figure 2F, bottom panel) (Kapoor et al., 2013). Moreover, the actin R256me1 mark in the R256C mutant was also abolished (Figure S1B). Furthermore, the observed nuclear defects in R256C point mutants were rescued by the transient expression of WT actin (Figure S1C). Together, these results suggest that the R256 residue is required for the nuclear actin functions and does not affect the transport of actin into the nucleus.
Actin R256me1 Is Conserved in Nuclear Actins of Yeast, Mouse, and Human Cells
Given that actin is evolutionarily conserved, similar to histones, and that the histone PTMs are well known to be conserved in different organisms from yeast to human (Fischle et al., 2003; Jenuwein and Allis, 2001), it is plausible that the actin R256 methylation might also exist in actins from other organisms. To address this, we first performed sequence alignment of actin residues around R256 in different actins. Our results suggest that the amino acid sequence around the actin R256 residue is indeed highly conserved in different actins and in human actin isoforms (Figures 3A and S1D). Given that the actin R256me1 antibody was raised against the peptide around the R256 residue of yeast actin (Act1) and the conservation of R256 in evolution, it is possible that this antibody can also detect the R256me1 mark in other actins. To test this possibility, we probed the whole-cell lysate from mouse fibroblast (NIH 3T3) and human embryonic kidney (HEK293) cell lines with the actin R256me1 antibody. Our results show that the actin R256me1 antibody detected actin, which is primarily β-actin, in the whole-cell lysates of NIH 3T3 and HEK293 cells (Figure 3B). A similar cross-reactivity among different organisms has been identified in conserved histone marks (Lin et al., 1989). Moreover, as shown in Figure 3C, the actin R256me1 antibody detected mostly nuclear actin from the nuclear fraction compared to the cytoplasmic actin from cytosolic fraction of mouse fibroblasts (NIH 3T3). To further confirm this finding, we performed immunostaining of NIH 3T3 cells by using the actin R256me1 antibody. Strikingly, as shown in Figure 3D, the actin R256me1 antibody selectively stained nuclear actin over cytoplasmic actin, suggesting R256 methylation as a conserved nuclear actin mark. A similar result was also observed with HEK293 cells (data not shown). It should be noted that a small amount of staining of cytoplasmic actin, particularly around the nucleus, was also detected by immunofluorescence microscopy, which may be either due to antibody background or the dynamic movement of a small fraction of actin to and from the nucleus.
Figure 3. Actin R256me1 Mark Is a Conserved Nuclear Actin Mark Regulated by PRMT5.
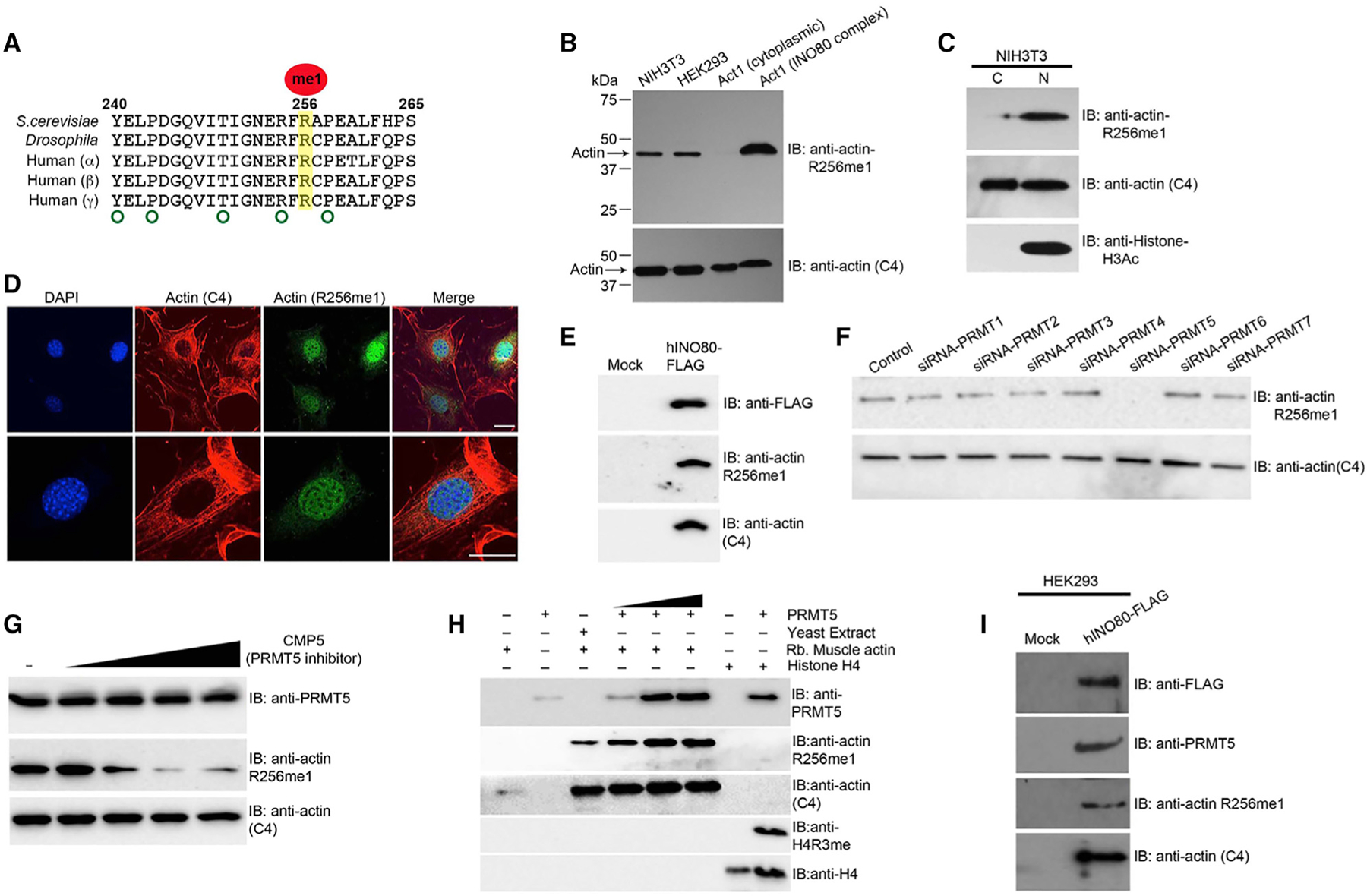
(A) Amino acid sequence alignment of actin homologs from different species showing high conservation of the R256 residue and sequence around it..
(B and C) Western blot analysis of the whole-cell extract, purified cytoplasmic actin, nuclear-actin-containing INO80 complex, cytosolic and nuclear fractions of NIH 3T3 cells using actin R256me1 antibodies, actin (C4) (loading control), and histone H3Ac (nuclear fraction marker), as indicated.
(D) Immunofluorescence confocal images of the NIH 3T3 cells, stained with actin (C4) and actin R256me1 antibodies, showing the preferential staining of nuclear actin over cytoplasmic actin by actin R256me1 antibodies. DAPI was used to stain the nucleus. Scale bar: 50 μm.
(E) Western blot analyses of the hINO80 complex purified from HEK293 cells after FLAG tagging hIno80 in the genome using antibodies as indicated.
(F and G) Western blot analyses of whole-cell extract of HEK293 cells transfected with siRNA against individual PRMTs as mentioned at the top, using actin (C4) (loading control) antibodies and actin R256me1 antibodies as indicated (F). Results shows that PRMT5 knockdown substantially affects actin the R256me1 level compared to the knockdown of other known PRMTs after adding the PRMT5 inhibitor (CMP5), using antibodies as indicated (G).
(H) Western blot analyses after in vitro methylation assay using purified recombinant PRMT5 and rabbit muscle actin (unmethylated actin substrate) using antibodies as indicated at the right. Actin (C4) is a loading control, and histone H4 methylation by PRMT5 was used as a positive control in the assay.
(I) Western blot analyses of hINO80 complex purified from HEK293 cells after FLAG tagging hIno80 in the genome using antibodies as indicated. Data presented are the representative of three independent biological repeats with similar results.
To further validate our results, we chromosomally FLAG-tagged the human INO80 gene in HEK293 cells by using CRISPR-Cas9 technique, followed by IP by using anti-FLAG M2 agarose beads with high salt washes (0.5 M KCl HEGN). The pull-down product was subjected to western blot analyses using the actin R256me1 antibody. As shown in Figure 3E, our results suggest that similar to yeast INO80, the human INO80 complex also contains an actin subunit with the R256me1 mark.
PRMT5 Is Required for Actin R256me1 in HEK293 Cells
Arginine methylation is known to be regulated by PRMTs. There are four known PRMTs in yeast (Low and Wilkins, 2012), whereas more than seven known PRMTs regulate arginine methylation in mammalian cells (Bedford and Clarke, 2009; Bedford and Richard, 2005). We have not been able to detect changes in actin R256me1 levels in yeast PRMT mutants, suggesting that either gene redundancy or a different PRMT is involved. In mammalian systems, we searched for actin R256me1 regulators by knocking down PRMTs in HEK293 cells individually using small interfering RNA (siRNA) and analyzing the levels of actin R256me1 in whole-cell extract by using the actin R256me1 antibody. Our results showed that knockdown of PRMT5 in HEK293 cells substantially affected the actin R256me1 level, whereas the knockdown of other PRMTs did not show significant effects (Figures 3F and S2). To further confirm our results, we used a cell-permeable specific inhibitor of PRMT5 activity, CMP5 (Alinari et al., 2015), on HEK293 cells and analyzed actin R256me1 levels in whole-cell extract. Consistent with the knockdown results, the inhibition of PRMT5 activity by using CMP5 also showed the substantial loss of actin R256me1 mark in HEK293 cells (Figure 3G). To validate our in vivo observations, we tested the in vitro activity of PRMT5 on unmethylated actin substrate (rabbit muscle actin) by using purified components. As shown in Figure 3H, the addition of purified PRMT5 on unmethylated substrate rabbit muscle actin led to the appearance of actin R256me1 mark in a concentration-dependent manner. Interestingly, in the hINO80 complex purified from HEK293 cells, which contains the actin R256me1 mark, we found that PRMT5 is also tightly associated with the hINO80 complex (Figure 3I). Together, these results suggest that PRMT5 is a key regulator for the actin R256me1 mark.
The Landscape of Nuclear Actin and Protein Interactions Correlates Actin R256me1 Mark with Active Transcription
Although nuclear actin has been linked to all three classes of RNA polymerases and is a subunit of several chromatin-modifying complexes, how nuclear actin is distributed in the genome remains poorly defined. Given that actin R256me1 is a conserved actin-PTM present in all the actin-containing chromatin-modifying complexes in yeast (Figure 2D), it offered an opportunity to survey the landscape of nuclear actin in the genome. To address the genome-wide distribution of actin R256me1, the Saccharomyces cerevisiae genome was surveyed for actin R256me1 at near-base-pair resolution using ChIP-exo (Rhee and Pugh, 2011). An example of its binding at the CCW12 gene is shown in Figure 4A, in which it follows the qualitative pattern of RNA Pol II. However, actin R256me1 showed different magnitudes of enrichment compared to Pol II, depending on its genic location. Nevertheless, this result is consistent with early studies for which nuclear actin has been shown to be associated with the RNA-pol-II-transcribed genes (Obrdlik et al., 2008).
Figure 4. Genome-wide Distribution of Actin R256me1 in Yeast.
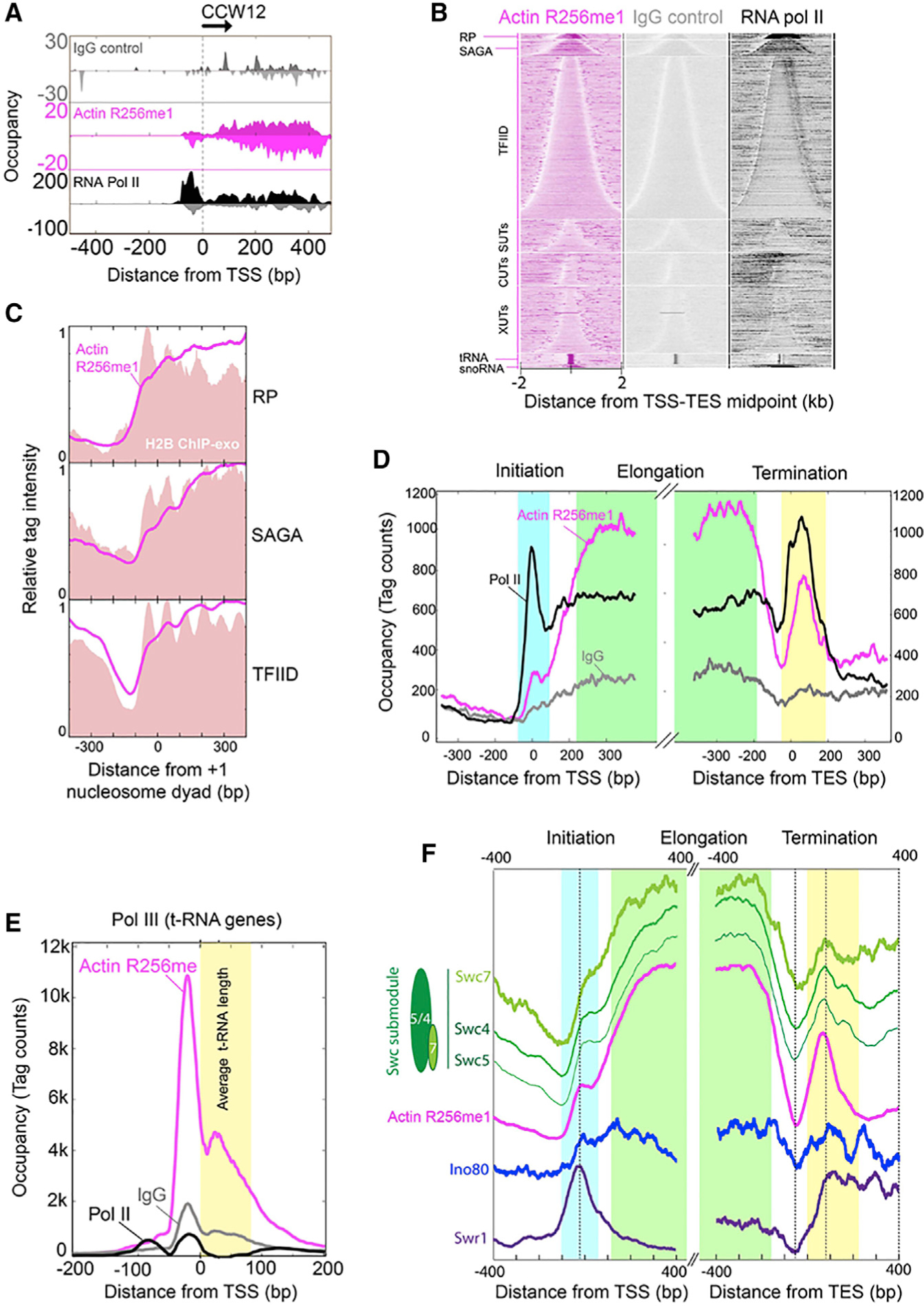
(A) Smoothed distribution of unshifted ChIP-exo tag 5′ ends on forward and reverse strands of CCW12 (YLR110C) gene as an example of a SAGA-regulated gene. Immunoglobulin G (IgG) control is shown in gray, actin R256me1 in magenta, and RNA Pol II in black.
(B) Heat maps showing tag 5′ ends (shifted in the 3′ direction by 6 bp, then strand information removed) for actin R256me1, IgG control, and RNA Pol II ChIP-exo, aligned by the midpoint of the transcript start and end point (TSS-TES), grouped into ribosomal protein (RP) genes, SAGA, TFIID, coding genes and SUTs, CUTs, XUTs, tRNA, and snoRNA noncoding genes and sorted by gene length in each group.
(C) Composite distribution (metagene analysis) of shifted tag 5′ ends for actin R256me1 (magenta trace) and H2B (red fill) around the +1 nucleosome dyad for RP, SAGA, and TFIID genes, oriented such that the direction of transcription is to the right. The y axis in each panel has been scaled from 0–1, and so quantitative levels cannot be compared between panels.
(D) Composite distribution of shifted tag 5′ ends for actin R256me1 (magenta), IgG control (gray), and RNA Pol II (black) around the TSS and TES. Light blue, green, and yellow boxes roughly delineate transcription initiation, elongation, and termination regions, respectively.
(E) Composite distribution of shifted tag 5′ ends of actin R256me1 (magenta), IgG control (gray), and RNA Pol II (black) around the TSS of tRNA genes, oriented such that the direction of transcription is to the right. The average tRNA length is indicated by yellow-filled region.
(F) Composite distribution of tag 5′ ends of Swr1 (purple), Ino80 (blue), actin R256me1 (magenta), and a Swc submodule (Swc5, Swc4, and Swc7 in different shades of green) around the TSS and TES. Each trace is separately scaled on y axis and vertically displaced for better visualization. Data presented are the representative of three biological repeats.
To address if actin R256me1 mark associates with RNA polymerases, we conducted multiple IP experiments by using the actin R256me1 antibody, followed by mass spectroscopy studies. The high-confidence mass spectrometry results showed that actin R256me1 mark indeed associates with RNA Pol II and RNA Pol III subunits and transcription regulators, such as TFIID and Sin3, in addition to the chromatin-modifying complexes (Table S2). These coIP studies suggest that actin R256me1 may also have roles outside chromatin remodeling complexes through interactions with basal transcription machinery and transcription regulators.
Distinct Distributions of Actin R256me1 Mark among RNA Pol II and Pol III Genes
We next examined the distribution of the actin R256me1 mark across all coding and noncoding genes (Figure 4B), separated by a variety of gene classes, including those that code for ribosomal proteins (RPs), regulated by factors such as SAGA or by TFIID, or are noncoding such as stable uncharacterized transcripts (SUTs), cryptic unstable transcripts (CUTs), Xrn1-sensitive antisense long noncoding transcripts (XUTs), tRNAs, and small nucleolar RNA (snoRNA). Remarkably, actin R256me1 was particularly enriched across the gene bodies of RPs and SAGA-regulated genes, tRNA genes, and snoRNA genes (Figures 4B and 4C), whereas it was not observed to be associated with telomeres or centromeres (data not shown) and was less enriched at other noncoding RNA (ncRNA) transcription units and TFIID-regulated mRNA genes. A composite plot of R256me1-actin binding across genes reveals that the actin R256me1 mark is present in relatively low amounts within the transcription pre-initiation complex, dramatically increasing in occupancy in gene bodies, particularly in relation to Pol II (Figure 4D). Actin R256me1 remains high throughout gene bodies, until the nucleosome-free end of the gene, where it essentially drops to baseline. Further downstream, both Pol II and actin R256me1 spike in occupancy over a ~150-bp region, in which a nucleosome resides. These data strongly support a positive role of nuclear actin in regulating Pol II transcription.
Interestingly, while actin R256me1 was generally coincident with Pol II, some regions like near the 3′ ends of RP genes had relatively low levels of Pol II but high levels of actin R256me1 and also relatively high levels of nucleosomes. In as much as Pol II and nucleosomes competitively occupy transcribed regions, actin R256me1 conceivably might associate with nucleosomes that have reassembled in the wake of Pol II, for which it tends to be positionally tied to H2B (Figure 4C, note that the y axis scale in each panel is adjusted from 0–1 and that there are two H2B peaks per nucleosome position). When the transcription units that were most highly bound by actin R256me1 were examined, actin R256me1 was found to largely (but not entirely) mirror RNA Pol II with respect to genes but not with respect to precise positioning (Figure S3A). This finding indicates that actin R256me1, although associated with highly transcribed genes, may be complexed with histones like H2B rather than to RNA Pol II itself. In sharp contrast to RNA-Pol-II-transcribed genes, actin R256me1 was highly enriched at the promoter region of tRNA genes, which are transcribed by RNA Pol III and are not encased in a nucleosome (compare Figures 4D and 4E). These results suggest that nuclear actin associates with the gene body nucleosomes of RNA-Pol-II-transcribed genes but associates with RNA Pol III pre-initiation factors at tRNA genes. This spatial separation suggests that the actin R256me1 mark might have distinct functions between RNA Pol II versus RNA-Pol-III-transcribed genes.
Given the association of actin R256me1 with three chromatin-modifying complexes (Figure 2D), it is plausible that actin-R256me1-containing complexes load and travel with Pol II but also translocate onto local nucleosomes in the wake of transcription. This seems difficult to reconcile in light of observations that place at least the INO80 and SWR1 complexes selectively at the 5′ ends of genes and not in gene bodies. However, a genomic study indicated that there might be a variety of submodules of at least SWR-C and INO80 complexes (Yen et al., 2013). For example, there is the SWC submodule that includes Swc4,5,7. Unlike SWR-C, the SWC complex tracks with nucleosomes across gene bodies (Figure 4F). Swc4 has been mentioned as a possible component of a complex containing nuclear actin along with Arp4, and there is some prior suggestion that at least Swc4 might be associated with nuclear actin (Kapoor and Shen, 2014). Interestingly, these SWC subunits are enriched where the actin R256me1 mark is enriched and have nearly identical genic distributions. Together, these results suggest that actin R256me1 might also be associated with the SWC submodule on nucleosomes in a transcription-linked manner. Actin and at least Swc4 are also part of NuA4, which might travel with RNA Pol II (Koerber et al., 2009; Rossetto et al., 2014). Taken together, the high-resolution genome-wide landscape of actin R256me1 and coIP studies suggests that the actin R256me1 mark may serve as a regulator of transcription, similar to certain histone PTMs, and may be involved in transcription regulation of highly transcribed genes through multiple mechanisms, either through chromatin remodeling complexes or interactions with transcription machinery.
RNA-Seq Studies Further Correlate Actin R256me1 with Active Transcription
To address how the expression of genes is affected upon the loss of actin R256me1 mark, we conducted RNA-seq followed by gene set enrichment analysis (GSEA) for the WT and actin R256C mutant in yeast to compare the differential gene expression profiles. Our results showed that there are 2,614 genes significantly and differentially expressed between the WT and the mutant strain, with 1,442 genes downregulated and 1,172 genes upregulated (Figures 5A and S3B). Among these differentially expressed genes, 381 genes have at least a 2-fold change, with 160 genes downregulated and 221 genes upregulated. GSEA results show the top enriched pathways in the downregulated genes are cell cycle processes, chromatid segregation, and DNA-templated processes such as DNA replication and double-strand break repair (Figures 5A; Table S3). For example, the gene set representing the DNA replication pathway was significantly downregulated in the R256C mutant (Figure 5B). Notably, as shown in Figure 5C, 80% of the deregulated genes are highly expressed genes, with 86% of the genes downregulated and 71% of the genes upregulated. These results further suggest that actin R256me1 is correlated with the active transcription process and is involved in regulating genome integrity pathways.
Figure 5. Gene Expression Profiling of Actin R256C Mutant of Yeast.
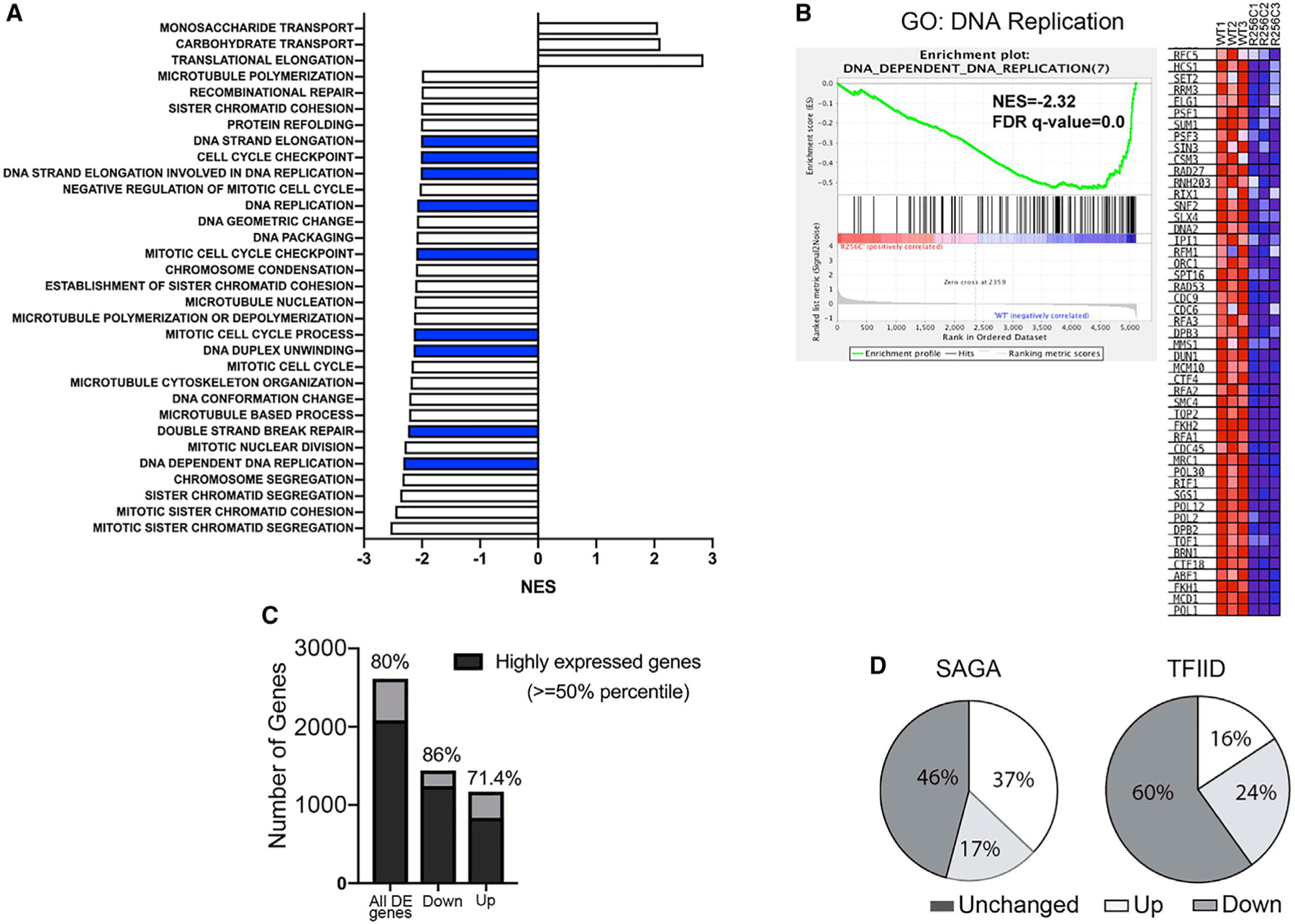
(A) Gene set enrichment analysis (GSEA) with compiled modules from Gene Ontology (GO). The bar graph represents only the gene sets that showed the normalized enrichment score (NES > 2.0) and false discovery rate (FDR) q value < 5%. Blue bars represent gene sets involved in the DNA repair process.
(B) Enrichment score diagram and heatmap illustrate the expression of the pathway-associated genes in DNA-dependent DNA replication in WT and actin R256C mutant cells.
(C) Bar graph showing the statistical analysis for the percentage of highly expressed genes in the differentially expressed genes and also in up- or downregulated genes in actin R256C mutant.
(D) Pie chart showing the gene expression changes in actin R256C mutant cells in the subcategories of either SAGA-regulated genes or TFIID-regulated genes. Data presented are the representative of three biological repeats of WT and actin R256C mutant yeast strains.
Given that actin R256me1 mark showed significant enrichment in SAGA-regulated genes compared to the TFIID-regulated genes (Figures 4B and S3A), we analyzed the percentage of genes that are affected in different classifications in terms of either SAGA- or TFIID-dominated genes. As shown in Figure 5D, more than half (>54%) of the SAGA-regulated genes are affected. In contrast, 40% TFIID-dominated genes are affected. Among the genes that are changed by at least 2-fold, there is a 5-fold enrichment of SAGA-regulated genes (15% SAGA versus 3% TFIID). This suggests that the actin R256C mutant mainly affects SAGA-regulated genes. This finding is consistent with the ChIP-exo data and suggests that the function of actin R256me1 is predominantly among the SAGA-regulated genes. Interestingly, for the SAGA-regulated genes, there are more genes upregulated than the genes downregulated (37% versus 17%). However, in the TFIID-regulated genes, there are more downregulated genes (24% versus 16%). It has been shown that SAGA-regulated genes are largely stress induced and are downregulated by the coordinated actions of a variety of chromatin modification complexes and RNA Pol II regulators (Geng and Laurent, 2004; Ghosh and Pugh, 2011; Huisinga and Pugh, 2004; Kremer and Gross, 2009; Zhang et al., 2008). Given that actin R256me1 is enriched at SAGA-regulated genes, this mark might be involved in recruiting repressors for its responsive genes. Interestingly, we identified Sin3 (the common subunit of histone deacetylase complex in Rpd3L and Rpd3S complex for repressing gene activation) in our coIP experiments by using the actin R256me1 antibody. Furthermore, we also identified the TAF5 subunit of the TFIID complex in these experiments. There-fore, actin R256me1 could affect the gene expression in both directions by disrupting or gaining interaction with activators or repressors in transcription.
Mutations at Actin PTM Sites Including Actin R256 Cause Human Diseases
Actin R256 residue is clinically relevant given that the mutations in the corresponding residue, R258, in human α-actin isoforms cause a number of human diseases (Rubenstein and Wen, 2014). For example, R258H/C mutations in α-smooth muscle actin cause TAAD. Excitingly, further analyses of other actin PTMs we identified showed a strong correlation of the sites of actin PTMs with human diseases such as R177 (human R179) methylation and K326 (human K328) methylation and acetylation (Table S1). These actin mutations of corresponding amino acid residues in human actin isoforms cause human diseases through multiple pathogenic mechanisms (Figure S4A). For example, mutations that disrupt R258 PTM (R258C or R258H) in the human smooth muscle-specific actin isoform, SM α-actin, encoded by ACTA2, predispose to both TAAD and early onset of cerebrovascular diseases (Guo et al., 2007). Similarly, mutations that disrupt R179 (R179L or R179C) in ACTA2 lead to patent ductus arteriosus and onset of cerebrovascular diseases (Figure S4A).
SM α-actin monomers polymerize to form actin filaments in the contractile unit of smooth muscle cells (SMCs), which provide contraction and force generation in SMCs (Yamin and Morgan, 2012). Missense mutations disrupting residues throughout the SM α-actin monomer predispose to TAAD. The conventional theory that explains the TAAD pathology is that these actin mutations cause “loss-of-function” cytoplasmic actin defects, leading to reduced contractility of vascular smooth muscle cells (Karimi and Milewicz, 2016; Lu et al., 2015). However, a subset of these mutations, including mutations disrupting R258 (R258C/H) and R179 (R179H/L/C), predispose patients to moyamoya-like cerebrovascular disease (Guo et al., 2009). Moyamoya disease is characterized by bilateral occlusion of the distal internal carotid artery and the formation of compensatory collateral vessels (Scott and Smith, 2009; Zipfel et al., 2005). The pathology associated with distal internal carotid artery occlusion or stenosis in these patients is characterized by neointimal lesions, which contain cells that stain for SMC markers but lack lipid deposition and inflammation typically seen in atherosclerotic lesions (Georgescu et al., 2015). Thus, we hypothesize that the subset of ACTA2 mutations such as R258C or R179H/L/C cause “gain-of-function” cell proliferation, leading to these occlusive lesions and alter the SMC phenotype such that these cells become hyperplastic and excessively migratory in response to vascular injury. The data on the pathology of the arterial lesions and from the Acta2−/− mouse support this hypothesis (Papke et al., 2013).
Given the strong correlation of sites of actin PTMs with the causal mutations in SM α-actin leading to human diseases with distinct pathology, we began to investigate the potential disease mechanisms through actin PTM angle, focusing on R256me1 as a proof-of-principle. We further hypothesize that the ACTA2 mutations disrupting corresponding R258 could alter the SMC phenotype through defects in the nuclear functions of SM α-actin (Figure S4B).
To begin testing this hypothesis, we first analyzed the presence of SM α-actin in the SMC nucleus. As shown in Figure S4C, SM α-actin was found to be present in both the nuclear and cytoplasmic fractions of SMCs. These results are similar to that of β-actin, which is known to be present in both the nucleus and cytoplasm of the mammalian cell. Next, to address whether nuclear SM α-actin contains the R258me1 mark, we stained the cytoplasmic and nuclear fractions of wild-type SMCs and SMCs deficient in SM α-actin (Acta2−/− SMCs) with the actin R256me1 antibody. Our results showed a reduction of the actin R256me1 (R258 in human α-actin) signal in the nuclear fraction of SMCs, despite the fact that previous studies found an increase of muscular γ-actin such that total cellular actin levels remained unchanged between the mutant and WT SMCs (Figure S4D). Given that in SMCs around 70% of total actin is SM α-actin and that the actin R256me1 antibody detects the R256me1 mark of β- and γ-actin as well, our results imply that actin in the SMC nucleus also contains R256me1 mark. Together, these results suggest that SM α-actin may also have nuclear functions and that the actin R258me1 mark is likely present in nuclear SM α-actin.
Implicating Nuclear Actin and Actin R256me1 in TAAD through Transcription Regulation
We further analyzed the potential role of the actin R256me1 mark in vascular diseases by monitoring the corresponding R258 methylation (R258me1) status of SM α-actin in dermal fibroblast cell lines derived from healthy volunteers and patients with heterozygous, disease-causing R258C mutation in ACTA2. SM α-actin expression was induced in healthy volunteer- and patient-derived dermal fibroblasts by infecting cells with Ad-MRTF-A (Kuwahara et al., 2010). This is a well-established system that mimics smooth muscle cells from patients to study the SM α-actin (Guo et al., 2007). In our study, these cells were subjected to fluorescence immunostaining and 2D western blot analyses to visualize the localization of actin R258me1 mark and to monitor the levels of actin R258me1 signal in different actin isoforms, respectively.
Interestingly, the fluorescence immunostaining of both induced and non-induced fibroblasts using actin R256me1 antibodies identified actin R258me1 mark preferentially in the nucleus of the cell (Figure S5A). This was consistent with the observations in NIH 3T3 cells (Figure 3D). Given that the fluorescence immunostaining with actin R256me1 antibodies detects R256me1 signals in all the actin isoforms in the nucleus, 2D western analysis of nuclear fractions from these cells was performed, which further demonstrate that all the actin isoforms, α-, β-, and γ-actins exist in the nucleus with R256me1 mark (Figure S5B). This is also consistent with our studies in yeast and mammalian cells (Figures 2 and 3) and further confirms that actin R256me1 is a conserved nuclear actin mark.
Importantly, the induction of SM α-actin reproducibly intensified R258me1 staining only in the SM α-actin isoform in 2D blots (Figure S5C). We used 2D western analyses as a tool to monitor changes in the R258me1 signal. In order to determine the mobility shift associated with R to C mutation in SM α-actin (pI 5.24 to 5.17), recombinant wild-type and mutant SM α-actin were purified from an insect cell expression system and subjected to 2D gel electrophoresis separately (Lu et al., 2015), followed by western analyses using actin R256me1 antibodies (Figure S5C). Notably, wild-type SM α-actin was stained by the actin R256me1 antibody, whereas R258C SM α-actin did not show staining (Figure S5C). Moreover, another recombinant SM α-actin mutant, ACTA2 R179H, also reacted with actin R256me1 antibodies (Figure S5C), further illustrating site specificity of the actin R256me1 antibodies.
To analyze the levels of R258me1 signal in SM α-actin of healthy volunteer- and patient-derived dermal fibroblasts, lysates from these cells were subjected to 2D western analyses using actin R256me1 antibodies. Strikingly, the actin R256me1 antibody stained all of the three isoforms of actin but did not stain the R258C variant SM α-actin (Figure S5D). These results show that the R258me1 signal is substantially reduced in the SM α-actin of patient-derived cells with the R258C mutation in ACTA2 compared to the healthy volunteers and suggest a possible nuclear role of SM α-actin and R258me1 mark in the cerebrovascular disease associated with disruption of R258.
Given that in yeast the conserved actin R256me1 correlates with active transcription (Figure 4), we hypothesized that the reduction of R258me1 mark in SM α-actin may affect the transcriptional profiles of R258C patient SMCs. Indeed, results of differential RNA expression analysis through RNA-seq revealed significant differences in a set of genes (Figure S5E; Table S4). Analysis of canonical pathways with very high probability (p < 0.0001) identified interferon signaling and DNA damage repair/cell cycle as the most affected pathways under both low and high stringency. These results are consistent with transcriptional defects as actin-containing chromatin modifying complexes have been implicated in the transcription of type I interferon-stimulated genes, and that these chromatin modifying complexes also participate in DNA damage repair and cell cycle regulation (Huang et al., 2002; Ivashkiv and Donlin, 2014; Liu et al., 2002; Morrison et al., 2004; Morrison and Shen, 2009; Price and D’Andrea, 2013; Yan et al., 2005). Given that actin is also implicated in transcription by all of the three classes of RNA polymerases, it is plausible that the changes in transcriptional profile may be related to defects in the RNA pol II-mediated transcription of genes involved in interferon signaling and genes involved in DNA damage repair. Nevertheless, these results suggest that the R258C mutation in SM α-actin cause distinct changes in the transcription profiles of the patient-derived cell lines, which might help explain the “gain-of-function” pathology of a subset of TAAD and cerebrovascular diseases. Albeit correlative, our human and yeast data collectively support roles of nuclear actin and actin R256me1 in human vascular diseases such as TAAD through gene regulation.
An Actin Code Hypothesis
The discovery of R256me1 provides a fundamental mechanism for distinguishing nuclear actin from cytoplasmic actin and offers initial validation for the context-specific actin PTM hypothesis. Interestingly, our yeast studies are consistent with the studies of mammalian cells, including those from patient-derived dermal fibroblast, both suggesting a role of R256me1 in transcription. How might the actin PTMs such as R256me1 change the property of actin? It is possible that a unique set of PTMs in nuclear actin may bring conformational changes that could lead to the distinct properties of nuclear actin compared to cytoplasmic actin. Our in silico MD simulation analyses of actin with R256me1 support this hypothesis (Figure 6 and Figure S6). Indeed, adding a single methyl group on R256 could lead to significant changes in actin conformation, such as opening the ATP-binding cleft. These structural changes discourage actin polymerization and are consistent with mechanisms of keeping nuclear actin in a monomeric state (Figure 6; Chik et al., 1996; Splettstoesser et al., 2009). It is also possible that R256me1 may regulate the dynamics of actin polymerization in the cell nucleus. Future direct studies on the effect of R256me1 mark and similar PTMs on actin dynamics will there-fore be important for determining the functional significance of such actin PTMs. As such, we propose that one class of actin PTMs may function through their abilities to change the structures of actin, leading to distinct mechanisms to regulate actin dynamics. On the other hand, as in the case of histone PTMs (Andrews et al., 2016a, 2016b; Jain and Barton, 2017; Li et al., 2014; Peña et al., 2006; Shi et al., 2006), another class of actin PTMs could serves as signals to recruit specific binders (readers) that may regulate distinct processes such as transcription. It should be noted that these two possibilities are not mutually exclusive.
Figure 6. Changes in Conformation of Actin after R256me1 of Yeast Actin: A 100-ns MD Simulation Analysis.
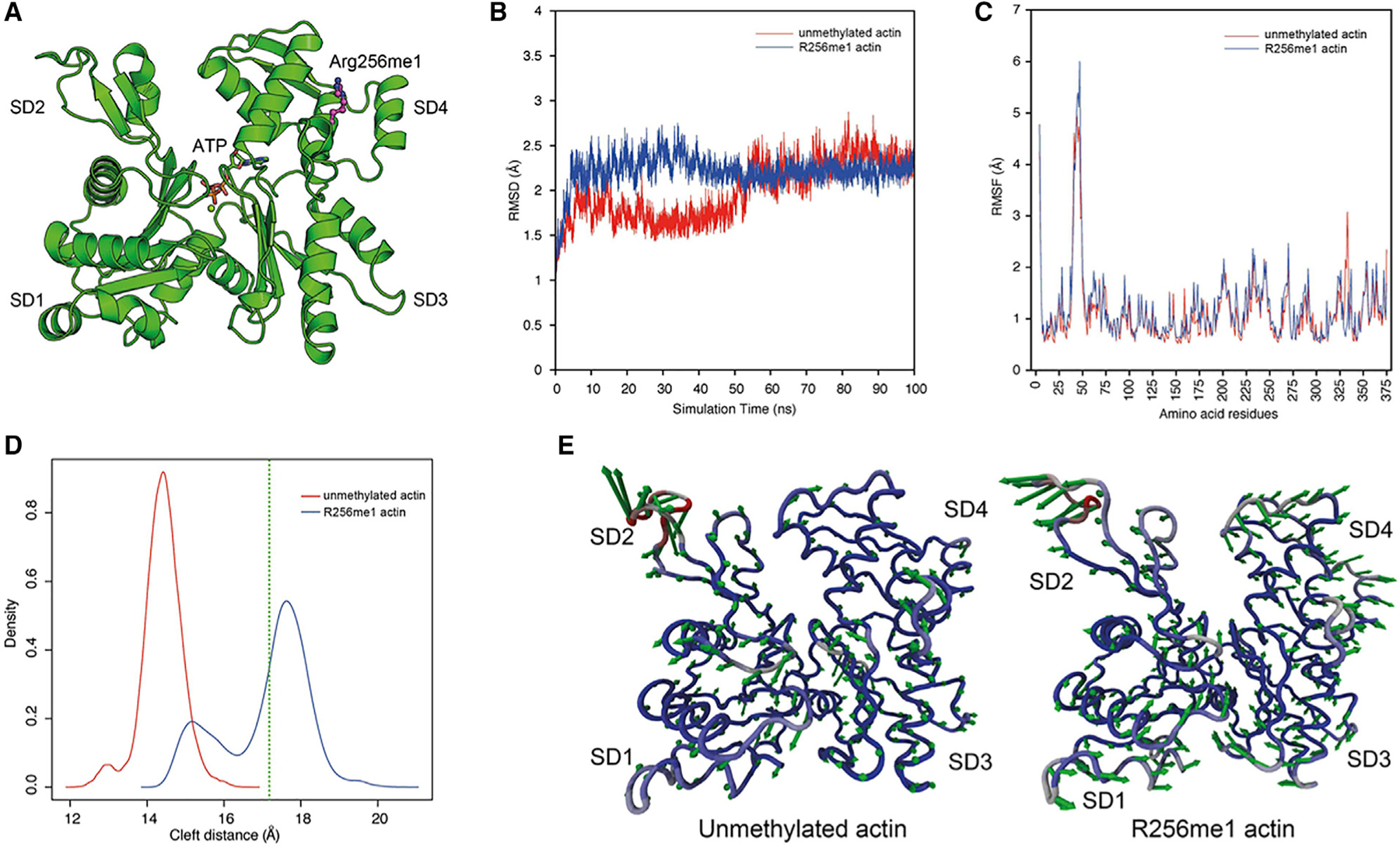
(A) Crystal structure of S. cerevisiae actin (PDB code: 1YAG) as determined in complex with human gelsolin segment 1. Actin subdomains (SD1–4) are annotated, and the bound ATP is shown in atom colored sticks. R256 is modified by adding a methyl group and presented as magenta atom colored ball and stick.
(B) Distribution of root-mean-square deviation (RMSD) of backbone atoms calculated over 100 ns for unmethylated actin and R256me1 actin monomer. R256me1 resulted in higher RMSD compared to unmethylated actin monomer.
(C) Atomic fluctuation of unmethylated actin and R256me1 actin residues calculated as RMSF of backbone atoms. Most of these dynamics arise from higher RMSF in the DNase I loop (residues 35–53).
(D) Distribution of distance between subdomains SD2 and SD4 (cleft distance) calculated over 100 ns for unmethylated actin and R256me1 actin monomer. The results shows open conformation in R256me1 actin compared to unmethylated actin monomer. Green dotted line represents the cleft distance in S. cerevisiae actin crystal structure (PDB code: 1YAG).
(E) Vector field representation of the first principal mode obtained from 100-ns MD trajectory for unmethylated actin (left) and (B) R256me1 actin. Color of ribbon represents the mobility from lower (blue) to higher (red). In contrast to unmethylated actin, comparatively fewer movements in SD2 and SD4 subdomain residues were observed in R256me1 actin, indicating the shift in equilibrium towards the open form. Please also see Figure S6 and STAR Methods.
The prevailing paradigm in actin biology focuses on actin regulation by ABPs. Many ABPs have been identified to date that govern actin dynamics in numerous cellular processes (Campellone and Welch, 2010; Revenu et al., 2004; Welch et al., 1997). In theory, in addition to ABPs, actin PTMs could also confer specific functions for actin, similar to histone PTMs. However, very few actin PTMs have been studied previously (Terman and Kashina, 2013). We hypothesize that context-specific actin PTMs could provide a new paradigm to regulate actin biology. As such, actin PTMs may form an Actin Code, similar to the Histone Code. Indeed, this proof-of-principle study of the R256me1 mark appears to support the Actin Code hypothesis through implication of an actin PTM in specific biological functions. Given that our actin PTM collection includes a wide range of PTMs on evolutionarily conserved residues in both nuclear and cytoplasmic actins, it is likely that the Actin Code hypothesis will also be applicable to actin biology in general. Importantly, the current actin ABP paradigm and the Actin Code paradigm are not mutually exclusive, and cross-talks between the two may exist. Our initial study of actin R256me1 reveals a fundamental mechanism for distinguishing nuclear actin from cytoplasmic actin and provides a platform for establishing actin biology through PTM regulations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to Xuetong Shen (xshen@mdanderson.org).
Materials Availability
All renewable reagents generated in this study are available upon request subject to ethical restrictions and with a completed material transfer agreement.
Data and Code Availability
Sequencing data and codes were deposited in online database. RNA-sequencing data were deposited in the Gene Expression Omnibus public database with accession number GSE156817. Further, data generated in this study are available from the corresponding authors upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All S. cerevisiae strains were in S288C background. A collection of act1 yeast mutants was a gift from David Drubin (Novick and Botstein, 1985; Wertman et al., 1992). ACT1-R256C, and ACT1-R256K yeast mutants were generated using standard plasmid shuffling as described elsewhere (Boeke et al., 1987). Plasmids with point mutations in ACT1 were generated using a pRS415-ACT1(LEU2) plasmid backbone and employing site directed mutagenesis (Agilent Technologies lnc.). Relevant ACT1 mutations were confirmed from PCR sequencing. Standard yeast genetic techniques were used to create gene deletions and epitope tagged yeast strains. Phenotypic analysis was done by plating yeast cells at five-fold serial dilutions. Plates were incubated at 30°C for 3–5 days, and then scored.
METHOD DETAILS
Purification of proteins and protein complexes, and generation of antibodies
For the purification of INO80, SWR1, and NuA4 complexes from yeast, INO80, SWR1, and ESA1 respectively were chromosomally tagged with triple FLAG tag at the C terminus, and protein complexes were purified from FLAG tagged strains as described previously (Shen, 2004). All complexes were purified using high salt washes (0.5M KCl), and further complexes were separated on a 5ml 17%–35% glycerol gradient in buffer H-0.3 (25mM HEPES KOH (pH 7.6), 1mM EDTA, 0.02% NP-40, and 0.3M KCl). Standard yeast actin purification protocols were employed and cytoplasmic actin was purified with a slight modification which involves the separation of nuclear and cytoplasmic fractions, and further processing cytoplasmic fractions for the purification of cytoplasmic actin using procedures described previously (Greer and Schekman, 1982; Rout and Blobel, 1993). Recombinant human SM-α-actin (WT, R258C and R179H) was expressed and purified using the baculovirus/Sf9 insect cell expression system as described previously (Lu et al., 2015). SDS-PAGE followed by silver staining was done to detect the purity of the proteins and protein complexes. Antibodies against actin R256me1 were raised in rabbit using yeast specific actin R256me1 peptide (VITIGNERFR(me1)APEAL). The serum was isolated after second booster and antibodies were purified using standard protocols.
IEF Analysis
INO80 complex, actin isoforms and mutation-induced charge variants of actin were separated by two-dimensional electrophoresis. Samples were prepared in Rehydration/Sample Buffer (BIO-RAD) and subjected to Agilent INGEL fractionation in a 24 cm, 4–7 or 3–10, immobilized pH gradient (IPG) gel strip (GE Healthcare). The gel strip was then subjected to SDS-PAGE followed by silver staining or transfer to nitrocellulose membrane. Membranes were reacted sequentially with antibodies to actin R256me1 and SM-α-actin after stripping. For fibroblast, control (WT) and R258C cells were lysed in Rehydration/Sample Buffer (BIO-RAD) followed by procedures described above.
Mass spectrometry analyses and identification of protein modifications
The protein complexes were separated by SDS-PAGE. Actin bands at 42 kDa were digested in-gel with trypsin and desalted using Millipore U-C18 ZipTip Pipette Tips following the manufacturer’s protocol. LC-MS/MS was conducted using the Dionex Ultimate 3000 RSLCnano LC coupled to the Thermo Orbitrap Elite. A 75 μm I.D. × 2.5 cm long trap packed with C18 3 μm material (Dionex Acclaim PepMap 100) was followed by two analytical columns in series: 75 μm I.D. × 20 cm, then 75 μm I.D. × 30 cm, packed with the same C18 material as the trap. Buffer A consisted of 0.1% formic acid in water and buffer B 0.1% formic acid in acetonitrile. The gradient started at 3% B and increased linearly to 27% B in 69 min, to 45% B at 80 min, then to 90% B at 82 min. After 8 min at 90% B, the solvent is returned to 5% B. The trap flow rate is 4 μl/min and the analytical column flows set to 300 nl/min. Each sample was run twice. The MS and MS/MS are acquired in the Orbitrap detection over the 98 min LC run, with full scan over 300–1700 m/z and resolution set to 60,000 for MS with AGC target at 1e6 and maximum ion time 500 ms. Top 10 MS/MS are acquired in CID mode at 15,000 resolution with AGC target at 5e4 and maximum ion time 500 ms. The isolation width is 3 Da, normalized collision energy 35, and minimum ion counts 10,000. Raw data is processed using SEQUEST HT embedded in Proteome Discoverer v1.4.1.14, searching the Uniprot yeast reference proteome (9/2014, 6559 entries). A decoy database is used for calculating false positive values. Mass tolerance is 10 ppm for MS and 0.02 Da for MS/MS. Tryptic digest with up to 2 missed cleavages and minimum 6 amino acids is used. Fixed modification is cysteine carbamidomethylation and variable modifications are methionine oxidation, lysine acetylation, lysine and arginine mono and dimethylation, lysine ubiquitinylation, and serine, threonine and tyrosine phosphorylation. A maximum of 4 dynamic modifications are allowed per peptide. Percolator is used for validation of peptide and protein identifications with high confidence filtering to achieve q < 0.01.
Immunofluorescence staining
Cells were seeded on coverslips overnight, followed by 20 min fixation with 4% paraformaldehyde in PBS at room temperature. Samples were blocked with 3% BSA/PBS for 20 min, permeabilized with 0.5% Triton X-100/PBS for 10 min, then incubated with primary antibodies overnight at 4°C. Coverslips were then washed 3X and incubated with secondary antibody for 1–2 h, followed by mounting on glass slides with Fluoromount-G. Cells were visualized by confocal microscopy.
Cell culture, human INO80 (hINO80) pulldown, and siRNA in HEK293 cells
The full length human INO80 transcript variant 1 (Accession: NM_017553.2) was used for 3X FLAG-tagging at C terminus using CRISPR-Cas9 system in HEK293 cells. The stable monoclonal population was verified by sequencing. HEK293 cells having INO80-3X FLAG were cultured in DMEM media (Corning #10013-CM) with 10% FBS (GIBCO # 26-140-095), 1X pen-strep-ampho (Biological Industries #03033-1B) in T25 flask and after 24 hr supplemented with 0.5μg/ml of puromycin for selection. Four million cells were subcultured in 150 mm plates (10 plates) until they reached full confluence by regularly changing the media every 2 days. At full confluence, the media was aspirated, and cells were washed twice with PBS. The cells were harvested in 0.3 HGN buffer containing the PI cocktail. The cell pellets were flash frozen and processed for pulldown using M2 affinity beads by slight modification of the protocol as described earlier (Shen, 2004). For siRNA experiments, HEK293 cells were transfected with siRNA against PRMTs using lipofectamine RNAiMAX Transfection Reagent (Thermo Scientific #13778030) at 60%–70% confluence. Cells were harvested after 24 hr in RIPA buffer (G Biosciences #786-490) to observe the knockdown effect using immunoblotting.
In vitro methylation assay
The lyophilized rabbit muscle actin (Cytoskeleton Inc, # AKL99-A) was dissolved in actin buffer (5mM Tris, 0.2mM CaCl2 pH 8.0). The methylation reaction was performed in Tris buffer (150mM NaCl, 20mM Tris pH 7.4) containing 0.5μM SAM (S-Adenosyl Methionine), 80ng actin and 100ng of purified recombinant PRMT5 (Sigma Aldrich #SRP0146-20UG) at 30°C for 1 hr.
ChIp-exo sequencing and DESeq analysis
S. cerevisiae S288C strains were grown to OD600 0.8 at 25°C in 500 mL of YPD (yeast peptone dextrose). Cells were cross-linked with 1% formaldehyde for 15 min. Followed by quenching with 0.125 M glycine. The crosslinked cells were ruptured, chromatin extracted and fragmented by sonication for desired DNA size. ChIP-exo experiments were carried out essentially as described (Rhee and Pugh, 2012). This included an immunoprecipitation step with desired antibody and magnetic beads, followed by DNA polishing, A-tailing, Illumina adaptor ligation (ExA2), lambda and recJ exonuclease digestion on the beads. After elution, a primer was annealed to EXA2 and extended with phi29 DNA polymerase, then A-tailed. A second Illumina adaptor was then ligated, and the products PCR-amplified and gel-purified. Sequencing was performed on an Illumina NextSeq500. Uniquely aligned sequence tags were mapped to the yeast genome (sacCer3) using BWA (version 0.5.9- r16) (Li and Durbin, 2009). Tags were shifted in the 3′ direction by 6 bp, and strand information removed, to better reflect the point of crosslinking. Reproducible biological replicates were merged together (n = 4). To compare actin R256me and IgG controls, data were normalized to make the total tags counts equal in both. This likely artificially inflated the IgG control relative to actin R256me1. For each gene the tags were summed over from the Transcription Start Site (TSS) to Transcription End Site (TES) for actin R256me1 and IgG control, and DESeq (Anders and Huber, 2010) was used was used to compute the significantly enriched genes over the IgG control. Reference +1 nucleosome dyads were from Zhang et al. (2011); TSS locations were from Xu et al. (2009).
Human fibroblast Culture, Transfection and Collection
Dermal fibroblasts from healthy volunteers or patients were derived from skin biopsies obtained with consent under a protocol approved by the Institutional Review Board (UTHSC Houston). Fibroblasts were immortalized by infection with retrovirus harboring genes for human telomerase catalytic subunit (hTERT) and puromycin resistance followed by selection in puromycin (Walter et al., 2004). For experiments, cells were infected for 48 hours with adenovirus directing the expression of myocardin-related transcription factor-A (Ad-MRTF-A; MOI 80) to induce myofibroblast differentiation and significant SM-α-actin expression (Velasquez et al., 2013). Cells were cultured under standard conditions in DMEM/10% FBS. Cultured fibroblasts were washed with PBS, scraped and transferred into a microcentrifuge tube. Proteins were precipitated with 10% TCA/10mM DTT and gently pelleted at 2000rpm for 2 min, washed with 500uL ethyl ether, dried for 5 min. Pellet was solubilized in Urea Sample Buffer for immunoblots, or solubilized in IEF Buffer for 2D Gel Electrophoresis. Nuclear/cytosolic fractionation was carried out following the manufacturer’s protocol (Catalog Number: AKR-171, CELL BIOLABS, INC). Briefly, cells infected with Ad-MRTF-A for 48h were released with trypsin and collected by centrifugation for 5 min at 4°C (600 g). The cell pellet was resuspended with ice cold Cytosol Extraction Buffer containing DTT/Protease Inhibitors. After incubation on ice for 10 min, cells were lysed in 25μl of Cell Lysis Reagent and vortexed for 10 s at the highest setting, followed by centrifugation for 10 min at 4°C (800 g). The supernatant (cytoplasmic fraction) was carefully transferred to a clean, chilled microcentrifuge tube and stored at −80°C. The wash step was repeated to reduce cross-contamination between fractions. The nuclear pellet was gently resuspended with 100 μl of Nuclear Extraction Buffer containing DTT/Protease Inhibitors, then maintained on ice for 30 min, vortexing for 10 s at the highest setting at 10 min intervals, followed by centrifugation for 30 min at 4°C (14000 g). Supernatant (nuclear fraction) was transferred to a clean, chilled microcentrifuge tube and stored at −80°C.
RNA sample preparation and RNA-Seq analysis
For RNA sample preparation from yeast strains, three biological replicates for each strain were inoculated overnight and diluted back to OD660 0.3 to grow in rich medium YPD at 30°C. 1.5×107 cells were collected by centrifugation when the culture reached early phase of exponential growth OD660 0.9. RNA was extracted using QIAGEN RNeasy plus Mini kit with extra enzymatic lysis step of the yeast cell wall using zymolyase 100T (100U per 107 cells). The quality of the RNA sample was assessed by bioanalyzer with RNA integrity number (RIN) all above 10. cDNA Libraries were prepared using the Illumina TruSeq Stranded mRNA (Cat. # RS-122-2101) kit according to the manufacturer’s protocol starting with 1 μg total RNA. Briefly, mRNAs were purified with oligo(dT) beads to capture polyA tails, then fragmented and converted to cDNA with reverse transcriptase. The resulting cDNAs were converted to double stranded cDNAs and subjected to end-repair, A-tailing, and adaptor ligation. The constructed libraries were amplified using 8 cycles of PCR. 1.5nM of pooled libraries were processed using a cBot (Illumina) for cluster generation before sequencing on an Illumina Hi Seq 3000 (2 × 76 bp run) (Chao et al., 2019). The gene differential expression analysis was performed with the DESeq2 Bioconductor package (Anders and Huber, 2010; Anders et al., 2013). The heatmap was generated with the differentially expressed genes and the pearson distance and the complete linkage clustering was utilized in the analysis. We have further performed gene set enrichment analysis using the GSEA tool developed by the Broad Institute that is available at https://www.broadinstitute.org/gsea (Mootha et al., 2003; Subramanian et al., 2005). GSEA utilizes a collection of differentially expressed gene sets annotated for the gene ontology (GO) biological process from the Molecular Signatures Database (MSigDB).
For RNA sample preparation from patient derived dermal fibroblasts, control and R258C cells were infected with Ad-MRTF-A for 48h, and then cells were trypsinized, centrifuged, resuspended and seeded on 6cm tissue culture dishes. After 24h in culture, cells were collected for RNA extraction. Total RNA was purified with RNeasy Protect Mini Kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. The quality of RNA used in cDNA library preparation was measured with an Agilent Bioanalyzer RNA 6000 Nano kit by Microarray Core at UT Southwestern Medical Center. Total RNA (1 μg) was used for production of the RNA-Seq cDNA library according to standard protocols that include cDNA synthesis and fragmentation, the addition of adaptors, size selection, amplification and quality control (Illumina). A HiSeq 2000 system (Illumina) was used for SE-50 sequencing (single-ended 50–base pair reads), with over 18 × 106 ‘reads’ per sample. Basic data analysis was done with CLC-Biosystems Genomic Work-bench (v7) to generate quantitative data for all genes, including reads per kilobase of exon per million mapped reads (RPKM). Network and pathway analyses were performed using Ingenuity Pathway Analysis (IPA) software (https://www.ingenuity.com). From a list of differentially-expressed genes, IPA produces networks according to Fisher’s exact test, and then provides the significance of each network, indicating the probability of association of molecules in the dataset with the pathway by random chance alone.
Phylogenetic analysis and sequence alignment
The sequences for 25 actin proteins [Acanthamoeba castellanii, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Tetrahymena thermophila, Dictyostelium discoideum, Trypanosoma brucei, Plasmodium falciparum, Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Xenopus laevis, Danio rerio cytoplasmic 1 (actb1), D. rerio cytoplasmic 2 (actb2), Mus musculus alpha skeletal muscle (ASkM), M. musculus alpha cardiac muscle (ACM), M. musculus aortic smooth muscle (ASmM), M. musculus cytoplasmic 1 (BC1), M. musculus cytoplasmic 2 (GC2), M. musculus gamma-enteric smooth muscle (GSM), Homo sapiens alpha skeletal muscle (ASkM), H. sapiens alpha cardiac muscle (ACM), H. sapiens aortic smooth muscle (ASmM), H. sapiens cytoplasmic 1 (BC1), H. sapiens cytoplasmic 2 (GC2), H. sapiens gamma-enteric smooth muscle (GSM)] were downloaded from Uniprot (Consortium, 2014) and aligned using ClustalW2 (Larkin et al., 2007). The phylogenetic relationship between human actin isoforms and actin from other species was inferred using the Neighbor-Joining algorithm (Saitou and Nei, 1987) as implemented in MEGA6 program (Tamura et al., 2013). The evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling, 1965). The bootstrap values were calculated using 1000 replicates.
Molecular Dynamic (MD) Simulation
All systems for MD simulations were prepared using S. cerevisiae actin crystal structure in complex human gelsolin segment 1 (PDB code 1YAG). Gelsolin segment was removed from the crystal structure and actin monomer was saved separately as input structure for MD simulations. Actin is an ATPase with four subdomains: SD1 (residues 1–32, 70–144, and 338–375), SD2 (residues 33–69), SD3 (residues 145–180 and 270–337) and SD4 (residues 181–269) (Kabsch et al., 1990). The nucleotide-binding site is located in a cleft between SD1–2 and SD3–4. Amino acid R256 is located in the SD4 domain of actin. To analyze the influence of R256me1 on the structure and dynamics of actin monomer, a 100 ns all-atom MD simulation of the actin with R256me1 mark was carried out and compared with the unmethylated actin. This is an established technique and has been used previously to predict changes in actin conformations upon nucleotide binding or changes in actin conformations in variant actin homologs (Chu and Voth, 2005, 2006; Kapoor et al., 2010; Kapoor et al., 2008; Pfaendtner et al., 2009). To investigate the effect of R256me1 on actin structure, methyl group was added to R256 NH1 atom of actin monomer and saved as R256me1 actin input structure. For MD simulations, both unmethylated actin and actin with R256me1 systems were solvated in a rectangular truncated octahedron filled with TIP4P-EW (Horn et al., 2004) water molecules. Na+ and Cl− ions were used to neutralize the simulation system. The protein, solvent and ion parameters were assigned using Amber99SB force field (Hornak et al., 2006). The parameters for mono-methylated arginine and ATP were derived from Papageorgiou (http://pc164.materials.uoi.gr/dpapageo/amberparams.php) and Bryce groups (https://www.pharmacy.manchester.ac.uk/bryce/amber) respectively. Initially, the water and ions were subjected to steepest-descent energy minimization to relax their initial configuration while restraining the protein atoms using harmonic position restraints of 10 kcal/mol/Å2. In the next step, all restraints were removed and the complete system was minimized for 2000 steps. The system was then gradually heated in the canonical ensemble from 0 to 300 K over the course of 500 ps and then equilibrated for a period of 5 ns. Finally, 100 ns production MD simulations were performed using Amber14 (Case et al., 2014). MD simulations were performed using NPT ensemble at a constant temperature of 300 K. SHAKE algorithm (Ryckaert et al., 1977) was used to constrain all hydrogen atoms. A 2 fs time step was used for the simulations. A cut-off of 10 Å was used for short-range interactions while Particle-Mesh Ewald (PME) (Darden et al., 1993) method was used to handle long-range interactions. The trajectory was analyzed using CPPTRAJ (Roe and Cheatham, 2013) and VMD (Humphrey et al., 1996). VMD’s NMWiz plugin and Prody program (Bakan et al., 2011) were utilized to perform principal component analysis. To explore the dynamics of the nucleotide cleft between the SD1–SD2 and SD3–SD4 domains, the distance between the center of mass of amino acid residues in SD2 (residues 31–34 and 56–69) and SD4 (residues 203–216) throughout the MD trajectory was calculated. The cleft distance between the SD1–SD2 and SD3–SD4 domains in the input crystal structure is 17.2 Å. To further decipher the dynamic properties and correlated movements of various subdomains, the principal component analysis was applied to the 100 ns MD trajectory. The graphics were prepared using Gnuplot4.6 (Williams and Kelley, 2012), PyMOL, R (Albert and Redon, 1998) and VMD (Humphrey et al., 1996).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analyses were performed using GraphPad Prism 7 software (GraphPad Software). Data expressed are mean ± SD, biological replicates (n number) are specified for each experiment in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-actin (clone C4) | EMD Millipore | Cat# MAB1501 |
| Mouse anti-β-actin | Abcam | Cat# ab8226 |
| Mouse anti-smooth muscle α-actin (ACTA2) | Abcam | Cat# ab220179 |
| Mouse anti-FLAG | Sigma-Aldrich | Cat# F3165 |
| Rabbit anti-Histone H3Ac | Active Motif | Cat# 61637 |
| Rabbit anti-Histone H4 | Abcam | Cat# ab7311 |
| Rabbit anti-Caspase-3 | Abcam | Cat# ab32042 |
| Rabbit anti-PRMT1 | Abcam | Cat# ab190892 |
| Rabbit anti-PRMT2 | Abcam | Cat# ab154154 |
| Rabbit anti-PRMT3 | Abcam | Cat# ab191562 |
| Rabbit anti-PRMT4 | Abcam | Cat# ab128851 |
| Rabbit anti-PRMT5 | Abcam | Cat# ab109451 |
| Rabbit anti-PRMT6 | Abcam | Cat# ab72205 |
| Rabbit anti-PRMT7 | Abcam | Cat# ab181214 |
| Rabbit anti-actin R256me1 | This study | N/A |
| Rabbit anti-actin K61me1 | This study | N/A |
| Rabbit anti-actin K61Ac | This study | N/A |
| Rabbit anti-actin K326me1 | This study | N/A |
| Rabbit anti-actin K326Ac | This Study | N/A |
| Biological Samples | ||
| Fetal Bovine Serum | GIBCO | Cat# 26-140-095 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rabbit muscle actin | Cytoskeleton Inc. | Cat# AKL99-A |
| Recombinant PRMT5 | Sigma-Aldrich | Cat# #SRP0146-20UG |
| ReadyPrep 2D-Starter Kit Rehydration/Sample Buffer | BIO-RAD | Cat# 1632106 |
| DMEM | Corning | Cat# 10013-CM |
| Pen-Strep-Ampho | Biological Industries | Cat# #03033-1B |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Scientific | Cat# #13778030 |
| RIPA Buffer | G Biosciences | Cat# 786-490 |
| S-Adenosyl Methionine | Sigma-Aldrich | Cat# A9384 |
| CMP5, PRMT5 inhibitor | Sigma-Aldrich | Cat# 5343560001 |
| FLAG peptide | Sigma-Aldrich | Cat# F3290-25MG |
| Critical Commercial Assays | ||
| Nuclear/Cytosolic Fractionation Kit | Cell Biolabs, Inc. | Cat# ARK-171 |
| TruSeq Stranded mRNA kit | Illumina | Cat# RS-122-2101t |
| Deposited Data | ||
| Raw data | This paper | GEO: GSE156817 |
| Experimental Models: Cell Lines | ||
| HEK293 | Sigma-Aldrich | Cat# 85120602-1VL |
| NIH 3T3 | Sigma-Aldrich | Cat# 93061524-1VL |
| Software and Algorithms | ||
| ClustalW2 | http://www.clustal.org/clustal2/ | Larkin et al., 2007 |
| MEGA6 | https://www.megasoftware.net | Tamura et al., 2013 |
| Gnuplot4.6 | http://www.gnuplot.info | Williams and Kelley, 2012 |
| PyMOL | Schrodinger, Inc. | The PyMOL Molecular Graphics System |
| R | https://www.r-project.org | Albert and Redon, 1998 |
| VMD | http://www.clustal.org/clustal2/ | Humphrey et al., 1996 |
| AMBER14 | http://ambermd.org | Case et al., 2014 |
| CPPTRAJ | http://ambermd.org | Roe and Cheatham, 2013 |
| NMWiz | http://prody.csb.pitt.edu | Bakan et al., 2011 |
| Prody | http://prody.csb.pitt.edu | Bakan et al., 2011 |
| DESeq2 Bioconductor package | https://github.com/mikelove/DESeq2 | Anders and Huber, 2010; Anders et al., 2013 |
| GSEA | https://www.broadinstitute.org/gsea | Mootha et al., 2003; Subramanian et al., 2005 |
| GraphPad Prism 7 | GraphPad Software | N/A |
Highlights.
Actin is post-translationally modified in the cell nucleus, similar to histones
Arginine 256 mono-methylation is a conserved mark mostly present in nuclear actin
A survey of actin R256me1 in yeast reveals a genome-wide landscape of nuclear actin
Actin R256me1 is implicated in transcription and linked to human diseases
ACKNOWLEDGMENTS
We would like to thank Joi Holcomb for help with the graphical abstract; Collene Jeter, James T. Stull, John Xue, Hironuri Funabiki, Mayuk Guha, Jacek Gaertig, Marvin Mercado, Andre Bui, and John O’Brien for their helpful suggestions; and Sarah Adai and Chris Contreras for reading of the manuscript. P.K. is a Scholar for Cancer Research at Cancer Prevention Research Institute of Texas (CPRIT). Funding to P.K. is supported by CPRIT: RR160067, University of Texas STARs Program, and Start-up funds from The University of Texas Health Science Center at Tyler, TX. X.S. is supported by funds and grants from the United States National Cancer Institute (K22CA100017), American Cancer Society, the United States National Institute of General Medical Sciences (2R01GM093104), CPRIT: RP160242, the Center for Cancer Epigenetics, and the IRG program at MD Anderson Cancer Center. K.M.T. is funded by NIH HL110869. NIH grant R01ES013768 to B.F.P. also supported this work. This study also made use of the Science Park Flow Cytometry and Cell Imaging Core as well as NGS Core, supported by CPRIT Core Facility Support Grant RP120348. We thank RIKEN for computing resources on the HOKUSAI-Great-Wave supercomputer used in this study.
Footnotes
DECLARATION OF INTERESTS
B.F.P. has a financial interest in Piconic, LLC, which uses the ChIP-exo technology implemented in this study, and could potentially benefit from the outcomes of this research.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108172.
REFERENCES
- Abe A, Saeki K, Yasunaga T, and Wakabayashi T (2000). Acetylation at the N-terminus of actin strengthens weak interaction between actin and myosin. Biochem. Biophys. Res. Commun 268, 14–19. [DOI] [PubMed] [Google Scholar]
- Albert P, and Redon C (1998). Efficient antibody generation using histone H1 subfractions purified from western blots. Anal. Biochem 261, 87–92. [DOI] [PubMed] [Google Scholar]
- Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, Smith EM, Quinion C, Smith PL, Kim L, Patton JT, et al. (2015). Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 125, 2530–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, and Robinson MD (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, Krajewski K, Shi X, Strahl BD, and Kutateladze TG (2016a). The Taf14 YEATS domain is a reader of histone crotonylation. Nat. Chem. Biol 12, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews FH, Strahl BD, and Kutateladze TG (2016b). Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol 12, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K, Liu X, Ferron F, Shu S, Korn ED, and Dominguez R (2008). Modulation of actin structure and function by phosphorylation of Tyr-53 and profilin binding. Proc. Natl. Acad. Sci. USA 105, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Meireles LM, and Bahar I (2011). ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, and Clarke SG (2009). Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, and Richard S (2005). Arginine methylation an emerging regulator of protein function. Mol. Cell 18, 263–272. [DOI] [PubMed] [Google Scholar]
- Berger EM, Cox G, Weber L, and Kenney JS (1981). Actin acetylation in Drosophila tissue culture cells. Biochem. Genet 19, 321–331. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, and Fink GR (1987). 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol 154, 164–175. [DOI] [PubMed] [Google Scholar]
- Campellone KG, and Welch MD (2010). A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol 11, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Berryman J, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, et al. (2014). AMBER 14 (San Francisco: University of California; ). [Google Scholar]
- Chao HP, Chen Y, Takata Y, Tomida MW, Lin K, Kirk JS, Simper MS, Mikulec CD, Rundhaug JE, Fischer SM, et al. (2019). Systematic evaluation of RNA-Seq preparation protocol performance. BMC Genomics 20, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chik JK, Lindberg U, and Schutt CE (1996). The structure of an open state of beta-actin at 2.65 A resolution. J. Mol. Biol 263, 607–623. [DOI] [PubMed] [Google Scholar]
- Chu JW, and Voth GA (2005). Allostery of actin filaments: molecular dynamics simulations and coarse-grained analysis. Proc. Natl. Acad. Sci. USA 102, 13111–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JW, and Voth GA (2006). Coarse-grained modeling of the actin filament derived from atomistic-scale simulations. Biophys. J 90, 1572–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium U (2014). Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42, D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, and Pedersen L (1993). Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- Fischle W, Wang Y, and Allis CD (2003). Histone and chromatin cross-talk. Curr. Opin. Cell Biol 15, 172–183. [DOI] [PubMed] [Google Scholar]
- Geng F, and Laurent BC (2004). Roles of SWI/SNF and HATs throughout the dynamic transcription of a yeast glucose-repressible gene. EMBO J 23, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu MM, Pinho Mda C, Richardson TE, Torrealba J, Buja LM Milewicz DM Raisanen JM and Burns DK (2015). The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathol. Commun 3, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, and Pugh BF (2011). Sequential recruitment of SAGA and TFIID in a genomic response to DNA damage in Saccharomyces cerevisiae. Mol. Cell. Biol 31, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer C, and Schekman R (1982). Actin from Saccharomyces cerevisiae. Mol. Cell. Biol 2, 1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet 39, 1488–1493. [DOI] [PubMed] [Google Scholar]
- Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, et al. (2009). Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet 84, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann WA, Arduini A, Nicol SM, Camacho CJ, Lessard JL, Fuller-Pace FV, and de Lanerolle P (2009). SUMOylation of nuclear actin. J. Cell Biol 186, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, and Head-Gordon T (2004). Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, and Simmerling C (2006). Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard PK, Sefton BM, and Firtel RA (1993). Tyrosine phosphorylation of actin in Dictyostelium associated with cell-shape changes. Science 259, 241–244. [DOI] [PubMed] [Google Scholar]
- Huang M, Qian F, Hu Y, Ang C, Li Z, and Wen Z (2002). Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nat. Cell Biol 4, 774–781. [DOI] [PubMed] [Google Scholar]
- Huisinga KL, and Pugh BF (2004). A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13, 573–585. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14 (33–38), 27–28. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AK, and Barton MC (2017). Bromodomain Histone Readers and Cancer. J. Mol. Biol 429, 2003–2010. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, and Holmes KC (1990). Atomic structure of the actin:DNase I complex. Nature 347, 37–44. [DOI] [PubMed] [Google Scholar]
- Kapoor P, and Shen X (2014). Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol 24, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Sahasrabuddhe AA, Kumar A, Mitra K, Siddiqi MI, and Gupta CM (2008). An unconventional form of actin in protozoan hemoflagel-late, Leishmania. J. Biol. Chem 283, 22760–22773. [DOI] [PubMed] [Google Scholar]
- Kapoor P, Kumar A, Naik R, Ganguli M, Siddiqi MI, Sahasrabuddhe AA, and Gupta CM (2010). Leishmania actin binds and nicks kDNA as well as inhibits decatenation activity of type II topoisomerase. Nucleic Acids Res 38, 3308–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Chen M, Winkler DD, Luger K, and Shen X (2013). Evidence for monomeric actin function in INO80 chromatin remodeling. Nat. Struct. Mol. Biol 20, 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR III, Mogilner A, Zebroski H, and Kashina A (2006). Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science 313, 192–196. [DOI] [PubMed] [Google Scholar]
- Karimi A, and Milewicz DM (2016). Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol 32, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi Y, Clements C, Mahadeo DC, Cotter DA, and Sameshima M (1998). High levels of actin tyrosine phosphorylation: correlation with the dormant state of Dictyostelium spores. J. Cell Sci 111, 2923–2932. [DOI] [PubMed] [Google Scholar]
- Koerber RT, Rhee HS, Jiang C, and Pugh BF (2009). Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol. Cell 35, 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer SB, and Gross DS (2009). SAGA and Rpd3 chromatin modification complexes dynamically regulate heat shock gene structure and expression. J. Biol. Chem 284, 32914–32931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K, Kinoshita H, Kuwabara Y, Nakagawa Y, Usami S, Minami T, Yamada Y, Fujiwara M, and Nakao K (2010). Myocardin-related transcription factor A is a common mediator of mechanical stress- and neurohu-moral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol. Cell. Biol 30, 4134–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, et al. (2014). AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Leone JW, Cook RG, and Allis CD (1989). Antibodies specific to acetylated histones document the existence of deposition- and transcription-related histone acetylation in Tetrahymena. J. Cell Biol 108, 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kang H, Liu R, Chen X, and Zhao K (2002). Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol. Cell. Biol 22, 6471–6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shu S, Hong MS, Levine RL, and Korn ED (2006). Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proc. Natl. Acad. Sci. USA 103, 13694–13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JK, and Wilkins MR (2012). Protein arginine methylation in Saccharomyces cerevisiae. FEBS J 279, 4423–4443. [DOI] [PubMed] [Google Scholar]
- Lu H, Fagnant PM, Bookwalter CS, Joel P, and Trybus KM (2015). Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci. USA 112, E4168–E4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, and Shen X (2009). Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol 10, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, and Shen X (2004). INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119, 767–775. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, Wehr TS, Hensley S, Bao Y, Shen J, Collins SR, et al. (2007). Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 130, 499–511. [DOI] [PubMed] [Google Scholar]
- Novick P, and Botstein D (1985). Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell 40, 405–416. [DOI] [PubMed] [Google Scholar]
- Obrdlik A, Kukalev A, Louvet E, Farrants AK, Caputo L, and Percipalle P (2008). The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol. Cell. Biol 28, 6342–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke CL, Cao J, Kwartler CS, Villamizar C, Byanova KL, Lim SM, Sreenivasappa H, Fischer G, Pham J, Rees M, et al. (2013). Smooth muscle hyperplasia due to loss of smooth muscle α-actin is driven by activation of focal adhesion kinase, altered p53 localization and increased levels of platelet-derived growth factor receptor-β. Hum. Mol. Genet 22, 3123–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, and Kutateladze TG (2006). Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaendtner J, Branduardi D, Parrinello M, Pollard TD, and Voth GA (2009). Nucleotide-dependent conformational states of actin. Proc. Natl. Acad. Sci. USA 106, 12723–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, and D’Andrea AD (2013). Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenu C, Athman R, Robine S, and Louvard D (2004). The co-workers of actin filaments: from cell structures to signals. Nat. Rev. Mol. Cell Biol 5, 635–646. [DOI] [PubMed] [Google Scholar]
- Rhee HS, and Pugh BF (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, and Pugh BF (2012). ChIP-exo Method for Identifying Genomic Location of DNA-Binding Proteins with Near-Single-Nucleotide Accuracy. Curr. Protoc. Mol. Biol Chapter 21, Unit21 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham TE III.. (2013). PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Rossetto D, Cramet M, Wang AY, Steunou AL, Lacoste N, Schulze JM, Côté V, Monnet-Saksouk J, Piquet S, Nourani A, et al. (2014). Eaf5/7/3 form a functionally independent NuA4 submodule linked to RNA polymerase II-coupled nucleosome recycling. EMBO J 33, 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, and Blobel G (1993). Isolation of the yeast nuclear pore complex. J. Cell Biol 123, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein PA, and Wen KK (2014). Insights into the effects of disease-causing mutations in human actins. Cytoskeleton (Hoboken) 71, 211–229. [DOI] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, and Berendsen HJC (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Physiol 23, 327–341. [Google Scholar]
- Saitou N, and Nei M (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Schweiger A, Mihalache O, Ecke M, and Gerisch G (1992). Stage-specific tyrosine phosphorylation of actin in Dictyostelium discoideum cells. J. Cell Sci 102, 601–609. [DOI] [PubMed] [Google Scholar]
- Scott RM, and Smith ER (2009). Moyamoya disease and moyamoya syndrome. N. Engl. J. Med 360, 1226–1237. [DOI] [PubMed] [Google Scholar]
- Shen X (2004). Preparation and analysis of the INO80 complex. Methods Enzymol 377, 401–412. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, and Wu C (2003). Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 12, 147–155. [DOI] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, et al. (2006). ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splettstoesser T, Noé F, Oda T, and Smith JC (2009). Nucleotide-dependence of G-actin conformation from multiple molecular dynamics simulations and observation of a putatively polymerization-competent superclosed state. Proteins 76, 353–364. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, and Kashina A (2013). Post-translational modification and regulation of actin. Curr. Opin. Cell Biol 25, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN, and Small EM (2013). Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl. Acad. Sci. USA 110, 16850–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, Forsyth NR, Wright WE, Shay JW, and Roth MG (2004). The establishment of telomerase-immortalized Tangier disease cell lines indicates the existence of an apolipoprotein A-I-inducible but ABCA1-independent cholesterol efflux pathway. J. Biol. Chem 279, 20866–20873. [DOI] [PubMed] [Google Scholar]
- Welch MD, Mallavarapu A, Rosenblatt J, and Mitchison TJ (1997). Actin dynamics in vivo. Curr. Opin. Cell Biol 9, 54–61. [DOI] [PubMed] [Google Scholar]
- Wertman KF, Drubin DG, and Botstein D (1992). Systematic mutational analysis of the yeast ACT1 gene. Genetics 132, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, and Kelley C (2012). Gnuplot 4.6: an interactive plotting program.
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, and Steinmetz LM (2009). Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamin R, and Morgan KG (2012). Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J. Physiol 590, 4145–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, and Wang W (2005). PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19, 1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, and Pugh BF (2013). SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kruk JA, and Reese JC (2008). Dissection of coactivator requirement at RNR3 reveals unexpected contributions from TFIID and SAGA. J. Biol. Chem 283, 27360–27368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, and Pugh BF (2011). A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332, 977–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel GJ, Fox DJ Jr., and Rivet DJ (2005). Moyamoya disease in adults: the role of cerebral revascularization. Skull Base 15, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, and Pauling L (1965). In Evolutionary divergence and convergence in proteins, Genes E, Bryson PV, and Vogel HJ, eds. (Academic Press; ), pp. 97–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data and codes were deposited in online database. RNA-sequencing data were deposited in the Gene Expression Omnibus public database with accession number GSE156817. Further, data generated in this study are available from the corresponding authors upon request.