

Abstract
Background
The leading cause of mortality in patients with Marfan syndrome (MFS) is thoracic aortic aneurysm and dissection. Notch signaling is essential for vessel morphogenesis and function. However, the role of Notch signaling in aortic pathology and aortic smooth muscle cell (SMC) differentiation in Marfan syndrome (MFS) is not completely understood.
Methods
RNA-sequencing on ascending aortic tissue from a mouse model of MFS, Fbn1mgR/mgR, and wild-type controls was performed. Notch 3 expression and activation in aortic tissue were confirmed with real-time RT-PCR, immunohistochemistry, and Western blot. Fbn1mgR/mgR and wild-type mice were treated with a γ-secretase inhibitor, DAPT, to block Notch activation. Aortic aneurysms and rupture were evaluated with connective tissue staining, ultrasound, and life table analysis.
Results
The murine RNA-sequencing data were validated with mouse and human MFS aortic tissue, demonstrating elevated Notch3 activation in MFS. Data further revealed that upregulation and activation of Notch3 were concomitant with increased expression of SMC contractile markers. Inhibiting Notch3 activation with DAPT attenuated aortic enlargement and improved survival of Fbn1mgR/mgR mice. DAPT treatment reduced elastin fiber fragmentation in the aorta and reversed the differentiation of SMCs.
Conclusions
Our data demonstrated that matrix abnormalities in the aorta of MFS are associated with increased Notch3 activation. Enhanced Notch3 activation in MFS contributed to aortic aneurysm formation in MFS. This might be mediated by inducing a contractile phenotypic change of SMC. Our results suggest that inhibiting Notch3 activation may provide a strategy to prevent and treat aortic aneurysms in MFS.
1. Introduction
Marfan syndrome (MFS) is a dominantly inherited disorder of connective tissue with prominent abnormalities in the ocular, skeletal, and cardiovascular systems. The leading causes of mortality in patients with MFS are progressive aortic root dilation, aneurysm formation, and aortic dissection and rupture [1]. Although β-blockers have been the primary medical therapy for MFS patients with aortic aneurysms, current therapies for MFS have limited efficacy. Therefore, new approaches are required to identify novel therapeutic targets.
MFS is caused by mutations in the gene encoding fibrillin-1, a large extracellular matrix (ECM) glycoprotein. Fibrillin-1 is a major constituent of ECM microfibrils, which act as a scaffolding protein for elastin deposition and formation of elastic fibers [2–5]. It also binds or interacts with a large variety of ligands to regulate the cellular behavior and control cell survival, differentiation, and response to injury [6–14]. It has been demonstrated that fibrillin-1 binds to a disintegrin-like and metalloprotease with thrombospondin type-1 motif (ADAMTS) and ADAMTS-like (ADAMTSL) proteins to modulate microfibril assembly [6, 7], interacts with bone morphogenetic proteins (BMPs) and integrins to regulate cell function and differentiation [8–14], and interacts with latent TGF-β binding proteins (LTBPs) to regulate and control the local TGF-β bioavailability [15, 16]. The cardiovascular manifestations associated with fibrillin-1 gene (FBN1) mutations have been linked to elevated TGF-β signaling [15, 16]. Therefore, fibrillin-1 not only serves as a structure protein but also regulates local growth factor signaling. Through studying murine models of MFS, it has been shown that the haploinsufficiency caused by FBN1 mutation plays an important role in the pathogenesis of MFS [17].
Notch signaling is essential for vessel morphogenesis and function. The notch genes encode large single-transmembrane receptors that mediate communication between neighboring cells that are crucial for cell fate decisions during organ development [18]. In mammals, four Notch receptors (Notch1–4) have been identified, all of which are composed of a large extracellular domain which mediates ligand interaction, a transmembrane domain, and an intracellular domain. Upon the binding of transmembrane ligands from Delta/Serrate/LAG-2 (DSL) family, DLL, and Jagged, Notch receptors undergo proteolytic cleavage by several proteinases, including γ-secretase [18]. This cleavage results in the release and translocation of the Notch intracellular domain (NICD) into the nucleus, where it interacts with the transcription factor CBF-1 to promote transcriptional activation and cell fate determination [19, 20]. In vascular smooth muscle cells (SMCs), the Notch2 and Notch3 receptors predominate [21–24]. Notch signaling is critical for the regulation of SMC contractile differentiation and extracellular matrix protein synthesis both in vitro and in vivo [21, 25, 26]. Studies by Crosas-Molist et al. showed that expression of SMC contractile protein markers was higher in the aortas of Marfan patients compared to healthy controls [27]. By using a murine model of MFS, Fbn1mgR/mgR mice, we have demonstrated that premature differentiation of SMCs contributes to aortic aneurysm pathology in MFS [28]. However, the role of Notch receptors in aneurysm formation and SMC differentiation in MFS remains unclear.
In this study, we observed increased Notch3 expression and activation in the aorta of Fbn1mgR/mgR mice. We investigated the impact of Notch3 in aortic aneurysm development in MFS. We found that inhibition of Notch signaling moderated VSMC contractile phenotype transition and attenuated aortic aneurysm development in Fbn1mgR/mgR mice.
2. Material and Methods
2.1. Mice
Heterozygous FBN1 mutant mice (Fbn1mgR/+) in a mixed C57Bl/6J:129 SvEv background were mated to generate homozygous mutant mice (Fbn1mgR/mgR) and wild-type (WT) littermates [28]. Both male and female mice were included in the study. Genotyping of mice was performed at postnatal day 7 (PD7) by PCR [28, 29]. All experiments were carried out in accordance with the guidelines of the University of Nebraska Medical Center Animal Care Committee for the use and care of laboratory animals. All mice were maintained in the pathogen-free animal facility. Human TAA samples (n = 10) were provided from the NIH GenTac Biobank. Age- and gender- matched control aortic samples (n = 10) were obtained from Nebraska Organ Recovery System.
2.2. Next-Generation RNA-Sequencing and Real-Time RT-PCR
RNA from ascending thoracic aorta of WT and Fbn1mgR/mgR mice was extracted using TRIzol reagent (Thermo Fisher Scientific). The mRNA was converted to cDNA by reverse transcription, and sequencing adaptors were ligated to the ends of cDNA fragments. Following amplification by PCR, the RNA-Seq library was sequenced using an Illumina NextSeq500 sequence analyzer (Illumina, san Diego, CA). The sequence reads were processed with a series of software [30–32]. The values of fragments per kilobase of transcript per million mapped reads (FPKM) were used for statistical comparison. The significant differential expression genes were defined by q value ≤ 0.05 [33]. A list of normalized differentially expressed genes were visualized with heatmap.
For real-time RT-PCR, aortic RNA was reverse transcribed into cDNA using iScript Reverse Transcription Supermix (Bio-Rad Laboratories, Inc.). Real-time RT-PCR was performed using SsoAdvanced Universal SYBR® Green Supermix according to the manufacturer's instruction (Bio-Rad Laboratories, Inc.) on an ABI StepOne machine (Thermo Fisher Scientific). Fold differences were calculated using mRNA expression normalized to 18 s rRNA and analyzed using the ΔΔCt relative quantification method.
2.3. Immunohistochemistry and Verhoeff-Van Gieson (VVG) Connective Tissue Staining
After perfusion-fixation with 10% neutral-buffered formalin, mouse ascending aortic tissues were embedded in paraffin and cut into 4 μm sections. For immunohistochemical staining, tissue sections were incubated with anti-Notch3 antibody (Santa Cruz Biotechnology, Dallas, TX) with a dilution of 1 : 250 for 32 min at 37°C. Discovery ChromoMap DAB kit (Roche, South San Francisco, CA) was used for antigen localization, and the slides were processed using Ventana Discovery Ultra instruments (Roche). For mouse tissue staining, microscopic fields (40X) of aorta were collected from each slide. Images were captured by Roche Ventana iScan HT (Roche). Definiens Tissue Studio software was used for the quantification of staining area and density. The mean density of Notch3 was determined for five regions of interest per aortic section. For human aortic sections, Notch3-positive cells were counted in each high power field (40x). The mean Notch3-positive cells were determined for five regions of each section. For VVG staining, mouse aortic sections were stained with Verhoeff's solution, ferric chloride, sodium thiosulfate, and Van Gieson's solution (Poly Scientific, Bay Shore, NY). Each staining cycle alternated between fixing and washing procedures. The slides were examined and photographed using light microscopy (20x; Nikon). The numbers of elastin breaks per field under 40x magnification were counted in each section. The mean values were determined for five regions of each section.
2.4. DAPT Treatment and Kaplan-Meier's Survival Curve
Beginning on PD10, WT (n = 10/group) and Fbn1mgR/mgR (n = 10/group) mice were treated with γ-secretase inhibitor, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) (Abcam, Cambridge, UK) until PD42. DAPT was suspended in DMSO. Mice were injected subcutaneously with 10 mg/kg of DAPT daily. A control group received DMSO only. Mice were randomly allocated in each group. WT littermates and Fbn1mgR/mgR mice without or with DAPT treatment were sacrificed at PD42. For histological studies, the ascending thoracic aortas were perfusion-fixed with 10% neutral buffered formalin and collected. For protein extraction, aortic samples were snap frozen in liquid nitrogen. To configure the Kaplan-Meier survival curves, mice (n = 15/group) were evaluated daily and survival recorded. Mice were followed up to 32 weeks, at which time all surviving mice were sacrificed.
2.5. High Frequency Ultrasound
Transthoracic ultrasound of WT and Fbn1mgR/mgR mice with or without DAPT treatment were performed with Vevo 3100 High Resolution In Vivo Micro-imaging system (VisualSonics, Toronto, Ontario, Canada) equipped with an integrated isoflurane-based anesthesia system. These studies were performed at 5, 8, and 12 weeks of age. Short-axis scans of the ascending aorta were performed using B-mode ultrasonography with the RMV 707 Scanhead. The aortic diameters were measured by M-mode in systole. Three independent measurements were obtained for each mouse. To define user variability, all echocardiographic studies were performed by 2 experienced individuals, and results were compared for agreement.
2.6. Isolation of Mouse SMC and Cell Culture
WT and Fbn1mgR/mgR mice (n = 6/group) were anesthetized and underwent laparotomy at PD28. Mouse thoracic aortas were isolated and minced. SMC isolation was described previously [28]. The cells were grown to confluence and passed after trypsinization with 0.25% trypsin. Aortic SMCs were maintained in vascular cell basal medium (ATCC, Manassas, VA) with 5% FBS. To examine the effect of DAPT, cells were incubated with serum-free medium and treated with 20 μM DAPT for 48 h. DMSO treatment was used as vehicle control. Cells were harvested for protein extraction.
2.7. Western Blot Analysis
Aortic proteins were extracted as previously described [29]. Briefly, the protein from the aortic tissue and cells was extracted with RIPA lysis and extraction buffer (Thermo Fisher Scientific, Waltham, MA). The protein concentration of aortic proteins was standardized with a Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA). Equal amounts (25-35 μg) of aortic extracts from WT and Fbn1mgR/mgR mice without or with DAPT treatment were loaded into 4–20% Criterion TGX precast gels (Bio-Rad Laboratories, Inc.). Following electrophoresis, the gel was transferred onto a 0.45 μm PVDF membrane (Bio-Rad Laboratories, Inc.). The membrane was incubated overnight at 4°C with antibodies directed against Notch3, α-actin, and β-tubulin (1 : 1,000) (Cell Signaling, Beverly, MA). Bound primary antibodies were detected with HRP-conjugated, species-specific, secondary antibodies (Cell Signaling) using the Clarity Western ECL system (Bio-Rad Laboratories, Inc.). The quantification was done using NIH ImageJ software and standardized by internal loading controls. The molecular sizes were determined using protein standards from Fermentas (Glen Burnie, MA).
2.8. Statistical Analyses
Data are presented as the mean ± the standard error (SE). Life table analysis was used for the Kaplan-Meier survival curve. For continuous variables, if the data were normally distributed, Student's t-test (comparison between two groups) or ANOVA with the appropriate post hoc test (comparison among groups of three or more) was used. Statistical significance was accepted at a P < 0.05.
3. Results
3.1. Notch3 Expression and Activation Were Increased in the Aorta of Both Humans and Mice with MFS
RNA-sequencing (RNA-Seq) analysis with aortic samples from WT and Fbn1mgR/mgR mice at PD28 (n = 4) was performed. The RNA-Seq data revealed an increased expression of Notch3 with upregulation of contractile proteins in Fbn1mgR/mgR mice (Figure 1(a)). This is consistent with our previous findings that aortic SMCs in MFS were prematurely differentiating into a contractile phenotype, i.e., upregulation of α-actin, SM22α, and calponin [28]. Increased Notch3 expression in the aortas of Fbn1mgR/mgR mice was confirmed with real-time PCR (Figure 1(b)). Immunohistochemical analysis of aortic tissue showed the protein levels of Notch3 were also increased in Fbn1mgR/mgR mice compared to WT controls at PD28 (Figures 1(c)–1(e)). Notch3 is a receptor that is composed of a large extracellular domain, a transmembrane domain, and an intracellular domain. Upon receptor-ligand binding at the cell surface, Notch3 undergoes proteolytic cleavage by proteinases, including γ-secretase [18]. This cleavage results in Notch3 activation and the release and translocation of the Notch3 intracellular domain (N3ICD) into the nucleus to promote transcriptional activation. We next tested activation of Notch3 in the aorta of WT and Fbn1mgR/mgR mice at PD28 by Western blot analysis. We found that active Notch3 (N3ICD) was significantly higher in Fbn1mgR/mgR mice compared to WT controls (Figures 2(a) and 2(b)). Furthermore, we sought to determine whether increased Notch3 activation is also seen in aortic samples from Marfan patients. The levels and activation of Notch3 in human aortic tissue were examined. Western blot analysis displayed that active Notch3 in the aorta was significantly higher in patients with MFS compared to matched controls (Figures 2(c) and 2(d)). Immunohistochemistry results showed that Notch3 levels were higher in Marfan patients than controls (Figures 2(e)–2(g)). These findings indicate that increased activation of Notch3 may play a role in the pathogenesis of human MFS and mouse model of MFS.
Figure 1.
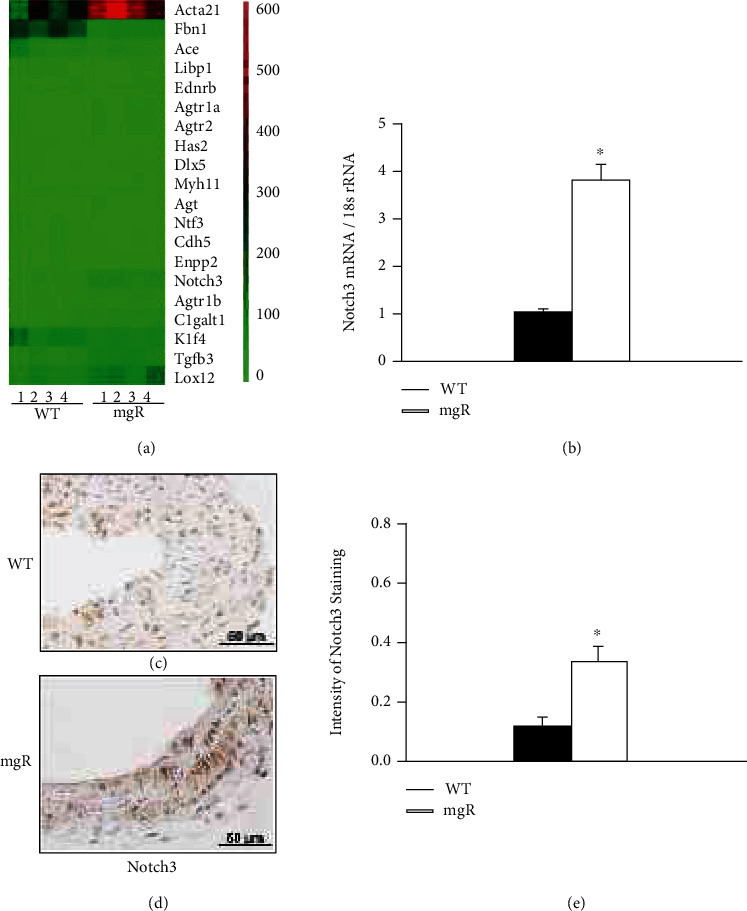
Genes were differentially expressed in the aorta of WT and Fbn1mgR/mgR (mgR) mice including Notch3, Fbn1, and α-actin. (a) Heatmap of the differentially expressed genes in the aorta of WT and mgR mice at PD28 (n = 4/group) was generated using data from RNA-Seq analysis. (b) Notch3 mRNA expression in the aorta of WT (n = 8) and mgR (n = 8) mice at PD28 was analyzed by real-time PCR. The bar graph (b) shows relative expression of Notch3 and 18 s rRNA. Immunohistochemical staining of Notch3 in the aortic sections from (c) WT and (d) mgR mice at PD28 (n = 4–6/group), respectively. Positive staining is shown in brown (DAB). Notch3-positive cells were quantitated by Definiens Tissue Studio software. Brown chromogen intensity is shown in (e) bar graph (n = 4–6/group). ∗P < 0.05 compared to WT controls; Student's t-test.
Figure 2.
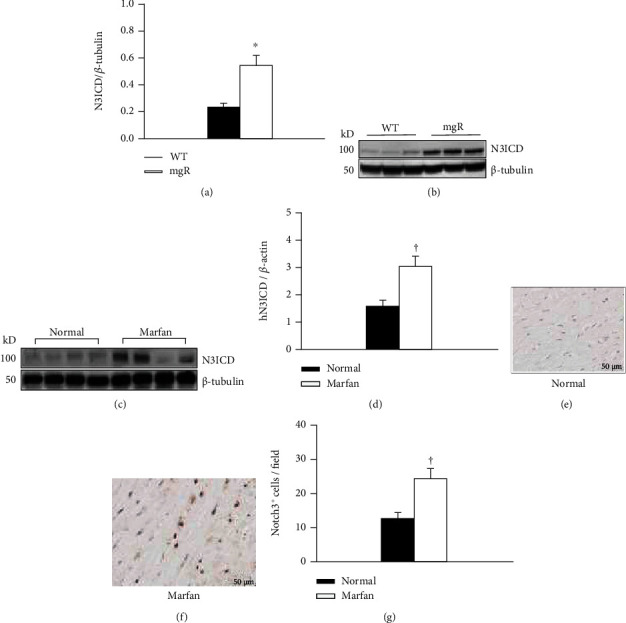
Notch3 activation was increased in the aorta of Marfan patients and Fbn1mgR/mgR (mgR) mice. Western blot analysis of active Notch3 (N3ICD) levels was performed on aortic protein from WT and mgR mice at PD28 (a) (n = 8/group) and Marfan patients and normal controls (n = 10/group) (c). The bar graphs show relative N3ICD levels in mouse (b) and human aortic tissue (d), respectively. ∗P < 0.05 compared to WT controls; †P < 0.01 compared to normal controls; Student's t-test. Immunohistochemical staining of human Notch3 in the aortic sections from normal control (e) and Marfan patients (f) (n = 6/group). Positive staining is shown in brown (DAB). Notch3-positive cells were counted (cells/high power field, 40x). (g) The values reflect the mean ± SE. †P < 0.01 compared to normal control; Student's t-test.
3.2. Inhibition of Notch3 Activation Preserved Elastic Fiber Integrity
Previous studies of Fbn1mgR/mgR mouse aortas demonstrated aortic elastic fiber disorganization and fragmentation [28, 34]. To determine the role of Notch3 activation in aortic elastin degradation and aneurysm formation of Fbn1mgR/mgR mice, WT and Fbn1mgR/mgR mice were treated with a γ-secretase inhibitor, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglucine t-butyl ester (DAPT). The treatment started at PD10 and stopped at PD42. The effects of DAPT treatment on aortic histology were evaluated by VVG staining. At PD42, while aortic lamellae in WT mice were intact and normal with treatment of DMSO or DAPT (Figures 3(a) and 3(c)), aortas of DMSO- (vehicle) treated Fbn1mgR/mgR mice showed elastic fiber disruption and medial hypertrophy (Figures 3(b)). However, DAPT-treated Fbn1mgR/mgR mice displayed a markedly lesser degree of aortic medial hypertrophy and elastin fragmentation (Figures 3(d) and 3(e)).
Figure 3.
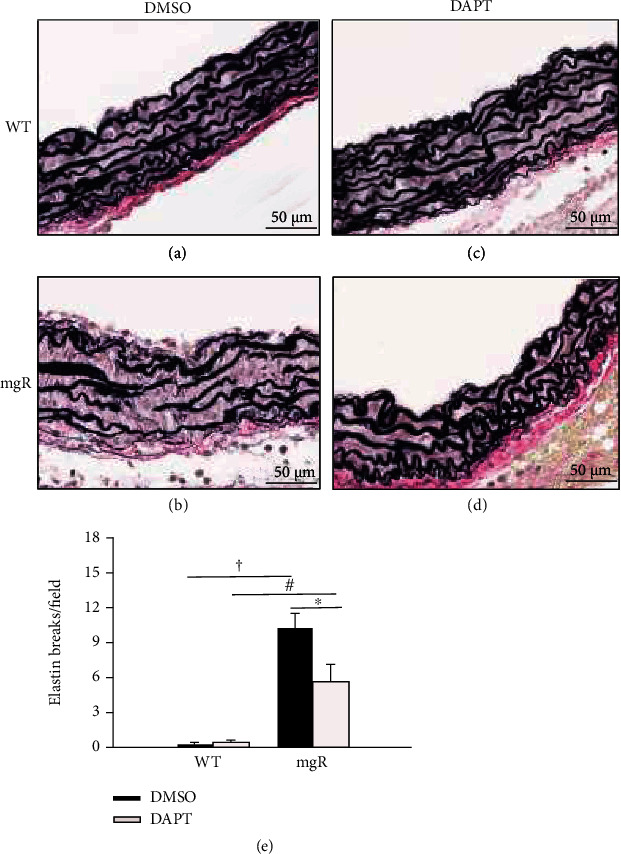
DAPT treatment, which inhibits Notch activation, prevented elastin fragmentation. (a–d) Verhoeff-van Gieson (VVG) staining of elastic fibers of the ascending aorta from WT and Fbn1mgR/mgR (mgR) mice at PD42 treated with DMSO (vehicle control) or DAPT; WT mice treated with DMSO (a) or DAPT (c); mgR mice treated with DMSO (b) or DAPT (d) at PD42. (e) Elastin breaks per field under 40x magnification in DMSO- or DAPT-treated WT and mgR mice (n = 5 aorta/groups). ∗P < 0.05, ǂP < 0.01, and †P < 0.001, ANOVA with Tukey-Kramer post hoc test.
3.3. DAPT Treatment Delayed Aortic Expansion and Improved Survival of Fbn1mgR/mgR Mice
The ascending aortic diameters of WT or Fbn1mgR/mgR mice receiving DAPT or DMSO were measured with high-frequency ultrasound at 5, 8, and 12 weeks of age (Figure 4(a)). Aortic diameters of DMSO-treated WT mice were significantly smaller than those of DMSO-treated Fbn1mgR/mgR mice at all time points (Figure 4(b)). At the 5-week time point, aortic diameters in DAPT-treated Fbn1mgR/mgR mice were significantly smaller than those in DMSO-treated Fbn1mgR/mgR mice (Figures 4(b) and 4(c)). The aortic diameters in DAPT-treated mice did not differ from DMSO-treated mice at 8- or 12-week time points. However, by 8 weeks of age, 3 out of 10 Fbn1mgR/mgR DMSO-treated mice died from aortic rupture. Furthermore, survival studies were performed in WT and Fbn1mgR/mgR mice treated with DAPT or DMSO (Figure 4(d)). The mice were followed up until death or to 32 weeks. Mice surviving to 32 weeks were euthanized. WT mice survived a normal lifespan; 2 out of 15 WT mice treated with DAPT died after 30 weeks of age. The mean survival of DMSO-treated Fbn1mgR/mgR mice (n = 15) was 138.4 ± 13.6 days, while Fbn1mgR/mgR mice treated with DAPT (n = 15) survived 199.9 ± 10.5 days. This result demonstrated that DAPT treatment significantly prolonged the lifespan of Fbn1mgR/mgR mice. Taken together, these data suggest that inhibition of Notch3 activation could play a role in preventing the early developmental abnormalities that occur in the Marfan aorta.
Figure 4.
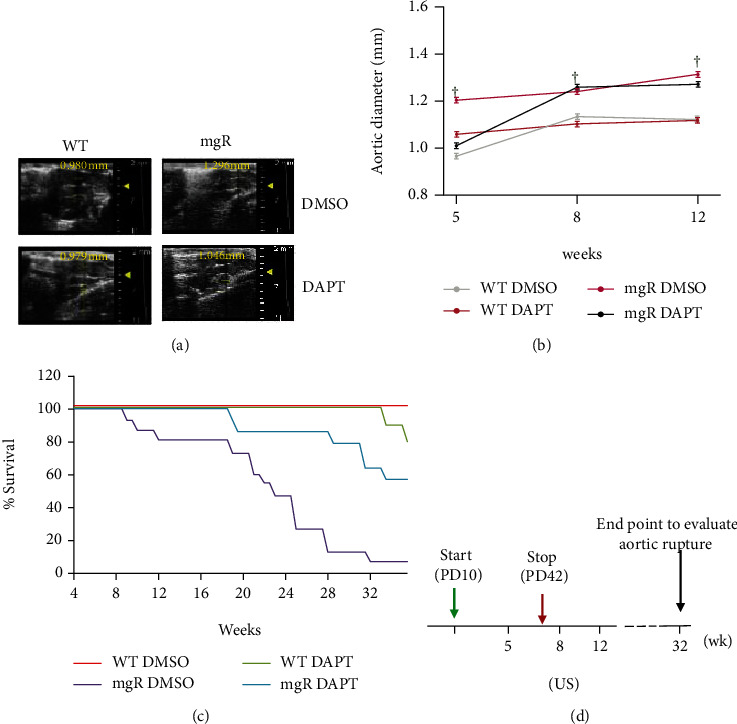
DAPT treatment delayed aortic expansion and improved survival of Fbn1mgR/mgR (mgR) mice. (a) Timeline of mouse experiments: WT and mgR mice were treated with DAPT (10 mg/kg) or DMSO by subcutaneous injection started at PD10 (start) and stopped at PD42 (stop). (b) Aortic diameters of mgR mice and WT littermates receiving DAPT or DMSO (n = 10/group) were measured by ultrasound (US) at 5, 8, and 12 weeks of age. The aortic diameters of DMSO-treated mgR mice were larger compared with WT controls at all time points (†P < 0.01 compared to WT controls). DAPT treatment inhibited aortic dilation of mgR mice at the 5-week time point (∗P < 0.01 compared to DMSO-treated mgR mice). (c) Representative ultrasound images of ascending aorta of mice at 5-week time point. (d) Life table analysis of WT (n = 10/group) and mgR (n = 15/group) mice treated with DAPT or DMSO from PD10 to PD42. Survival rate of mgR mice treated with DAPT was improved compared with DMSO-treated mgR mice (P < 0.01).
3.4. DAPT Treatment Reversed Abnormal SMC Phenotypic Switch in Fbn1mgR/mgR Mice
Our previous studies demonstrated that aortic SMCs of Fbn1mgR/mgR mice prematurely switched to a more contractile phenotype which contributed to aneurysm formation [28]. We sought to determine whether pharmacological inhibition of Notch3 activation in Fbn1mgR/mgR mice had effect on SMC differentiation. The levels of a SMC marker, α-actin, in the aorta were examined by Western blot (Figure 5(a)). While DAPT treatment inhibited aortic Notch3 activation in Fbn1mgR/mgR mice, it also reduced the expression of α-actin (Figures 5(a) and 5(b)). To confirm the effect of Notch3 activation on SMC differentiation, aortic SMCs were isolated from WT and Fbn1mgR/mgR mice. Consistent with the results from mouse aorta, N3CID levels were significantly higher in SMCs from Fbn1mgR/mgR mice than in SMCs from WT mice (Figures 6(a) and 6(b)). Furthermore, DAPT treatment reduced Notch3 activation and expression of contractile markers, α-actin and SM22α, in SMCs from Fbn1mgR/mgR mice. These results indicate that contribution of enhanced Notch3 activity to aneurysm formation in Fbn1mgR/mgR mice may be associated with aortic SMC differentiation. Taken together, these data suggest that inhibition of Notch3 activation could play a role in preventing the developmental abnormalities that occur in the Marfan aorta.
Figure 5.
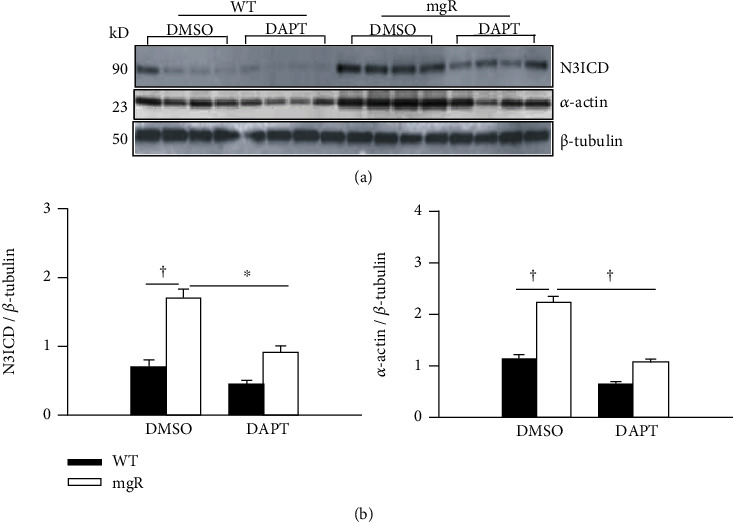
DAPT treatment blocked Notch3 activation and reversed SMC phenotype in the aorta of Fbn1mgR/mgR (mgR) mice. (a) Aortic protein from WT and mgR mice treated with DMSO and DAPT (n = 5-6/group) was extracted at PD 42. The levels of N3ICD and the smooth muscle cell differentiation marker, α-actin, were examined by Western blot analysis. (b) The bar graph shows relative active Notch3 (N3ICD) and α-actin levels in the aortic tissue. ∗P < 0.01 and †P < 0.001; ANOVA with Tukey-Kramer post hoc test.
Figure 6.

DAPT treatment inhibited Notch3 activation and reversed SMC phenotype of aortic SMCs from Fbn1mgR/mgR (mgR) mice. Aortic SMCs were isolated from WT and mgR mice and treated with DMSO or DAPT (20 mM) for 48 hrs. Protein from cells was extracted. (a) The levels of Notch3 activation (N3ICD), α-actin, and SM22α were examined by Western blot analysis. (b) The bar graph shows relative N3ICD, α-actin, and SM22α levels in the cells. ∗P < 0.05 compared to DMSO-treated mgR SMCs.
4. Discussion
We have shown that Notch3 expression and activation are increased in the aorta of MFS. During aortic development of MFS (Fbn1mgR/mgR) mice, increased Notch3 activation contributes to aortic aneurysm formation. Inhibition of Notch3 activation by γ-secretase inhibitor, DAPT, attenuates aortic elastic fiber fragmentation and aortic enlargement and improves mouse survival. The mechanism is due, in part, to mediation of aortic SMC phenotype modulation. These findings indicate that Notch3 is an important regulator of aortic SMC function and differentiation during aortic development and suggest that therapy modulating Notch3 signaling in the aorta may be beneficial in inhibiting aortic aneurysm formation in MFS.
The Fbn1mgR/mgR mice are one of commonly used murine models of MFS with a hypomorphic mutation of FBN1. Homozygous Fbn1mgR/mgR mice display clinical features and manifestations similar to classic patients of MFS. They die naturally at an average age of 2-3 months [34]. Previous studies showed that dysregulation of TGF-β activation contributed to pathogenesis of MFS [35, 36]. Pharmacological inhibition of TGF-β with TGF-β-neutralizing antibody attenuated aneurysm formation in MFS mice [16]. However, later results with the Fbn1mgR/mgR mouse model indicated that TGF-β could exert opposite effects on thoracic aortic aneurysm (TAA) pathology that broadly correlated with the early and late stages of TAA progression [37]. It was demonstrated that early treatment (PD16) with TGF-β-neutralizing antibodies exacerbated TAA formation, while later treatment (PD45) had a contrasting beneficial effect [37]. Our previous study also supported that initial consequence of FBN1 mutation was not accompanied by a significant increase in TGF-β activation [28]. Differential contributions of TGF-β signaling to aortic physiology early and later after birth in MFS prompt us to identify new biomarkers and therapies independent of the TGF-β signaling pathway to better understand aortic pathogenesis in MFS.
It is demonstrated that aortic development in mice occurs primarily between embryonic day (ED)14 and postnatal day (PD)14 and is typically complete by PD28 [38, 39]. During this time, aortic SMCs are highly proliferative and express structural matrix proteins that are important for vascular strength and compliance. After that, aortic SMCs shift out of the matrix phase and express the spectrum of contractile proteins to prepare for their unique contractile function. In patients with MFS, aortic tissue and aortic SMCs had increased expression of contractile protein markers (α-actin, SM22α, and calponin-1) and collagen [27]. Our previous studies using a murine model of MFS, Fbn1mgR/mgR mice, supported these findings [28]. We demonstrated that aortic SMCs in MFS mice prematurely differentiated into a contractile phenotype, which contributed to the pathology in MFS [28]. We also showed that abnormal phenotypic switching of aortic SMC led to reduced elastin synthesis in Marfan mice [28]. However, the mechanisms modulating SMC phenotypic switching and matrix protein production in MFS are not completely understood. In this study, we have found that expression and activation of Notch3 are increased in the aortas of mice and humans with MFS, which is consistent with the findings from other investigators [40, 41]. Notch signaling is critical for cell growth regulation and cell fate determination in many cell types, including aortic SMCs [42, 43]. The regulation of contractile differentiation and extracellular matrix synthesis by Notch activity has been well studied, but results have shown that Notch signaling has both anti-differentiation and prodifferentiation functions in vascular SMCs, suggesting context dependence and/or spatial-temporal regulation during development [44–46]. The Notch signaling pathway plays an important role in the developing cardiovascular system. Notch1 is the primary expressed receptor in endothelial cells. Notch1 mutation is linked to bicuspid aortic valve and plays an important role in aortic root dilation [47, 48]. Notch2 and 3 are mainly expressed in vascular SMC and required for mural cell expansion and maturation. Notch4 is an endothelial cell-specific mammalian Notch gene [49]. In addition, Notch ligands, such as Jagged 1, also play important roles in normal arterial development [50–52].
Notch3 is predominantly expressed in vascular SMC in humans [25, 53–55]. It is a key regulator of vascular SMC phenotypes and is required for arterial identity and vascular SMC maturation [25, 53, 56]. Expression of the constitutively active Notch3 resulted in cell shape changes and an increase in α-actin [25]. Our studies showed similar results that levels of contractile protein, α-actin, were higher in the aorta of Fbn1mgR/mgR mice compared to control mice [28]. To determine the impact of increased Notch3 activity on the phenotypic state of VSMC in MFS, we administered DAPT, a γ-secretase inhibitor, to Fbn1mgR/mgR mice starting at PD10. DAPT inhibited Notch3 signaling by preventing the proteolysis of Notch3. DAPT treatment also reduced the expression of SMC contractile marker, α-actin, in the aorta of Fbn1mgR/mgR mice, indicating that DAPT treatment could reverse aortic SMC phenotype in Fbn1mgR/mgR mice. Furthermore, we investigated the effects of DAPT treatment on the aortic development and aneurysm formation/rupture of MFS. Blocking Notch3 activation with DAPT in the early aortic development of Fbn1mgR/mgR mice attenuated aneurysm enlargement and preserved elastic fiber integrity, therefore prolonging the lifespan of Fbn1mgR/mgR mice.
In summary, our data demonstrate that the early and potentially important SMC phenotypic switch and the associated matrix abnormalities in the Marfan aorta are associated with increased Notch3 activation. Increased Notch3 activation in MFS contributed to perturbation of early aortic development in MFS through inducing contractile phenotypic switch of SMC. The aortic pathology can be attenuated by treatment with a drug that blocks Notch3 activation. Our results suggest that inhibition of the effect of Notch3 signaling in the early aortic development of MFS may be a useful strategy to prevent and treat aortic aneurysms in MFS.
Acknowledgments
This work was supported by the National Marfan Foundation (to WX) and the NHLBI of the National Institutes of Health under award numbers R01HL130623 (to WX) and R01HL062400 (to BTB). The Fbn1mgR/+ mice were provided by the laboratory of Francesco Ramirez, the Mount Sinai School of Medicine. We thank Lijun Sun for the assistance with histological analysis.
Abbreviations
- SMC:
Aortic smooth muscle cell
- MFS:
Marfan syndrome
- DAPT:
N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- ECM:
Extracellular matrix.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Murdoch J. L., Walker B. A., Halpern B. L., Kuzma J. W., McKusick V. A. Life expectancy and causes of death in the Marfan syndrome. The New England Journal of Medicine . 1972;286(15):804–808. doi: 10.1056/NEJM197204132861502. [DOI] [PubMed] [Google Scholar]
- 2.Reinhardt D. P., Keene D. R., Corson G. M., et al. Fibrillin-1: organization in microfibrils and structural properties. Journal of Molecular Biology . 1996;258:104–116. doi: 10.1006/jmbi.1996.0237. [DOI] [PubMed] [Google Scholar]
- 3.Kuo C. L., Isogai Z., Keene D. R., et al. Effects of fibrillin-1 degradation on microfibril ultrastructure. The Journal of Biological Chemistry . 2007;282(6):4007–4020. doi: 10.1074/jbc.M606370200. [DOI] [PubMed] [Google Scholar]
- 4.Kewley M. A., Williams G., Steven F. S. Studies of elastic tissue formation in the developing bovine ligamentum nuchae. The Journal of Pathology . 1978;124:95–101. doi: 10.1002/path.1711240205. [DOI] [PubMed] [Google Scholar]
- 5.Carta L., Pereira L., Arteaga-Solis E., et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. The Journal of Biological Chemistry . 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kutz W. E., Wang L. W., Bader H. L., et al. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. The Journal of Biological Chemistry . 2011;286:17156–17167. doi: 10.1074/jbc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubmacher D., Apte S. S. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biology . 2015;47:34–43. doi: 10.1016/j.matbio.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiedemann K., Sasaki T., Gustafsson E., et al. Microfibrils at basement membrane zones interact with perlecan via fibrillin-1. The Journal of Biological Chemistry . 2005;280:11404–11412. doi: 10.1074/jbc.M409882200. [DOI] [PubMed] [Google Scholar]
- 9.Whitelock J. M., Melrose J., Iozzo R. V. Diverse cell signaling events modulated by perlecan. Biochemistry . 2008;47:11174–11183. doi: 10.1021/bi8013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerever A., Mercier F., Nonaka R., et al. Perlecan is required for FGF-2 signaling in the neural stem cell niche. Stem Cell Research . 2014;12:492–505. doi: 10.1016/j.scr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thisse B., Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Developmental Biology . 2005;287:390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Wohl A. P., Troilo H., Collins R. F., Baldock C., Sengle G. Extracellular regulation of bone morphogenetic protein activity by the microfibril component fibrillin-1. The Journal of Biological Chemistry . 2016;291:12732–12746. doi: 10.1074/jbc.M115.704734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengle G., Tsutsui K., Keene D. R., et al. Microenvironmental regulation by fibrillin-1. PLoS Genetics . 2012;8, article e1002425 doi: 10.1371/journal.pgen.1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregory K. E., Ono R. N., Charbonneau N. L., et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. The Journal of Biological Chemistry . 2005;280:27970–27980. doi: 10.1074/jbc.M504270200. [DOI] [PubMed] [Google Scholar]
- 15.Massague J., Chen Y. G. Controlling TGF-β signaling. Genes & Development . 2000;14(6):627–644. doi: 10.1101/gad.14.6.627. [DOI] [PubMed] [Google Scholar]
- 16.Neptune E. R., Frischmeyer P. A., Arking D. E., et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nature Genetics . 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 17.Judge D. P., Biery N. J., Keene D. R., et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. The Journal of Clinical Investigation . 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopan R., Ilagan M. X. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell . 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iso T., Hamamori Y., Kedes L. Notch signaling in vascular development. Arteriosclerosis, Thrombosis, and Vascular Biology . 2003;23:543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- 20.Iso T., Kedes L., Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. Journal of Cellular Physiology . 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 21.Liu H., Zhang W., Kennard S., Caldwell R. B., Lilly B. Notch3 is critical for proper angiogenesis and mural cell investment. Circulation Research . 2010;107:860–870. doi: 10.1161/CIRCRESAHA.110.218271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.High F. A., Lu M. M., Pear W. S., Loomes K. M., Kaestner K. H., Epstein J. A. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proceedings of the National Academy of Sciences of the United States of America . 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.High F. A., Zhang M., Proweller A., et al. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. The Journal of Clinical Investigation . 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y., Takeshita K., Liu P. Y., et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation . 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domenga V., Fardoux P., Lacombe P., et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes & Development . 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gridley T. Notch signaling in vascular development and physiology. Development . 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 27.Crosas-Molist E., Meirelles T., Lopez-Luque J., et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology . 2015;35:960–972. doi: 10.1161/ATVBAHA.114.304412. [DOI] [PubMed] [Google Scholar]
- 28.Dale M., Fitzgerald M. P., Liu Z., et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan syndrome. PLoS One . 2017;12(10, article e0186603) doi: 10.1371/journal.pone.0186603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jespersen K., Liu Z., Li C., et al. Enhanced Notch3 signaling contributes to pulmonary emphysema in a murine model of Marfan syndrome. Scientific Reports . 2020;10, article 10949 doi: 10.1038/s41598-020-67941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C., Roberts A., Goff L., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nature Protocols . 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapnell C., Williams B. A., Pertea G., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology . 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C., Pachter L., Salzberg S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics . 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey J. D., Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America . 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pereira L., Lee S. Y., Gayraud B., et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proceedings of the National Academy of Sciences of the United States of America . 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habashi J. P., Judge D. P., Holm T. M., et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science . 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm T. M., Habashi J. P., Doyle J. J., et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science . 2011;332(6027):358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cook J. R., Clayton N. P., Carta L., et al. Dimorphic effects of transforming growth Factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arteriosclerosis, thrombosis, and vascular biology . 2015;35(4):911–917. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagenseil J. E., Mecham R. P. New insights into elastic fiber assembly. Birth Defects Research. Part C, Embryo Today . 2007;81:229–240. doi: 10.1002/bdrc.20111. [DOI] [PubMed] [Google Scholar]
- 39.Kelleher C. M., McLean S. E., Mecham R. P. Vascular extracellular matrix and aortic development. Current Topics in Developmental Biology . 2004;62:153–188. doi: 10.1016/S0070-2153(04)62006-0. [DOI] [PubMed] [Google Scholar]
- 40.Pedroza A. J., Koyano T., Trojan J., et al. Divergent effects of canonical and non-canonical TGF‐β signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. Journal of Cellular and Molecular Medicine . 2020;24(3):2369–2383. doi: 10.1111/jcmm.14921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cesarini V., Pisano C., Rossi G., et al. Regulation of PDE5 expression in human aorta and thoracic aortic aneurysms. Scientific Reports . 2019;9, article 12206 doi: 10.1038/s41598-019-48432-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fouillade C., Monet-Lepretre M., Baron-Menguy C., Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovascular Research . 2012;95:138–146. doi: 10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- 43.Manabe I., Owens G. K. CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo. The Journal of Clinical Investigation . 2001;107:823–834. doi: 10.1172/JCI11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morrow D., Cullen J. P., Liu W., Cahill P. A., Redmond E. M. Alcohol inhibits smooth muscle cell proliferation via regulation of the Notch signaling pathway. Arteriosclerosis, Thrombosis, and Vascular Biology . 2010;30:2597–2603. doi: 10.1161/ATVBAHA.110.215681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boucher J. M., Peterson S. M., Urs S., Zhang C., Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. The Journal of Biological Chemistry . 2011;286:28312–28321. doi: 10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Y., Urs S., Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circulation Research . 2008;102:661–668. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garg V., Muth A. N., Ransom J. F., et al. Mutations in NOTCH1 cause aortic valve disease. Nature . 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 48.Koenig S. N., LaHaye S., Feller J. D., et al. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. Insight . 2017;2(21) doi: 10.1172/jci.insight.91353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uyttendaele H., Marazzi G., Wu G., Yan Q., Sassoon D., Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development . 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- 50.Raas-Rothschild A., Shteyer E., Lerer I., Nir A., Granot E., Rein A. J. Jagged1 gene mutation for abdominal coarctation of the aorta in Alagille syndrome. American Journal of Medical Genetics . 2002;112(1):75–78. doi: 10.1002/ajmg.10652. [DOI] [PubMed] [Google Scholar]
- 51.Robert-Moreno A., Guiu J., Ruiz-Herguido C., et al. Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. The EMBO Journal . 2008;27:1886–1895. doi: 10.1038/emboj.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manderfield L. J., High F. A., Engleka K. A., et al. Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation . 2012;125:314–323. doi: 10.1161/CIRCULATIONAHA.111.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krebs L. T., Xue Y., Norton C. R., et al. Notch signaling is essential for vascular morphogenesis in mice. Genes & Development . 2000;14:1343–1352. doi: 10.1101/gad.14.11.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joutel A., Andreux F., Gaulis S., et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. The Journal of clinical investigation . 2000;105:597–605. doi: 10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villa N., Walker L., Lindsell C. E., Gasson J., Iruela-Arispe M. L., Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mechanisms of Development . 2001;108:161–164. doi: 10.1016/S0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 56.Ruchoux M. M., Domenga V., Brulin P., et al. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. The American Journal of Pathology . 2003;162:329–342. doi: 10.1016/S0002-9440(10)63824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.