

Abstract
Cancer vaccines harness the host immune system to generate antigen-specific antitumor immunity for long-term tumor elimination with durable immunomodulation. Commonly investigated strategies reintroduce ex vivo autologous dendritic cells (DCs) but have limited clinical adoption due to difficulty in manufacturing, delivery and low clinical efficacy. To combat this, we designed the “NanoLymph”, an implantable subcutaneous device for antigen-specific antitumor immunomodulation. The NanoLymph consists of a dual-reservoir platform for sustained release of immune stimulants via a nanoporous membrane and hydrogel-encapsulated antigens for local immune cell recruitment and activation, respectively. Here, we present the development and characterization of the NanoLymph as well as efficacy validation for immunomodulation in an immunocompetent murine model. Specifically, we established the NanoLymph biocompatibility and mechanical stability. Further, we demonstrated minimally invasive transcutaneous refilling of the drug reservoir in vivo for prolonging drug release duration. Importantly, our study demonstrated that local elution of two drugs (GMCSF and Resiquimod) generates an immune stimulatory microenvironment capable of local DC recruitment and activation and generation of antigen-specific T lymphocytes within 14 days. In summary, the NanoLymph approach can achieve in situ immunomodulation, presenting a viable strategy for therapeutic cancer vaccines.
Keywords: local controlled release, oncoimmunotherapy, in situ delivery, immunomodulation, subcutaneous implant, cancer vaccine
Introduction
Although immune checkpoint blockade antibodies have revolutionized cancer treatment, only 13% of patients respond, with limited success in most solid tumors1-3. Further, patients often experience relapse due to immunotherapy resistance2, 4. Consequently, there is a crucial unmet need to increase immunotherapy response rate across all cancer types and patient populations. Further, recent clinical success with oncoimmunotherapeutics have pushed towards developing patient-centric therapeutic approaches to eradicate solid tumors5-7. These methods harness the host immune system to generate antigen-specific targeted responses capable of overcoming an immunosuppressive tumor microenvironment8, 9. To this end, therapeutic cancer vaccines aim at mobilizing patient- and tumor-specific antitumor immune response10. By increasing immunogenicity and maintaining specificity, therapeutic cancer vaccines can serve as a potentially facile approach to eliminate tumors, evade tumor suppressive mechanisms and generate in situ immunological memory for long-term tumor elimination11-13.
Cancer vaccines must be capable of identifying tumorigenic cells without inducing autoimmunity14 and generating widespread antitumor immunity while limiting systemic toxicity and off-target effects. Various approaches in clinical and preclinical studies often target dendritic cells (DCs), the most powerful antigen-presenting cell. DCs can efficiently capture, internalize and process antigens for presentation to both CD4+ and CD8+ T cells after migration to secondary lymphoid organs15. Ex vivo protocols, in which autologous or allogeneic DCs are isolated, maturated and transfused into patients16, have low clinical adoption rate due to difficulty in manufacturing, delivery and minimal efficacy17, 18, and cost. Further, ex vivo strategies are limited by short lifespan of transfused cells, immune rejection, immunosuppressive tumor and low tissue uptake19-21. Moreover, ex vivo DCs requires repeated administration at high doses to achieve adequate biodistribution and tissue penetrance. High degree of patient adherence is often difficult to achieve18 even with implementation of technology-based outreach efforts22-24. Thus far, limited clinical impact is observed with ex vivo DC approaches16.
To combat these limitations, biomaterial-based cancer vaccines have demonstrated enhancement of the cancer-immunity cascade by generating antigen-specific responses in preclinical models25-28. Specifically, scaffold-based technologies were demonstrated to generate an immunostimulatory microenvironment to modulate the host immune system in situ29-32. While a number of these systems have been developed, they are often administered once and cannot be precisely tuned to patient responses25, 33, 34.
Fundamental to therapeutic cancer vaccines is the development of a self-contained approach to recruit, prime and initiate antitumor immune cells without external actuation or complex manipulation. In this work, we developed an immunostimulatory implant capable of continuously recruiting and activating immune cells locally for systemic-reaching immunomodulatory effects. Our technology, termed the “NanoLymph”, is a subcutaneously (SQ) implantable device with a dual-reservoir structure for both immune stimulants (drug) and antigens. We envision clinical deployment of the NanoLymph to occur in a four-step process (Figure 1). First, the NanoLymph is implanted SQ with a minimally invasive procedure similar to insertion of long-acting drug delivery implants35. The implantation incision and surrounding tissues is allowed to heal for two weeks to permit any potential acute foreign body response to resolve36, 37 (Figure 1A). Thereafter, the NanoLymph is transcutaneously (transQ) loaded with immunostimulants and hydrogel-encapsulated antigen in the drug and antigen reservoir, respectively (Figure 1B). Immune stimulants are eluted in a sustained manner across a nanoporous membrane into the interconnected antigen reservoir, which forms a drug-antigen gradient within the NanoLymph proximity. This gradient of immunostimulants autonomously triggers continuous recruitment of DCs locally. Recruited DCs interface directly with antigens encapsulated within the hydrogel (Figure 1C). DCs are activated against presented antigenic milieu and home to local lymph nodes to generate sustained systemic effects (Figure 1D). By recruiting and activating DCs within NanoLymph, we aim to harness their co-stimulatory properties to induce T cell immunomodulatory effects. Distinct from current vaccine approaches, the NanoLymph is designed as a long-term platform, with considerations for minimally invasive refilling for continued immune modulation in tune with patient needs.
Figure 1: NanoLymph deployment strategy.

(A) NanoLymph is subcutaneously implanted in for two weeks to allow foreign body reaction to equilibrate. (B) Transcutaneous loading of immunostimulants or antigens into the NanoLymph drug and antigen reservoir, respectively. Loading and venting needles for the drug reservoir are advanced into the NanoLymph through self-sealing silicone ports (visible in Figure 2). Venting needle permits sufficient loading of drug reservoir and removal of any entrapped air or excess fluids. Loading needle attached to antigen-loaded syringe for antigen reservoir filling is advanced through skin and nylon mesh. (C) Release of immunostimulants from drug reservoir into antigen reservoir and surrounding subcutaneous microenvironment creates stimulatory gradient for local recruitment of dendritic cells (DC) to NanoLymph. (D) Recruited DC are activated upon interaction with hydrogel-encapsulated antigens. Activated DC exfiltrate NanoLymph and home to secondary lymphoid organs.
Materials and Methods
NanoLymph fabrication and assembly
NanoLymph devices were fabricated with a stereolithography 3D printer (Form 3B, Formlabs, Massachusetts, United States of America (USA)) using biocompatible BioMed Clear resin (Formlabs). A solid modeling software (SolidWorks®, Dassault Systèmes, Vélizy-Villacoublay, France) was used to create a 3D dataset for the fabrication process. The D-shaped NanoLymph was designed with dimensions of 11.2 mm x 11.1 mm x 2.6 mm, presenting drug reservoir volume of 45 μL and antigen reservoir volume of 80 μL. Devices were cleaned and cured according to manufacturer instructions and sterilized using autoclave at 121°C, 15 psi for 30 minutes.
Devices were then assembled in aseptic manner under a laminar-flow hood in a stepwise fashion. Silicone plugs devised from medical-grade silicone and corresponding primer (NuSil, Avantor®, Pennsylvania, USA) were affixed and cured at 60°C for 10 minutes. Nanoporous membranes and nylon meshes were cut using a laser cutter (VLS 2.30, Universal Laser Systems, Arizona, USA), sterilized in 70% ethanol, affixed using the same silicone used for the plugs and cured overnight at room temperature. Assembled devices were incubated at 37°C in RPMI 1640 media with L-glutamine (Cytiva HyClone, Massachusetts, USA) for 48 hours to ensure sterility via no observed bacterial growth. Lastly, devices were filled using sterile phosphate buffered saline (PBS, Fisher Scientific, Massachusetts, USA) and 20% w/v pluronic F-127 (PF-127, Sigma-Aldrich, Missouri, USA) using aseptic technique.
In vitro material degradation
Resin, nanoporous membranes and nylon meshes (n=6 per group) were weighed and each placed in individual 1.5 mL Eppendorf tubes with sink solution of sterile 1X PBS with 2% fetal bovine serum (FBS, Sigma-Aldrich, Missouri USA) and incubated at either 37°C or 67°C (accelerated conditions). Samples were dried and weighed weekly (37°C) to evaluate material degradation; sink solution was fully replaced with fresh buffer at each time point. Material degradation was assessed via weight change percentage relative to day 0 using the following equation:
where wi is the original weight of each material and wt is the weight at each time point.
At predetermined endpoints, materials incubated in 67°C were removed, dried, fixed in 10% formalin, dehydrated in ethanol and sputtered with 7 nm iridium. Imaging was performed using Nova NanoSEM 230 under high vacuum setting and 5 kV electron beam at Houston Methodist Research Institute Scanning Electron Microscopy and Atomic Force Microscopy Core.
In vitro leakage
NanoLymph prototypes (n=3) were 3D printed as described above without drug elution holes. Prototypes were cleaned and silicone ports affixed. Two needles (23G and 30G) were inserted in either opposing silicone ports. A 1 mL solution of 10 mg/mL rhodamine B dye solubilized in PBS within a 1 mL syringe was attached to the larger bore needle. Refill of rhodamine B was visually assessed within venting needle. Implants were incubated at 37°C in 2 mL PBS, shaking continuously. Sink solutions were replenished according to predetermined schedule and analyzed via fluorescent spectrophotometry for presence of rhodamine B.
Cell proliferation assay
To gain insight on biocompatibility of assembled NanoLymph, MTT assays were performed using murine dendritic cells (DC2.4), murine fibroblasts (L929) and murine macrophage (RAW 264.7) cell lines (ATCC, Virginia, USA) in duplicate. DC2.4 cells were cultured in RPMI-1640 with L-glutamine and non-essential amino acids media supplemented with 10% FBS, 100 U penicillin (Gibco, Texas, USA), 100 U streptomycin (Gibco), 1X HEPES (Sigma-Aldrich, Missouri USA) and 0.0054x β-mercaptoethanol (Gibco). L929 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Corning, Massachusetts, USA) supplemented with 10% horse serum (ATCC, Virginia, USA), 100 U penicillin (Gibco), 100 U streptomycin (Gibco). RAW 264.7 cells were cultured in DMEM media supplemented with 10% FBS, 100 U penicillin (Gibco), 100 U streptomycin (Gibco). Media was replenished every 48-72 hours and cells were passaged at 70-80% confluency as assessed by visual inspection via light microscopy. NanoLymph extract was generated by incubation of each material utilizing a volume/surface area ratio of 0.8 mL/cm2 in appropriate media condition for each cell line at 37°C on shaker for 72 hours. This 100% extract was then diluted to 50% and 25% using complete appropriate media pre-incubated at 37°C for 72 hours.
For MTT proliferation assay, cells were seeded at 10,000 cells per well in a flat bottom 96 well plate and incubated overnight to allow cell adherence. The next day, culture medium was aspirated and appropriate media was replenished in duplicate with negative control as media only. Twenty-four hours later, MTT assay was performed using TACS MTT cell proliferation assay (R&D Systems, Minnesota, USA) following manufacturer instructions. Absorbance was measured at OD 570/690 nm with Synergy™ HT Microplate Reader (Bio-Tech Instruments, Inc., Vermont, USA).
In vitro drug release
NanoLymph prototypes (n=6 per condition) with drug reservoir only were printed and affixed with 30 nm polyethersulfone (PES) nanomembranes (Sterlitech, Washington, USA) following similar protocol as described above. Devices were loaded with 3 μg granulocyte-macrophage colony-stimulating factor (GMCSF, PeproTech, New Jersey, USA) solubilized in PBS via silicone ports. Devices were immersed in 200 μL sterile PBS sink solution at 37°C, each in individual microcentrifuge tubes. Sink solution was collected and changed daily for 15 days. Drug quantification was assessed via commercially available mouse GM-CSF enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, Massachusetts, USA) according to manufacturer’s protocol. Absorbance was measured at OD 450/620 nm with Synergy™ HT Microplate Reader.
To quantify resiquimod (R848) release, NanoLymph prototypes were assembled and loaded with 30 μg of small molecule cyanine7 carboxylic acid (Cy7-CA), a surrogate for R848 due to similar physical properties38. Devices were similarly immersed in sink solution and changed daily as described above. Daily absorbance was measured at OD 747 nm with UV-Vis Spectrophotometer (Cary 50; Agilent Technologies).
In vitro drug bioactivity
To evaluate drug stability over time, primary bone marrow derived DC (BMDC) harvested were isolated in accordance to established protocols39. BMDC were labeled with CFSE (CellTrace, Life Technologies, California, USA) at 5 μM per 1 × 106 cells for 20 minutes at 37°C, quenched with media, washed twice and allowed to incubate overnight starved of GMCSF. The next day, 20ng/mL of GMCSF from collected sink solutions were added. Negative control wells received equivalent volumes of sterile PBS while positive control wells received fresh 20ng/mL of stock GMCSF (PeproTech). After 48 hours, cells were isolated, fluorescently labeled with CD11c and Live/Dead staining (Supplementary Table 1) and assessed using LSR-II flow cytometer (BD Biosciences, California, USA). FlowJo v10.7.2 (FlowJo, Oregon, USA) was used for analysis.
Animals
8-13 week old female C57BL/6 and BALB/c mice (Taconic Biosciences, New York, USA) were housed at the Houston Methodist Research Institute (HMRI) Comparative Medicine Facility in Houston, TX. All animal experiments were carried out in accordance with provisions of Animal Welfare Act and principles of the National Institutes of Health Guide for Care and Use of Laboratory Animals with protocols approved by the Institutional Animal Care and Use Committee. All animals had access to clean, fresh water at all times and a standard laboratory diet and were monitored daily. All procedures were performed under anesthesia with inhaled isofluorane. Euthanasia was accomplished in a humane manner (carbon dioxide inhalation, confirmed by cardiac puncture) by techniques recommended by the American Veterinary Medical Association Guidelines on Euthanasia.
Minimally invasive SQ implantation
A small incision (approximately 1 cm) was made on dorsal right lateral side caudal to cervical spine. Blunt dissection was used to separate dermal layer from SQ fascia to generate SQ pocket on right flank. Assembled sterilized devices were placed into this pocket. The incision site was closed with wound clips (Roboz, Maryland, USA). All animals received five days of oral analgesic pre- and post-surgery as well as SQ buprenorphine SR (0.5 mg/kg), and bupivacaine (5 mg/kg) as a splash block during surgery for postsurgical pain.
In vivo NanoLymph biocompatibility assessment
To assess biocompatibility and toxicity of the NanoLymph, we implanted sterile NanoLymph in the flanks of C57BL/6 mice as described above. Mice body condition and vital signs (weight and rectal temperature) were assessed every two days. At predetermined endpoints, mice were euthanized as described above. Organs including liver, kidney, NanoLymph and surrounding fibrotic capsule were harvested for histological analysis as described below.
Histological analysis
C57BL/6 mice were each implanted with either one NanoLymph or one titanium control and euthanized as described above at predetermined endpoints. Fibrotic capsule immediately surrounding the SQ implant site, liver and kidney were isolated, fixed in 10% formalin, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin (H&E) or Masson’s Trichrome at the Research Pathology Core of Houston Methodist Research Institute (HMRI).
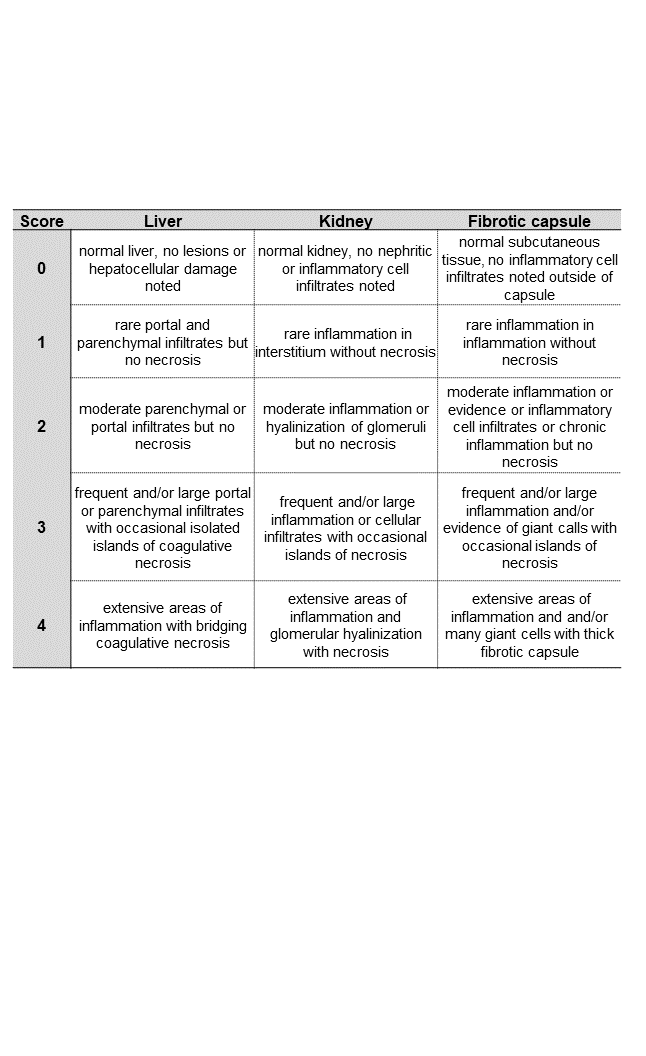
Sections were imaged using EVOS Cell Imaging Systems (ThermoFisher, Massachusetts, USA) mounted with 4x and 20x non-oil objective and 10x ocular attached to digital camera. Semiquantitative histopathological assessment was evaluated in accordance to inflammatory scoring system40, which was adopted from published standards41, 42. Sections were evaluated by two trained pathologists blinded to treatment groups following scoring guidelines (Supplementary Table 2). Numerical scores were aggregated and averaged to provide overall score.
Serum ALT analysis
Quantification of serum alanine transaminase (ALT) was used to assess impact of NanoLymph implantation on liver function. ALT quantification was assessed via commercially available mouse ALT Activity kit (Sigma Aldrich, Massachusetts, USA) according to manufacturer’s protocol. Briefly, serum was mixed with a coupled enzyme/substrate mix, resulting in a colorimetric product at OD 570 nm proportional to pyruvate generated at 37 °C. Quantification was assessed as per kit instructions as amount of enzyme that generates 1.0 μmole of pyruvate per minute at 37 °C.
NanoLymph refillability
Using Cadmium Telluride Quantum Dots 705 (Qdot 705; Sigma Aldrich), we evaluated the drug reservoir refill capability of the NanoLymph. NanoLymph were SQ implanted 10-week old BALB/c mice either 2 weeks or 22 weeks prior to refilling procedure, modeling short- and long-term use, respectively. Mice were shaved on right flank with 20 mm diameter circle extending from middle of implant prior to baseline imaging to avoid potential signal obscurance by fur. Mice were anesthetized with isofluorance and placed on left lateral decubitus position on imaging stage of the in vivo imaging system (IVIS, PerkinElmer, Massachusetts, USA). For quantum dot detection, we used an excitation wavelength of 640 nm and emission wavelength of 700 nm.
After baseline imaging, the drug reservoir was transcutaneously filled with Qdot 705 solubilized in PBS. Briefly, two needles (23G and 30G) were each inserted in either opposing silicone ports. A 1 mL solution of 1 mg/mL quantum dots solubilized in PBS within a 1 mL syringe was attached to the larger bore needle. Refill was visually assessed by evidence of vented PBS and excess Qdot 705 through 30G needle in vitro. Analogous procedure was used to refill antigen reservoir with 21G needle loaded with 20% w/v PF-127 in PBS in vitro. We imaged the mice using IVIS as described above to assess reservoir filling. To confirm that loaded cargo can be removed transcutaneously, we flushed the drug reservoir with 3 mL sterile PBS following refilling procedure described above. Mice were reimaged using IVIS as described above.
Living Image Software 4.5.2 (PerkinElmer) was used to acquire images. Images were qualitatively assessed by scaling all images on one consistent scale for comparison. Images were quantitatively assessed by drawing region of interest (ROI) around the NanoLymph and measuring fluorescent signal in radiance (p sec−1 cm−2 sr−1).
Biodistribution of drug and antigen from NanoLymph
To assess drug and antigen biodistribution from the NanoLymph, two cohorts of mice were used. For the NanoLymph group, BALB/c or C57BL/6 mice were shaved, anesthetized, imaged and refilled as described above 2 and 22 weeks after implantation. For the SQ injection group, equal concentration of either mock drug or antigen were injected SQ on right flank of BALB/c mice after baseline imaging. SQ injection of mock drug was delivered in 50 μL of PBS while antigen was delivered in 20% w/v PF-127 in 50 μL of PBS. SQ conditions were chosen to mimic cargo loaded within NanoLymph. Cyanine7 carboxylic acid (Cy7-CA, Abcam, Cambridge, United Kingdom), and Ovalbumin-AlexaFluor 647 (OVA-AF647, Invitrogen, Massachusetts, USA) were used to model R84838 and ovalbumin, respectively. Cy7-CA was solubilized in sterile PBS and delivered to the drug reservoir. OVA-AF647 was solubilized in sterile PBS and mixed in a 1:1 ratio with 40% PF-127 to prepare final solution of encapsulated OVA-AF647 in 20% PF-127. For Cy7-CA detection, we used an excitation wavelength of 710 nm and emission wavelength of 760 nm. For OVA detection, we used an excitation wavelength of 640 nm and emission wavelength of 680 nm. Mice were imaged over period of 14 days and fluorescent signal analyzed as described above.
Following euthanasia at predetermined endpoints, NanoLymph, surrounding fibrotic capsule, adjacent inguinal lymph node, liver, spleen, kidneys and lung were collected. For SQ groups, surrounding skin and tissue were collected in lieu of NanoLymph. Blood, collected via cardiac puncture, was transferred to serum separation tubes (BD Biosciences) for serum isolation following centrifugation at 2,000 x g for 15 minutes. Serum was collected and stored at −80°C. Ex vivo fluorescence imaging was performed on organs using IVIS as described above. Using Living Image 4.5.2 software, ROI were drawn and radiance values were measured as described above. Normalization was performed relative to fluorescent imaging at 1 hour.
Immune cell homing and general multicolor flow cytometry assessment
To assess efficacy of immune cell modulation, NanoLymphs were implanted SQ in the flank of C57BL/6 mice as previously described. After two weeks, mice were randomized into four groups: PBS control, drug only, antigen only and drug + antigen. Reservoirs were transcutaneously filled as described above. For groups receiving drug, mice were refilled with 3 μg GMCSF and 30 μg R848 solubilized within 50 μL PBS. For conditions receiving antigen, 1 μg OVA (257-264) SIINFEKL peptide labeled with FAM (AnaSpec, California, USA) was solubilized in 100 μL of 20% w/v PF-127 in PBS. Mice were euthanized 14 days following transQ filling. NanoLymph and adjacent inguinal lymph node were collected for flow cytometry analysis. Cells were isolated from NanoLymph by aspiration through 1mL syringe. Adjacent inguinal lymph nodes were suspended to single-cell suspensions by mechanical filtration in 70 μm cell strainer. Single cells were first blocked with antimouse CD16/CD32/Fc block (eBioscience, California, USA) and stained using either myeloid, T cell or OVA tetramer panel (Supplementary Table 1) for extracellular staining at 4°C for 30 minutes. H-2K(b) SIINFEKL PE-labeled appropriate tetramer was provided by NIH Tetramer Core Facility (Emory University, Georgia, USA). After washing with staining buffer twice, cells were fixed overnight with fixation buffer (eBioscience) and washed prior to data acquisition. Fluorescent data were collected using LSR-II flow cytometer (BD Biosciences, California, USA) and analyzed using FlowJo v10.7.2 (FlowJo, Oregon, USA). Analysis was performed after exclusion of debris, doublets and non-viable cells. Analysis was performed on a population-level relative to respective parent population via FlowJo43.
Statistical analysis
Results are expressed as mean ± standard deviation. All statistical analyses were performed using GraphPad Prism 9.1.1 software (GraphPad Software Inc., San Diego, CA, USA). Student t-test, one-way ANOVA or two-way ANOVA were performed when appropriate to determine statistical significance between experimental groups. P-values less than 0.05 were considered statistically significant with following symbols indicating significance as follows: n.s. not significant, * P≤0.05, ** P≤0.01, *** P≤0.001, **** P≤0.0001.
Results
Development, fabrication and stability of the NanoLymph
We developed the NanoLymph to serve as a local immunomodulatory niche capable of DC recruitment and activation as well as downstream T cell priming. The NanoLymph is a dual reservoir system presenting a “D-shape” with two connected reservoirs, each for drug and antigen cargo (Figure 2A-D). The drug reservoir is connected to the antigen reservoir via two square windows, each measuring 0.8 × 0.8 mm, upon which a nanoporous PES membrane is affixed using biocompatible silicone glue (Figure 2C). Drug sustainably elutes from the drug reservoir through the nanoporous membrane into the antigen reservoir and thereafter into the SQ space, creating a concentration gradient and an immune stimulatory microenvironment. The antigen reservoir contains a hydrogel capable of housing recruited cells and is enclosed via dual nylon meshes (Figure 2B). The inner 100 μm × 100 μm mesh provides cell and hydrogel retention, while the outer 300 μm × 300 μm mesh allows for mechanical support44. Both drug and antigen reservoirs can be accessed transcutaneously in a minimally-invasive procedure previously established in our group44. Self-sealing silicone plugs located on either end of the drug reservoir serves as loading and venting ports, respectively, for transQ refill. The antigen reservoir is accessed via simple advancement of needle through the skin and nylon meshes to reach the reservoir.
Figure 2: NanoLymph design and characterization.

(A) Rendering of NanoLymph cross-section with view of drug and antigen reservoir as well as self-sealing silicone loading ports on opposing sides. (B) SEM image of nylon woven mesh membrane; scale bar 500 μm. (C) SEM image of polyethersulfone (PES) nanoporous membrane; scale bar 10 μm. (D) NanoLymph shown to scale in comparison with commercially available M&M™; scale bar 500 mm. (E) Degradation of NanoLymph components in vitro as function of change in weight from baseline. Horizontal dotted line indicates 75% weight change. NanoLymph in purple, nylon mesh in teal, nanoporous membrane in orange. (F) SEM image of nylon woven mesh (scale bar: 500 μm) and PES nanoporous membrane (scale bar: 10 μm) 55 days after incubation at 67 °C (G) Percent of rhodamine B leaked from in vitro implants after 18 weeks
The NanoLymph is fabricated using stereolithography 3D printing in biocompatible resin. Stereolithography allowed us to create the complex hollow structure with high dimensional tolerances and accuracy and smooth surface morphology.45 Currently used in dental implants, stereolithography offers rapid scalability and customizable flexibility46. The NanoLymph presented here is optimized for murine models and measures 11.2 × 11.1 × 2.6 mm3 (height × width × depth), smaller in size than a M&M™ candy (Figure 2D).
As the NanoLymph is intended for long-term deployment, it is critical to assess material stability of all components. Thus, we assessed degradation of the resin, nanoporous membrane and nylon meshes though specimen weight change in vitro over time at 37°C in simulated interstitial fluid44, 47. Simulated interstitial fluid, composed of PBS with 1% FBS, was prepared to mimic fluid, salt and protein composition found in the SQ space. We observed a minimal change in weight; all materials remained at or above 70% of original weight throughout duration of study (Figure 2E)47. The resin material encompassing the NanoLymph exhibited less than 1% decrease in weight, indicating no substantial degradation over time. Early time points with the PES nanomembrane observed between 5-18% increase in weight attributable to initial water absorption. This finding is consistent with previous reports for similar polyesters48, 49. Further, SEM imaging of woven meshes and nanoporous membranes showed intact material integrity for 55 days in vitro at 67°C, corresponding to over 300 days in vivo50 (Figure 2F). Our laboratory has previously demonstrated similar material retained integrity in a rat animal model for up to 10 weeks44. Additionally, it is critical to ensure no drug will leak through resin structure or silicone ports. We previously demonstrated that silicone ports remain patent after repeated probing via a small-gauge needle with no significant leakage of drug51. To test potential leakage, rhodamine B dye was loaded via resealable silicone ports in NanoLymph prototypes without drug elution membranes and incubated in a sink solution of 1X PBS for up to 18 weeks. The presence of rhodamine B in sink solution was assessed via fluorescent spectrophotometry. We observed no significant leakage throughout the study up to 18 weeks (Figure 2G). Consistent with this report, we have previously demonstrated
NanoLymph is non-cytotoxic and biocompatible
We evaluated NanoLymph cytotoxicity via MTT cell viability assay with murine dendritic cells (DC2.4), fibroblasts (L929) and macrophages (RAW 264.7). These cell types would interact with the NanoLymph throughout the duration of SQ implantation and thus would be the most relevant to our study. Cells incubated with varying concentrations of NanoLymph extract maintained viability above the 70% threshold52 comparable to control (complete media only) and showed no difference in viability across all cell lines (Figure 3A).
Figure 3: NanoLymph cytotoxicity and biocompatibility.

(A) Viability of murine dendritic cells (DC2.4), fibroblasts (L929) and macrophages (RAW) after incubation with NanoLymph extract in media or complete media alone control (Veh) assessed via MTT assay. Horizontal dotted line represents 70% cell viability (B) Changes in weight (black) and rectal temperature (blue) assessed thrice weekly in mice after NanoLymph implantation. (C) Thickness of fibrotic capsule in resin (gray) and titanium (blue) samples across endpoints. (D) Pathological scoring of histological sections of fibrotic capsule. (E) Hematoxylin and Eosin (H&E) and Masson’s Trichrome stained fibrotic capsule at 7, 14, and 28 weeks post implantation with NanoLymph versus titanium control. Scale bar 100 μm. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Graphs are plotted as mean ± SD. Statistical significance determined via one-way ANOVA.
To evaluate NanoLymph biocompatibility in mice, we implanted NanoLymph SQ in the flank of C57BL/6 mice. To monitor for signs of device-induced toxicity, we assessed mice overall well-being, weight and rectal temperature thrice weekly (Figure 3B) over 28 days. Under observation, mice were active, well-groomed and in good health consistent with no indication of pain or distress53. The implantation site remained healthy with no evidence of drainage or abscess formation throughout the study. Further, mice had no notable weight loss compared to pre-implantation baseline and non-significant variability in core body temperature. Similarly, in a long-term implantation study of 19 weeks, the mice had healthy weight gain (Supplementary Figure 1A) and no significant variation in body temperature (Supplementary Figure 1B).
Implanted materials undergo a multi-step orchestrated cascade termed the “foreign body response” (FBR) encompassing release of inflammatory cytokines that recruit neutrophils, macrophages and fibroblasts leading to fibrotic encapsulation37. Although this is an expected physiological response, unresolved FBR may pose a significant challenge to implant function54. Here, we evaluated NanoLymph-induced FBR via histologic analysis at the implantation site. We used hematoxylin and eosin (H&E) and Masson’s Trichrome to evaluate cellular infiltration and collagen deposition, respectively. Specifically, we assessed the NanoLymph-induced fibrotic response in early (7, 14 and 28 days) and late (154 days) post-implantation period, compared to titanium, a material commonly used in medical implants. We demonstrated that the fibrotic capsule remained thin throughout the investigation. Although the resin material initially generated a thicker fibrotic capsule than that of titanium control, the two conditions equilibrated by 14 days (Figure 3C). In both materials, fibrotic capsule thickness steadily increased over time. Fibrotic capsule thickness steadily increased in both resin and titanium implants (Figure 3C). By day 154 time point, we noted no significant increase in thickness in the resin implants compared to day 28. Further, histological assessment of cellular infiltrates, necrosis and tissue damage55 in the fibrotic capsule and surrounding tissue of NanoLymph exhibited similar characteristics to that of titanium at all endpoints. The NanoLymph presented minimal inflammatory response localized to implant site with underlying healthy surrounding SQ tissue and muscle (Figure 3D, E) comparable to control titanium implants. This response is typical of FBR to medical implants without evidence of chronic or ongoing inflammation.
To assess potential device-induced adverse effects, we assessed signs of liver toxicity or inflammation in mice SQ implanted with NanoLymph. Serum ALT enzyme levels were analyzed as a measure of hepatic damage at 17, 21 and 28 days post-implantation. All mice exhibited no significant increase in ALT levels at any endpoint (Supplementary Figure 2A). Further, across all endpoints, there was minimal nephritic and hepatic inflammation comparable to titanium control by histological evaluation (Supplementary Figure 2B, 2C). These results demonstrate that NanoLymph is biocompatible and develops minimal FBR comparable to known biocompatible titanium.
Constant and sustained release of drug from NanoLymph
To study the immune response triggered by the NanoLymph, we used two immunostimulants for their DC recruitment and maturation properties. Our drug cocktail is composed of granulocyte-macrophage colony stimulating factor (GMCSF) and a Toll-Like Receptor 7/8 agonist, Resiquimod (R848). GMCSF is a commonly used cytokine in ex vivo DC-based vaccines in both preclinical and clinical trials16 with positive immunogenic responses. GMCSF is an endogenous cytokine produced by multiple immune cell types in response to inflammation or infection. It promotes DC generation, recruitment and survival56, 57. R848, an imidazoquinolone compound, enhances DC survival, induces cytokine production and promotes tumor-antigen T cell priming58-63. We hypothesized that GMCSF with R848 can synergistically generate an immunostimulatory microenvironment for continuous DC recruitment and activation at the NanoLymph to achieve antigen-specific T cell responses.
To evaluate drug release, we performed in vitro assays in which the drug reservoir mounted either 30nm or 100nm PES nanoporous membranes. PES was chosen for its high dimensional stability, resistance to oxidizing agents, resistance to biofouling, thermal stability and strong mechanical properties64-66. Implants were loaded with 3 μg of GMCSF and 30 μg Cy7-CA. Cy7-CA is a fluorophore used as a surrogate for R848 due to their similar physical and molecular properties and ease of detection via UV-Vis absorbance38. GMCSF release from 30 nm nanoporous membranes exhibited a sustained release, with an average release rate of 2.5 ng/day over 14 days (Figure 4A). There was a moderate burst release the first three days with an average of 4.5 ± 2.8 ng GMCSF released per day. As expected, GMCSF release from larger membrane sizes of 100 nm demonstrated rapid near-complete release within first 4 days and thereafter was undetectable. GMCSF release via 100 nm membranes averaged 5.6 ± 2.9 ng per day for first 4 days. Once devices were exhausted, release rate dropped to average 0.5 ± 0.1 ng for remainder 10 days. Cy7-CA release showed a typical exponentially decaying concentration-driven diffusion profile presenting higher release rates at first (3392 ng/day for the first 5 days) followed by lower diffusion rates (257 ng/day after day 5) using membrane sizes of 30 nm. Release using 100 nm followed comparable profile with initial higher release rates (4009 ng/day) for first 5 days followed by 215 ng/day after day 5 (Figure 4B).
Figure 4: Drug release from NanoLymph.

(A) Cumulative release of GMCSF through 30 nm (blue, circle) and 100 nm (gray, triangle) nanoporous membrane. (B) Cumulative release of Cy7-Ca through 30 nm and 100 nm nanoporous membrane. (C) Viability of primary bone marrow derived dendritic cells (BMDC) cultured in released GMCSF (blue) compared to stock GMCSF (grey). Graphs are plotted as mean ± SD.
To assess drug bioactivity after release from the NanoLymph, we cultured primary murine bone marrow derived DC (BMDC) with released GMCSF compared to fresh stock. These BMDCs require daily supplementation with GMCSF for optimal growth in culture. There was no significant difference in BMDC viability between fresh or NanoLymph-released GMCSF across all time points (Figure 4C). These results demonstrated that the NanoLymph can release drugs in a constant manner while maintaining its biological activity, critical for continuous DC recruitment.
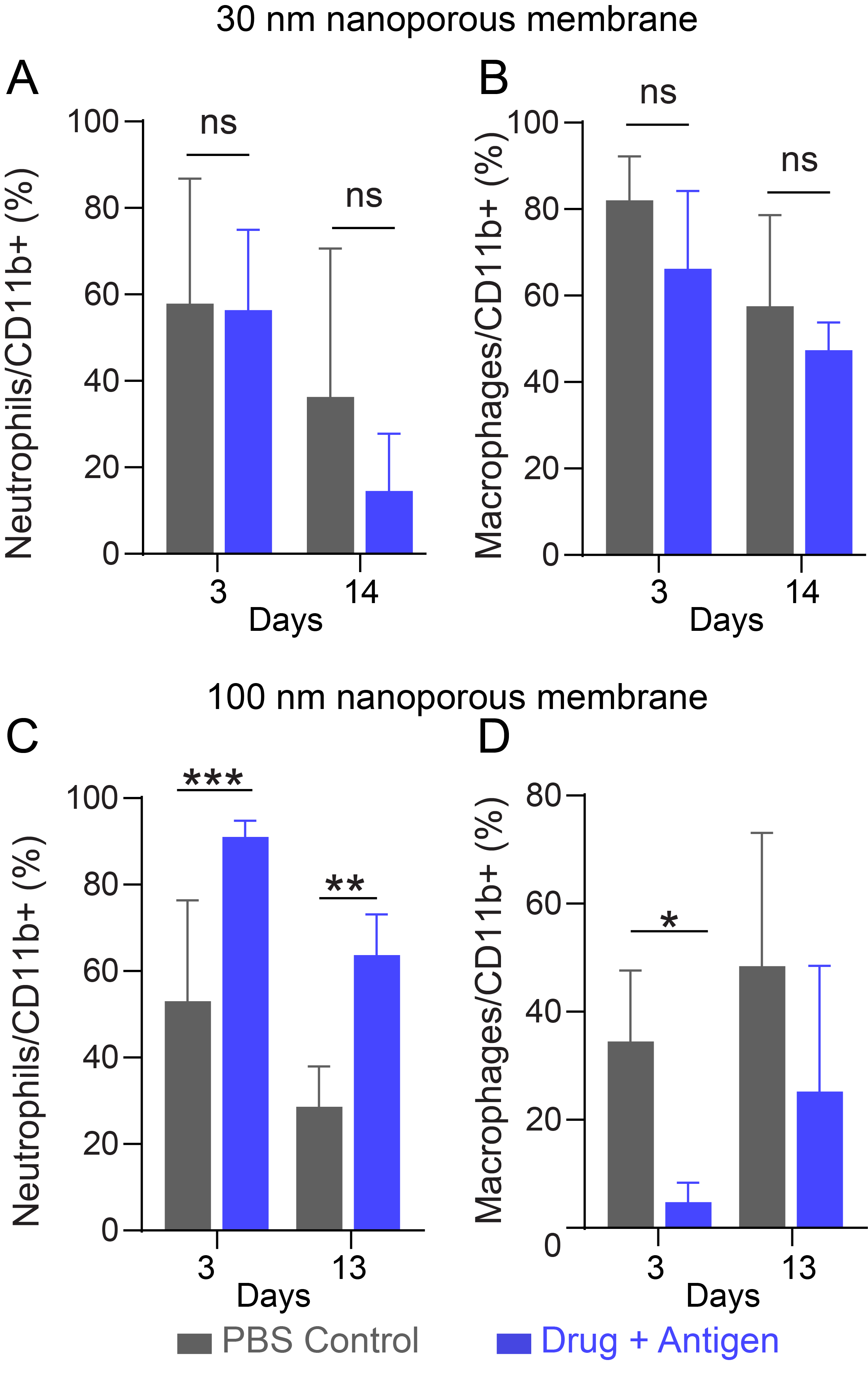
In a pilot in vivo study, we investigated cellular inflammatory responses to SQ implanted NanoLymph. We compared NanoLymph mounted with either 30 nm or 100 nm nanoporous membranes. After implantation, we allowed two weeks for potential acute FBR to subside. Thereafter, NanoLymph was transcutaneously loaded with GMCSF and R848 in the drug reservoir and ovalbumin peptide (OVAp) in the antigen reservoir. OVAp was encapsulated in 20% w/v PF-127 hydrogel following previously established protocols by our laboratory44. Use of OVAp was included to ensure activation of possible antigen cross-presentation by myeloid cells67, 68. For the control implants, PBS and 20% PF-127 hydrogel was used in the drug and antigen reservoir, respectively.
In the 30 nm membrane NanoLymph cohort, we did not observe significant neutrophil or macrophage influx on either days 3 or 14 post-drug and antigen loading (Supplementary Figure 3A, B). On the contrary, we observed a statistically significant neutrophil infiltration from NanoLymph with 100 nm nanoporous membranes compared to PBS control (Supplementary Figure 3C). This strong acute inflammatory response was observed on both days 3 and 13 post-drug and antigen loading. Thus, we conclude that rapid bolus dosage of GMCSF likely triggered a local neutrophil influx consistent with literature reports69, 70. We observed no drug-dependent effect of local macrophage recruitment at either time point, indicating no evidence of chronic inflammation (Supplementary Figure 3D). In fact, we noted a statistically significant decrease in macrophages locally recruited to NanoLymph eluting drug and antigen compared to control at day 3. Therefore, in consideration of small drug molecule sizes and to avoid unwanted acute inflammatory responses, we selected the 30 nm nanoporous membrane for the following studies.
In vivo NanoLymph transcutaneous refilling and drug release
The NanoLymph is designed for minimally invasive transQ loading of both the drug and cell reservoir. As a proof-of-concept, we used quantum dots (Qdot 705), which allowed for visualization via IVIS live animal imaging, to demonstrate transQ refillability of NanoLymph in mice. NanoLymph drug reservoir was transcutaneously filled with Qdot 705 two weeks after SQ implantation. Briefly, two needles were each inserted in either opposing silicone ports, which were easily palpable44. A 1 mL syringe with Qdot 705 solution was attached to the loading needle, while the other needle was left open for venting excess fluids. Successful drug reservoir refill was visually confirmed using IVIS imaging (Figure 5A). Fluorescent signal intensity of drug disappeared upon flushing the reservoir with sterile saline. Further, we demonstrated similar results 22 weeks after SQ implantation, indicating the reservoir remained patent and intact, suggestive of NanoLymph capability for long-term implantation (Figure 5A).
Figure 5: NanoLymph refillability and drug retention in vivo.

(A) IVIS analysis of SQ implanted NanoLymph showing pre-procedure (baseline), transcutaneous refilling of drug reservoir using quantum dots 705 (Qdot 705) and flushed out reservoir. (B) Normalized relative radiance of IVIS of Qdot 705 either injected subcutaneously (SQ, gray, circle) or within drug reservoir (NanoLymph, blue, square) across time points in vivo. Age-matched mice were implanted with NanoLymph for either 2 weeks (solid) or 22 weeks (dashed). (C) IVIS analysis of Qdot 705 injected SQ or within NanoLymph at various time points in mice at weeks post implantation. (D) Ex vivo imaging of skin overlying SQ injection or implanted NanoLymph. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Graphs are plotted as mean ± SD. Statistical significance determined via two-way ANOVA.
To assess the capability for sustained drug elution, we compared mice implanted for 2 weeks with Qdot 705 filled-NanoLymph (solid, blue) to a one-time SQ bolus injection (solid, gray) via IVIS imaging. Mice were assessed via IVIS imaging 1 hour after drug administration and on days 1, 3, 7, 11 and 14 thereafter. At hour 1, mice in both the NanoLymph and SQ group demonstrated high fluorescent signal (Figure 5B, C). Compared to the NanoLymph, mice receiving a SQ bolus dose had an immediate significant decrease in signal intensity, which was undetectable by day 11. In contrast, Qdot 705 eluted from NanoLymph showed sustained signal over 14 days, indicative of sustained progressive drug elution. Ex vivo imaging of SQ tissue showed completely cleared signal by day 14, whereas NanoLymph had drug retention and residual drug (Figure 5D).
To assess the capability for sustained drug elution after long-term implantation, we repeated the aforementioned experiment after 22 weeks of NanoLymph implantation. Age-matched mice were either transQ loaded with Qdot 705 within NanoLymph drug reservoir (dashed, blue) or administered one-time SQ bolus injection (dashed, grey) and imaged via IVIS as above. Elution from the NanoLymph exhibited similar rate of release in both short- and long-term implantation. Similarly, compared to NanoLymph, mice receiving a SQ bolus dose displayed analogous trend in both time points, with nearly undetectable signal by day 11 (Figure 5B, Supplementary Figure 4). These similar release profiles can be attributable to non-significant increase in fibrotic capsule (Figure 3C). Overall, these results demonstrated NanoLymph refillability and sustained drug release in vivo in both short- and long-term implantation.
To study in vivo drug biodistribution, we used Cy7-CA as a surrogate model for R848 via NanoLymph affixed with 30 nm nanomembranes, SQ implanted in mice. Cy7-CA was transcutaneously filled into NanoLymph drug reservoir as aforementioned. Payload delivered through the NanoLymph exhibited an extended release duration compared to a one-time SQ bolus injection (Figure 6A). By day 3 and thereafter, mice receiving SQ bolus dose showed consistent significant decrease in Cy7-CA signal compared to NanoLymph group. After day 7, almost all of Cy7-CA signal disappeared at the administration site in SQ injection group compared to the NanoLymph cohort (Figure 6A, B). Ex vivo imaging analysis demonstrated that NanoLymph released cargo into fibrotic capsule and skin milieu generating a local immunostimulatory niche (Figure 6C-F). At the overlying skin, NanoLymph retained local signal for longer than the one-time SQ bolus, which exhibited a significant decrease rapidly after day 1 (Figure 6C, F).
Figure 6: Drug retention within NanoLymph.

(A) IVIS analysis of Cy7-Ca in drug reservoir of implanted NanoLymph compared to injected subcutaneous (SQ) control in mice. (B) Normalized relative radiance analysis by IVIS of Cy7-CA comparing SQ (gray, circle) to NanoLymph drug reservoir (teal, sqare) across time points in vivo. (C) Ex vivo IVIS images of Cy7-Ca in organs comparing NanoLymph (NL) to bolus injected SQ. Relative radiance analysis by IVIS of Cy7-CA in (D) NanoLymph, (E) fibrotic capsule and (F) skin surrounding implant. (F) Relative radiance analysis by IVIS of Cy7-CA in (G) inguinal lymph node, (H) kidney and (I) liver. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Graphs are plotted as mean ± SD. Statistical significance determined via two-way ANOVA.
To evaluate systemic biodistribution of cargo, we explanted organs of relevance at predetermined endpoints for ex vivo IVIS imaging (Figure 6C). Payload released by NanoLymph exhibited a similar distribution to inguinal lymph node as SQ cohort at all time points (Figure 6C, G). Lastly, drug is cleared by the kidney and liver. SQ delivery resulted in rapid bolus clearance to the kidney and liver on day 1 in contrast to the NanoLymph cohort (Figure 6C, H-I). Payload delivery from either the NanoLymph or SQ group did not distribute to either the spleen or lung, which are typical of monoclonal antibodies71, 72(Figure 6C). Overall, we showed that the NanoLymph is capable of sustained drug elution across a period of at least 14 days with minimal biodistribution to distant organs.
Next, we evaluated the ability of the NanoLymph to control the release and retention of antigen over time. Antigen release and retention within the NanoLymph occurs via hydrogel degradation, which is widely studied44, 73-76. We used ovalbumin conjugated to Alexa Fluor 647 (OVA-AF647) to model our proposed antigen payload61, 62. OVA647-encapsulated within PF-127 was transQ loaded by simply advancing an injection needle through the skin and nylon mesh into the antigen reservoir44.
Strikingly, OVA-AF6467 delivered via SQ administration was almost immediately cleared out from the injection site with significant orders-of-magnitude decrease within one day, compared to NanoLymph (Supplementary Figure 5A). This effect is quantified via relative radiance of observed OVA-AF647 using IVIS imaging (Supplementary Figure 5B). To evaluate systemic biodistribution of cargo, we explanted organs of relevance at predetermined endpoints for ex vivo IVIS imaging (Supplementary Figure 5C). Similar to Cy7-Ca, OVA-AF647 eluted from the NanoLymph into fibrotic capsule and local skin tissue in a gradient-like manner over time (Supplementary Figure 5C-F). In contrast, signal at surrounding skin rapidly disappeared after day 1 in the one-time SQ bolus injection group (Supplementary Figure 5C, F). In the inguinal lymph node, kidney and liver, OVA-AF647 released by NanoLymph exhibited a similar distribution as SQ bolus at all time points (Supplementary Figure 5C, G-I). Further, antigen did not distribute to either the spleen or lung (Supplementary Figure 5C). This indicates that loaded antigens are confined locally to the immediate local NanoLymph microenvironment and exhibit minimal biodistribution to distant organs.
NanoLymph can recruit and activate dendritic cells in vivo leading to generation of antigen-specific lymphocytic response
We hypothesized that sustained delivery of immunostimulants in combination with an antigenic target via the NanoLymph can recruit and activate DC locally, which subsequently migrate to the adjacent inguinal lymph nodes for cross-presentation to T cells77. Therefore, we assessed immune responses in the NanoLymph and inguinal lymph node proximal to the NanoLymph site. To test this hypothesis, we SQ implanted mice with NanoLymph affixed with 30 nm nanomembranes. Fourteen (14) days after SQ implantation, mice were divided into control, drug only (GMCSF/R848), antigen only (OVAp) or combinatorial drug/antigen treatment. We transQ loaded the NanoLymph with drug combination and antigen in the drug and antigen reservoir, respectively, according to the groups. Controls were transQ filled with PBS and 20% PF-127 hydrogel in their respective reservoirs. After 14 days, we evaluated immune cell recruitment locally to the NanoLymph and the inguinal lymph node. We observed a statistically significant drug-dependent recruitment of CD11c+ DCs locally compared to control (Figure 7A). Further, mice receiving both drug and antigen had significantly higher proportion of CD80+ DCs recruited to the NanoLymph, indicating that the combinatorial elution is required for complete cellular activation (Figure 7B). This effect was recapitulated in adjacent inguinal lymph nodes with a significant drug-dependent increase of DCs (Figure 7C). Further, mice receiving both drug and antigen had a significantly higher proportion of OVA+DC, indicating effective NanoLymph-induced DC homing to the lymph node (Figure 7D). Specifically, DCs were recruited to the NanoLymph, where they interfaced with OVAp and migrated to the adjacent inguinal lymph node.
Figure 7: NanoLymph induced immunomodulation.

(A) DC recruitment at NanoLymph (B) Activated DCs in NanoLymph (C) DC migration to adjacent inguinal lymph node. (D) OVA+ DC localized at draining LN. (E) CD4+ T cell in LN. (F) Proportion of OVA tetramer+ CD4+ T cell in LN. (G) CD8+ T cell in LN. (H) Proportion of Ova tetramer+ CD8+ T cell in LN. Control NanoLymph in gray, Antigen only in orange, Drug only in green, Drug antigen combination in blue. All endpoints evaluated 14 days after transQ loading. Graphs are plotted as mean ± SD. Statistical significance determined via one-way ANOVA.
Antigen-presenting DC migration from the NanoLymph to local lymph nodes is crucial for initiating T cell activation cascade. To investigate this, subpopulations of T cells were assessed for antigen specificity by utilizing OVA tetramer staining. Although NanoLymph eluting both drug and antigen generated significantly fewer CD4+ T cell in adjacent inguinal lymph node (Figure 7E), there was a significant increase of tetramer+ CD4+ T cell compared to control (Figure 7F). Similarly, significantly higher number of CD8+ T cells were localized to inguinal lymph nodes in the combinatorial drug/antigen NanoLymph cohort (Figure 7G) with a corresponding robust tetramer+ CD8+ T cell response (Figure 7H). Activation and proliferation of cell-mediated immunity, comprising of CD4+ and CD8+ T cells, is responsible for orchestrating and directing a targeted immune response78. These results support our hypothesis, demonstrating that NanoLymph delivering both drug and antigen can activate DC locally and induce DC cross-presentation to generate antigen-specific cellular responses.
Discussion
Optimal material and design of therapeutic vaccines can enhance antigenic immunogenicity, generating either cell-mediated or humoral immunity. Past work from other groups in generating biomaterial-based therapeutic vaccine often are limited by encapsulation and degradation kinetics25, 34 , rapid burst release of drug79, 80 and lack of patient-centric design (i.e. minimally invasive, refillable)33. They are often single-administration strategies that cannot be tailored for future patient needs and have high incidence of adverse effects81. Our SQ implantable platform, the NanoLymph, was designed to address the aforementioned limitations and the critical need for a patient-centered vaccine strategy with minimal extracorporeal manipulation. Through sustained and constant elution of drugs and antigens, the NanoLymph achieves an enriched local microenvironment conducive for DC localization, activation and cross-presentation to T cells in distant lymphoid organs. Further, in considerations for future clinical translation, we demonstrate that the NanoLymph is biocompatible, mechanically stable and minimally invasive.
Current clinical trials for cancer vaccines often hinge on repeated administration of ex vivo activated autologous DC over period of weeks to months. Conventional techniques require invasive DC isolation from patients and ex vivo manipulation with GMCSF. Such cellular manufacturing utilizes immunostimulants in orders of magnitude higher than that released via the NanoLymph82-84. GMCSF’s potential for adverse effects is dose-dependent leading to immune-related adverse eventns85. Further, high-dose formulations are required due to minimal lymph node infiltration of DC, presumably contributing to limited clinical benefit and narrowed clinical utility18, 86. Lastly, such strategies rely on complicated isolation and dosing schedules, necessitating high degree of patient adherence. We posit that the NanoLymph can generate equivalent antigen-specific lymphocytic populations in situ compared to other approaches. It provides a local microenvironment conducive for systemic immune activation. Further, spatiotemporal cargo release from the NanoLymph can significantly influence immune cell responses. The capacity for tunable hierarchical release of multiple factors can be essential to achieving desired oncotheraputic outcomes without inducing lymphocytic exhaustion29.
In contrast to polymeric vaccine delivery approaches, the NanoLymph is drug- and antigen-agnostic and can be easily and rapidly be optimized for a variety of therapeutic targets without drug reformulation87. For example, FMS-related tyrosine kinase 3 ligand (FLT3-L) could be loaded in the NanoLymph to further potentiate immunomodulatory activity. FLT3-L promotes DC generation, recruitment and survival and could be used to synergize with GMCSF to augment DC responses88, 89. As an oncotherapeutic, in addition to activating an antigen-specific T cell responses, the NanoLymph can be used to simultaneously combat tumor-induced immunosuppression. The drug reservoir of the NanoLymph can be used to induce sustained local elution of immune checkpoint inhibitors (e.g. anti-PD-1 or anti-PD-L1). Local release of PD-L1 inhibitors will inhibit both the PD-1/PD-L1 axis as well as the PD-L1/CD80 interactions of DC. Blocking PD-L1 on DC increases free CD80 in circulation, maximizing co-stimulatory CD28 interaction on T cells to enhance lymphocyte priming90. Thus, use of PD-1 may further synergistically enhance CD80 molecules to enhance T cell priming91. Combinatorial local therapy of immune checkpoint inhibitors and immunostimulants could achieve robust systemic antitumor T cell responses while alleviating the risk of acquired resistance and treatment-associated toxicities92. Likewise, the NanoLymph can be loaded with either autologous or allogeneic immunodominant tumor antigens. Autologous antigens such as immunogenic neoantigens isolated from tumor biopsies93, 94 or whole-cell tumor lysates can deliver specific and non-specific antigens95, 96. The use of patient-derived antigens offers a personalized oncotherapeutic strategy to generate robust responses while limiting alloimmune reactivity.
These capabilities of the NanoLymph offer significant flexibility for management of various diseases influenced by the immune system. Previous work in our laboratory have demonstrated in situ delivery of immunosuppressants to protect allogeneic cell transplants from host immune rejection44, 76. Along the same lines, the immunomodulator potential of the NanoLymph can be leveraged for allergy immunotherapy to desensitize patients against common life-threatening allergens. Specifically, the NanoLymph could be used to elute cytokines (e.g., TGFβ and IL-2)97, 98 or mTOR inhibitors (e.g., rapamycin)99, 100, with house dust mite allergoid serving as the antigen101, 102. In this context, the NanoLymph can induce allergen desensitization by tolerogenic DCs suppressing Th2 mediated immune responses.
Most available medical implants lack drug refillability and therefore rely on either complete structure degradation or explanation and re-implantation for continued care, posing a significant barrier to care35. Minimally invasive transQ accessibility of the NanoLymph allows for drug replenishment, replacement or dosage tuning, if needed. Although we have optimized drug dosage here in a murine model, clinical translation may require further personalized dose tuning dependent on patient’s immune responses. For example, known sex-based differences103, 104 and interactions with gut microbiota105, 106 can influence maturation and function of immune cells. Therefore, the NanoLymph allows for therapeutic optimization on a per-patient basis in consideration of a myriad of external variables, allowing for patient-centric therapy. Further, the flexibility of NanoLymph device placement could be leveraged for disease-specific indications. In the context of cancer, considering that strong antitumor immune responses are initiated intra- and peritumorally55, 107, ideal implant placement could be in proximity to the primary tumor or draining lymph nodes108 55. Future studies will characterize the functionality of antigen-specific lymphocytes to generate strong intratumoral antitumor local and memory immune responses. Additionally, such studies will characterize impact and cargo release efficiency after multiple transQ refillings for generation of strong therapeutic antitumor immune responses. Lastly, it is necessary to evaluate the efficacy of the NanoLymph against current conventional vaccine techniques. Thus, future experiments will assess the immune response generated in mice receiving NanoLymph versus repeated bolus SQ administration.
The nondegradable NanoLymph can be easily manipulated for continual immune modulation. In contrast, biodegradable vaccines present non-retrievable systems slowly eluting cargo over time dependent on polyester degradation kinetics. Although such polyesters have a long history of durable safety in the clinic109, accidental release of entire payloads may be possible in unique clinical settings or through system malfunction. High dose delivery of GMCSF and other cytokines are linked to serious adverse effects including arterial thrombosis, vascular leak syndrome, bone pain and hematopoietic malignancies110. The NanoLymph presents a self-sealing design, passively controlling the rate of drug release through nanoporous membranes. Additionally, the NanoLymph uses a minimal concentration of drug to exert immunomodulatory effects, diminishing the possibility of severe adverse reactions. To further mitigate adverse effects, the NanoLymph structure can be retrieved entirely, if needed, to meet patient requirements.
Overall, we present a proof-of-concept study demonstrating that locally released drug and antigen from the NanoLymph in a spatiotemporal manner successfully recruits and activates DC to induce an antigen-specific immune response. Future studies will extrapolate on this effect by evaluating the prophylactic and therapeutic efficacy of the antitumor immune response for cancer treatment. Taken together, this work provides the foundation for the development of an immunomodulatory niche adaptable for multiple disease etiologies beyond cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Funding support for this work was provided by U54CA210181, The Nancy Owens Breast Cancer Foundation, the Houston Methodist Research Institute. D.I.V. received joint funding support from Texas A&M University MD/PhD Program and Houston Methodist Research Institute. The authors would like to thank Dr. Andreana Rivera and Yuelan Ren from the research pathology core of Houston Methodist Research Institute (HMRI) for their invaluable histopathological support. We would like to thank Dr. Michael Ittmann from Baylor College of Medicine and Dr. Andreana Rivera for histopathological scoring. Additionally, we would like to thank Dr. Jianhua “James” Gu from the electron microscopy core of HMRI for device SEM imaging. We thank the NIH Tetramer Core Facility for the provision of H-2K(b) SIINFEKL PE-labeled tetramer. The authors also thank Nathaneal Hernandez for valuable technical support, Nicola Di Trani and Jesus Paez Mayorga for useful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Dixita Ishani Viswanath: Conceptulization, Methodology, Formal analysis, Investigation, Writing-original draft, Writing-review & editing, Visualization. Hsuan-Chen Liu: Investigation, Writing-review & editing, Visualization. Simone Capuani: Investigation. Robin Vander Pol: Investigation. Shani Saunders: Investigation. Corrine Ying Xuan Chua: Conceptulization, Methodology, Writing-review & editing, Supervision. Alessandro Grattoni: Conceptulization, Methodology, Writing-review & editing, Supervision, resources, Project administration, Funding acquisition.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The raw/processed data required to reproduce these findings cannot be shared at this time due to legal limitations for patent applications. The data that support the findings of this study are available on request from the corresponding author.
References
- 1.Haslam A & Prasad V Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fares CM, Van Allen EM, Drake CG, Allison JP & Hu-Lieskovan S Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book 39, 147–164 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Tahmasebi S, Elahi R & Esmaeilzadeh A Solid Tumors Challenges and New Insights of CART Cell Engineering. Stem Cell Rev Rep 15, 619–636 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Wei SC, Duffy CR & Allison JP Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newick K, O'Brien S, Moon E & Albelda SM CAR T Cell Therapy for Solid Tumors. Annu Rev Med 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Atezolizumab Combo Approved for PD-L1-positive TNBC. Cancer Discov 9, OF2 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Gu L & Mooney DJ Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer 16, 56–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KG, Stromnes IM & Greenberg PD Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vermaelen K Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Frontiers in Immunology 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irvine DJ, Swartz MA & Szeto GL Engineering synthetic vaccines using cues from natural immunity. Nat Mater 12, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gajewski TF, Schreiber H & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Groot AS et al. Immune camouflage: relevance to vaccines and human immunology. Hum Vaccin Immunother 10, 3570–3575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiteside TL The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos PM & Butterfield LH Dendritic Cell-Based Cancer Vaccines. J Immunol 200, 443–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sprooten J et al. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 8, e1638212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheever MA & Higano CS PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res 17, 3520–3526 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Jaroslawski S & Toumi M Sipuleucel-T (Provenge((R)))-Autopsy of an Innovative Paradigm Change in Cancer Treatment: Why a Single-Product Biotech Company Failed to Capitalize on its Breakthrough Invention. BioDrugs 29, 301–307 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Mellman I, Coukos G & Dranoff G Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yaguchi T et al. The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol 93, 294–300 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Nayar S, Dasgupta P & Galustian C Extending the lifespan and efficacies of immune cells used in adoptive transfer for cancer immunotherapies-A review. Oncoimmunology 4, e1002720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kini V & Ho PM Interventions to Improve Medication Adherence: A Review. JAMA 320, 2461–2473 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Lam WY & Fresco P Medication Adherence Measures: An Overview. Biomed Res Int 2015, 217047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torres-Robles A et al. Comparison of Interventions to Improve Long-Term Medication Adherence Across Different Clinical Conditions: A Systematic Review With Network Meta-Analysis. Front Pharmacol 9, 1454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bencherif SA et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun 6, 7556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calmeiro J et al. Biomaterial-based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J Immunother Cancer 7, 238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones KS Biomaterials as vaccine adjuvants. Biotechnol Prog 24, 807–814 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Zhang R, Billingsley MM & Mitchell MJ Biomaterials for vaccine-based cancer immunotherapy. J Control Release 292, 256–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adu-Berchie K & Mooney DJ Biomaterials as Local Niches for Immunomodulation. Acc Chem Res 53, 1749–1760 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Bencherif SA et al. Injectable preformed scaffolds with shape-memory properties. Proc Natl Acad Sci U S A 109, 19590–19595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bookstaver ML, Tsai SJ, Bromberg JS & Jewell CM Improving Vaccine and Immunotherapy Design Using Biomaterials. Trends Immunol 39, 135–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leach DG, Young S & Hartgerink JD Advances in immunotherapy delivery from implantable and injectable biomaterials. Acta Biomater 88, 15–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T et al. A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat Commun 9, 1532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol 33, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pons-Faudoa FP, Ballerini A, Sakamoto J & Grattoni A Advanced implantable drug delivery technologies: transforming the clinical landscape of therapeutics for chronic diseases. Biomed Microdevices 21, 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higgins DM et al. Localized immunosuppressive environment in the foreign body response to implanted biomaterials. Am J Pathol 175, 161–170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenneth Ward W A review of the foreign-body response to subcutaneously-implanted devices: the role of macrophages and cytokines in biofouling and fibrosis. J Diabetes Sci Technol 2, 768–777 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park CG et al. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci Transl Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Cree ML, Stocker CF, Tu QM & Scaini LF Adherence to standard medication infusion concentrations and its impact on paediatric intensive care patient outcomes. Aust Crit Care 31, 213–217 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Pons-Faudoa FP et al. Preventive efficacy of a tenofovir alafenamide fumarate nanofluidic implant in SHIV-challenged nonhuman primates. Adv Ther (Weinh) 4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su JT et al. A Subcutaneous Implant of Tenofovir Alafenamide Fumarate Causes Local Inflammation and Tissue Necrosis in Rabbits and Macaques. Antimicrob Agents Chemother 64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.WHO and WHO Expert Committee on Biological Standardization WHO Expert Committee on Biological Standardization: Sixty-sixth Report. World Health Organization, Geneva: 999 (2016). [Google Scholar]
- 43.Herzenberg LA, Tung J, Moore WA, Herzenberg LA & Parks DR Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol 7, 681–685 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Paez-Mayorga J et al. Neovascularized implantable cell homing encapsulation platform with tunable local immunosuppressant delivery for allogeneic cell transplantation. Biomaterials 257, 120232 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Kumar H & Kim K Stereolithography 3D Bioprinting. Methods Mol Biol 2140, 93–108 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Della Bona A, Cantelli V, Britto VT, Collares KF & Stansbury JW 3D printing restorative materials using a stereolithographic technique: a systematic review. Dent Mater 37, 336–350 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farina M et al. Transcutaneously refillable, 3D-printed biopolymeric encapsulation system for the transplantation of endocrine cells. Biomaterials 177, 125–138 (2018). [DOI] [PubMed] [Google Scholar]
- 48.de Tayrac R et al. In vitro degradation and in vivo biocompatibility of poly(lactic acid) mesh for soft tissue reinforcement in vaginal surgery. J Biomed Mater Res B Appl Biomater 85, 529–536 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Weir NA, Buchanan FJ, Orr JF & Dickson GR Degradation of poly-L-lactide. Part 1: in vitro and in vivo physiological temperature degradation. Proc Inst Mech Eng H 218, 307–319 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Connors KA Chemical kinetics: the study of reaction rates in solution. (Wiley-VCH Verlag GmbH, 1990). [Google Scholar]
- 51.Di Trani N et al. Extending Drug Release from Implants via Transcutaneous Refilling with Solid Therapeutics. n/a, 2100214 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chua CYX et al. Carbon fiber reinforced polymers for implantable medical devices. Biomaterials 271, 120719 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Burkholder T, Foltz C, Karlsson E, Linton CG & Smith JM Health Evaluation of Experimental Laboratory Mice. Curr Protoc Mouse Biol 2, 145–165 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aghajani M & Esmaeili F Anti-biofouling assembly strategies for protein & cell repellent surfaces: a mini-review. J Biomater Sci Polym Ed, 1–20 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Liu HC et al. Potentiating anti-tumor efficacy through radiation and sustained intratumoral delivery of anti-CD40 and anti-PDL1. Int J Radiat Oncol Biol Phys (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greter M et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36, 1031–1046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becher B, Tugues S & Greter M GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 45, 963–973 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Novak N, Yu CF, Bieber T & Allam JP Toll-like receptor 7 agonists and skin. Drug News Perspect 21, 158–165 (2008). [PubMed] [Google Scholar]
- 59.Vuylsteke RJ et al. Local administration of granulocyte/macrophage colony-stimulating factor increases the number and activation state of dendritic cells in the sentinel lymph node of early-stage melanoma. Cancer Res 64, 8456–8460 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Zagon IS, Donahue RN, Rogosnitzky M & McLaughlin PJ Imiquimod upregulates the opioid growth factor receptor to inhibit cell proliferation independent of immune function. Exp Biol Med (Maywood) 233, 968–979 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Hurst J et al. TLR7 and TLR8 ligands and antiphospholipid antibodies show synergistic effects on the induction of IL-1beta and caspase-1 in monocytes and dendritic cells. Immunobiology 214, 683–691 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Hackstein H et al. The TLR7/8 ligand resiquimod targets monocyte-derived dendritic cell differentiation via TLR8 and augments functional dendritic cell generation. Cell Immunol 271, 401–412 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Kaczanowska S, Joseph AM & Davila E TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol 93, 847–863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsehaye MT, Velizarov S & Van der Bruggen B Stability of polyethersulfone membranes to oxidative agents: A review. Polymer Degradation and Stability 157, 15–33 (2018). [Google Scholar]
- 65.Albisa A, Espanol L, Prieto M & Sebastian V Polymeric Nanomaterials as Nanomembrane Entities for Biomolecule and Drug Delivery. Curr Pharm Des 23, 263–280 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Krieter DH & Lemke HD Polyethersulfone as a high-performance membrane. Contrib Nephrol 173, 130–136 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Muntjewerff EM, Meesters LD & van den Bogaart G Antigen Cross-Presentation by Macrophages. Front Immunol 11, 1276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hotta C, Fujimaki H, Yoshinari M, Nakazawa M & Minami M The delivery of an antigen from the endocytic compartment into the cytosol for cross-presentation is restricted to early immature dendritic cells. Immunology 117, 97–107 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feghali CA & Wright TM Cytokines in acute and chronic inflammation. Front Biosci 2, d12–26 (1997). [DOI] [PubMed] [Google Scholar]
- 70.McDermott AJ et al. Role of GM-CSF in the inflammatory cytokine network that regulates neutrophil influx into the colonic mucosa during Clostridium difficile infection in mice. Gut Microbes 5, 476–484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chua CYX et al. Intratumoral Nanofluidic System for Enhancing Tumor Biodistribution of Agonist CD40 Antibody. 3, 2000055 (2020). [Google Scholar]
- 72.Park JW et al. (89)Zr anti-CD44 immuno-PET monitors CD44 expression on splenic myeloid cells and HT29 colon cancer cells. Sci Rep 11, 3876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ashour AE et al. Flt3 ligand delivered in a pluronic formulation prolongs the survival of mice with orthotopic pancreatic adenocarcinoma. Cancer Biother Radiopharm 22, 235–238 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Diniz IM et al. Pluronic F-127 hydrogel as a promising scaffold for encapsulation of dental-derived mesenchymal stem cells. J Mater Sci Mater Med 26, 153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Escobar-Chavez JJ et al. Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J Pharm Pharm Sci 9, 339–358 (2006). [PubMed] [Google Scholar]
- 76.Paez-Mayorga J et al. Enhanced In Vivo Vascularization of 3D-Printed Cell Encapsulation Device Using Platelet-Rich Plasma and Mesenchymal Stem Cells. Adv Healthc Mater 9, e2000670 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Embgenbroich M & Burgdorf S Current Concepts of Antigen Cross-Presentation. Front Immunol 9, 1643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Esser MT et al. Memory T cells and vaccines. Vaccine 21,419–430 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Schwendener RA Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2, 159–182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cha BG, Jeong JH & Kim J Extra-Large Pore Mesoporous Silica Nanoparticles Enabling Co-Delivery of High Amounts of Protein Antigen and Toll-like Receptor 9 Agonist for Enhanced Cancer Vaccine Efficacy. ACS Cent Sci 4, 484–492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singer CF et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur J Cancer 132, 43–52 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Borrello IM et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood 114, 1736–1745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim VM et al. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yarchoan M et al. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med 9, 1485–1494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stern AC & Jones TC The side-effect profile of GM-CSF. Infection 20 Suppl 2, S124–127 (1992). [DOI] [PubMed] [Google Scholar]
- 86.Viswanath DI, Liu HC, Huston DP, Chua CYX & Grattoni A Emerging biomaterial-based strategies for personalized therapeutic in situ cancer vaccines. Biomaterials 280, 121297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lybaert L et al. Cancer Cell Lysate Entrapment in CaCO3 Engineered with Polymeric TLR-Agonists: Immune-Modulating Microparticles in View of Personalized Antitumor Vaccination. Chemistry of Materials 29, 4209–4217 (2017). [Google Scholar]
- 88.Chakravarty PK et al. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res 59, 6028–6032 (1999). [PubMed] [Google Scholar]
- 89.Manz MG, Weissman IL, Cozzio A, Merad M & Karsunky H Flt3 Ligand Regulates Dendritic Cell Development from Flt3+ Lymphoid and Myeloid-committed Progenitors to Flt3+ Dendritic Cells In Vivo. Journal of Experimental Medicine 198, 305–313 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen L & Flies DB Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13, 227–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mayoux M et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med 12 (2020). [DOI] [PubMed] [Google Scholar]
- 92.Bagchi S, Yuan R & Engleman EG Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol 16, 223–249 (2021). [DOI] [PubMed] [Google Scholar]
- 93.Gubin MM, Artyomov MN, Mardis ER & Schreiber RD Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 125, 3413–3421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu Z, Ott PA & Wu CJ Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 18, 168–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonzalez FE et al. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccin Immunother 10, 3261–3269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Joshi VB et al. Tumor lysate-loaded biodegradable microparticles as cancer vaccines. Expert Rev Vaccines 13, 9–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ & Driscoll JJ Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol 14, 55 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mojsilovic S, Mojsilovic SS, Bjelica S & Santibanez JF Transforming growth factor-beta1 and myeloid-derived suppressor cells: A cancerous partnership. Dev Dyn (2021). [DOI] [PubMed] [Google Scholar]
- 99.Shin S, Lee JH, Lee HJ, Chang SY & Chung SH Rapamycin attenuates Th2-driven experimental allergic conjunctivitis. Clin Immunol 190, 1–10 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Yamaki K & Yoshino S Preventive and therapeutic effects of rapamycin, a mammalian target of rapamycin inhibitor, on food allergy in mice. Allergy 67, 1259–1270 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Jutel M, Bruggenjurgen B, Richter H & Vogelberg C Real-world evidence of subcutaneous allergoid immunotherapy in house dust mite-induced allergic rhinitis and asthma. Allergy 75, 2050–2058 (2020). [DOI] [PubMed] [Google Scholar]
- 102.Shin JU et al. Allergen-specific immunotherapy induces regulatory T cells in an atopic dermatitis mouse model. Allergy 73, 1801–1811 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Kovats S Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 294, 63–69 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kovats S Estrogen receptors regulate an inflammatory pathway of dendritic cell differentiation: mechanisms and implications for immunity. Horm Behav 62, 254–262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aindelis G & Chlichlia K Modulation of Anti-Tumour Immune Responses by Probiotic Bacteria. Vaccines (Basel) 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nelson MH, Diven MA, Huff LW & Paulos CM Harnessing the Microbiome to Enhance Cancer Immunotherapy. J Immunol Res 2015, 368736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weiden J, Tel J & Figdor CG Synthetic immune niches for cancer immunotherapy. Nat Rev Immunol 18, 212–219 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Chua CYX et al. Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer. J Control Release 285, 23–34 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Ding D & Zhu Q Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater Sci Eng C Mater Biol Appl 92, 1041–1060 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Vial T & Descotes J Clinical toxicity of cytokines used as haemopoietic growth factors. Drug Saf 13, 371–406 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The raw/processed data required to reproduce these findings cannot be shared at this time due to legal limitations for patent applications. The data that support the findings of this study are available on request from the corresponding author.