

Abstract
Cells release extracellular vesicles (EVs) that can be detected both in vivo and in cell culture medium. Among EVs, exosomes are 50–150 nm vesicles that are systematically packaged into multivesicular bodies for release into the external environment. In cancer, these intentionally packaged exosomes carry a payload of proteins such as RNAs and surface receptors that facilitate the reprogramming of proximal cells to assemble a protumor microenvironment. Exosomes have been implicated as an important intermediary extracellular communication pathway between cells, including in melanoma. Human melanoma-derived exosomes (HMEX) have been demonstrated to modulate the extracellular environment and inhibit immune cell activation. There are many methods to isolate and enrich for exosomes and the method applied can impact yield and purity of the isolates. In this chapter we describe the REIUS (rapid exosome isolation using ultrafiltration and size exclusion chromatography) method to isolate HMEX from melanoma cell cultures and then demonstrate their enrichment using molecular and microscopic approaches.
Keywords: Extracellular Vesicles, EV, Isolation Techniques, Melanoma, Exosomes, Size Exclusion Chromatography, Ultrafiltration, REIUS
1. Introduction
Inter-communication between cells in a tumor microenvironment (TME) is multifactorial. Release of soluble signaling factors such as cytokines and chemokines that attract or repulse cells (particularly immune cells) control the tissue microenvironment providing a homeostatic environment down to the cellular level. Malignant cells manipulate extracellular signaling systems that disrupt the normal stromal environment by promoting cell plasticity and creating a tumor niche in the TME [1, 2]. Cancer cells do this in part by releasing extracellular vesicles (EVs) that are bilipid layer vesicles containing a payload of proteins, RNA, and surface receptors capable of modulating both the immediate microenvironment and the macroenvironment [3–6]. Amongst many subsets of EVs, exosomes are intentionally packaged vesicles ranging from 50–150 nm in size and are generated through the endocytic pathway via the endosomal sorting complexes required for transport (ESCRT) complex. The control of the microenvironment by tumor through exosome release is swift and sustained, and some cancer cells can release many fold more exosomes than normal stromal cells [7]. In this context, the study of exosomes and how they contribute to tumor control of the microenvironment is an active area of research interest [8].
Melanoma-derived exosomes are rapidly taken up by stromal cells and control the metabolic programming in those cells to generate a protumorigenic niche [9]. The exact mechanisms by which exosomes affect their target cells remain insufficiently understood [10, 11]. The field is further complicated by the variety of various exosome isolation methods employed to date which result in different levels of recovery and purity [12, 13]. Exosome isolation methods have advanced significantly over the past decade, and deeper investigations into exosomes and their control over the tumor microenvironment may reveal their use as targets for manipulation and therapeutic intervention [14, 15].
In this chapter, the REIUS method for isolating exosomes from cell cultures is introduced in detail [1, 9]. We have taken efforts to optimize the centrifugation speed to match most 50 ml tube benchtop centrifuges while retaining the same efficiency as that in enrichment and isolation of human melanoma-derived exosomes (HMEX). Isolation of exosomes using the REIUS method is simple, effective, and easily scalable to accommodate larger experiments (Fig. 1). We have also included the minimum molecular and microscopic methods necessary to validate a successful exosome isolation through the use of western blot, nanoparticle tracking analysis (NTA), and transmission electron microscopy (TEM) [14].
Fig. 1.
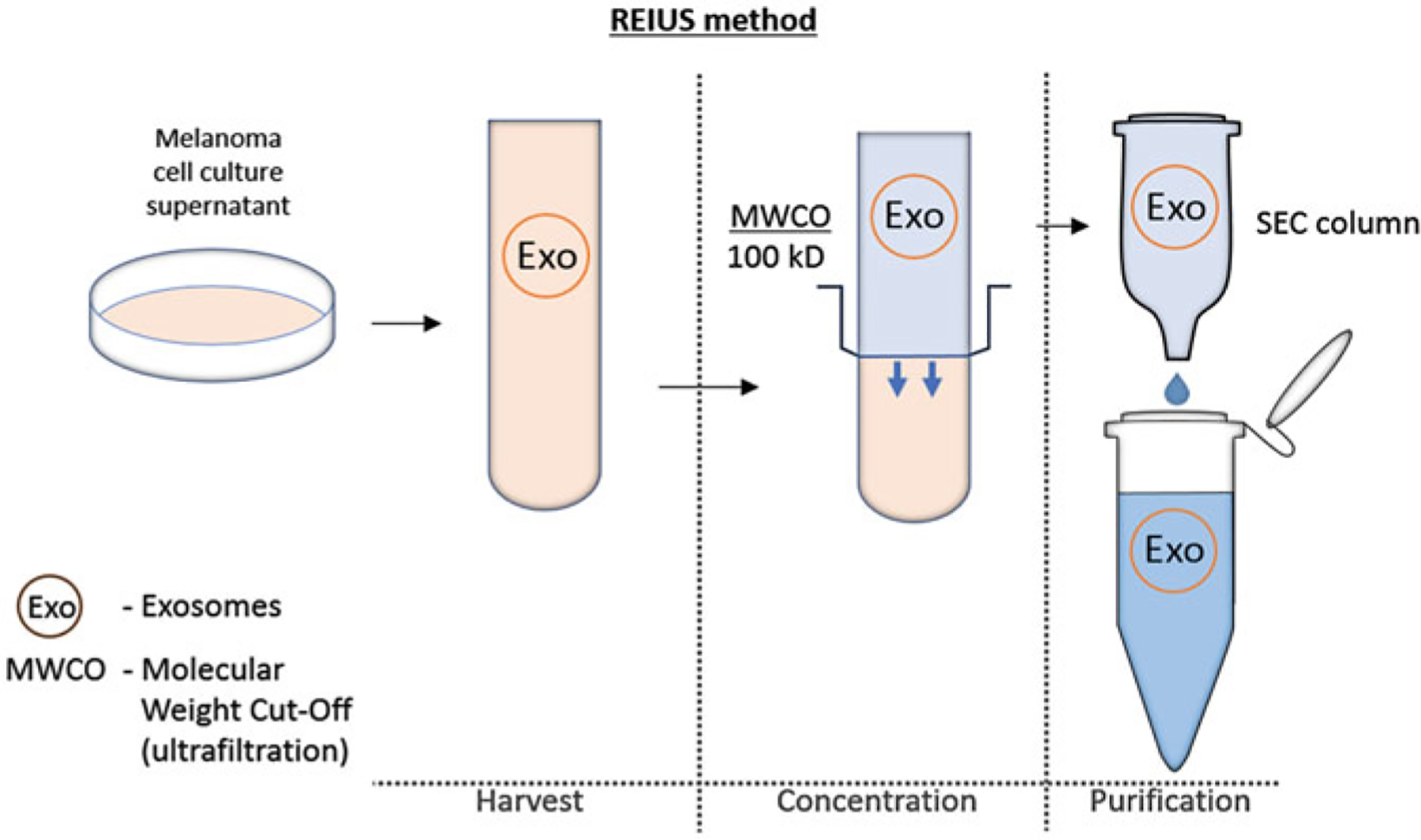
The REIUS method has three general steps performed in the following order: 1. Harvest: supernatants are collected from cell cultures, spun using a benchtop centrifuge to remove cells and debris and then particulates are removed for size stringency by passing through a 0.2 μm syringe-based filter. This restricts the EV to under 200 nm in size, consistent with the physiological exosome size limit. 2.Concentration: cleared supernatants are concentrated using an ultrafiltration column to reduce volumes to those appropriate for the SEC step. 3. Purification (SEC): exosomes are isolated using SEC columns and are used immediately or frozen for future use. To scale up, combine exosome isolates after the SEC step
2. Materials
2.1. Cell Culture
Melanoma cell lines (e.g., 2183-Her4, 526-mel, or 888-mel: all high exosome-producing lines).
150 mm tissue culture plates or T175 flasks
Complete cell culture medium: RPMI-1640 supplemented with antibiotics, GlutaMAX or l-glutamine, and 5–10% fetal bovine serum (FBS).
Dulbecco’s phosphate-buffered saline (DPBS): 1× solution.
0.4% trypan blue
Cell dissociation reagent (e.g., TrypLE Express Enzyme).
RPMI-1640 cell culture medium without supplements.
Exosome-depleted cell culture medium: complete cell culture medium containing 5–10% exosome-depleted FBS (see Note 1).
2.2. Harvesting of Supernatants and Exosome Isolation
Sterile 50 ml conical tubes.
Benchtop centrifuge capable of spinning 50 ml conical tubes at 3000 × g (e.g., Eppendorf 5810/5810R with A481 rotor or Sorvall Legend X1/X1R with TX-400 rotor).
Polyethersulfone (PES) syringe filters, 0.20 μm.
Sterile 30 ml syringes.
100 kDa MWCO centrifugal ultrafilter devices (e.g., Amicon Ultra-15 Centrifugal Filter devices, 100 K MWCO).
Size exclusion chromatography (SEC) columns (e.g., Exo-spin columns—EX03).
0.20 μm filter-sterilized 1× PBS (fresh).
1.5–1.7 ml low protein and nucleic acid-binding microcentrifuge tubes.
Microcentrifuge or benchtop centrifuge capable of spinning at 50 × g (e.g., Eppendorf 5810R with bucket adapters for microcentrifuge tubes).
2.3. Counting of Exosomes by NTA
Nanoparticle tracking analyzer (see Note 2).
100 nm beads (for instrument calibration)
0.20 μm filter-sterilized 1× PBS (freshly sterilized)
Sterile 1 cc syringes.
2.4. Western Blot
Ice-cold 1× DPBS.
Protease and phosphatase inhibitors.
RIPA buffer: 25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS). Just prior to use, add Halt™ Protease Inhibitor Cocktail (100×) protease/phosphatase inhibitors (Aprotinin, Bestatin, E-64, Leupeptin, Sodium Fluoride, Sodium Orthovanadate, Sodium Pyrophosphate and β-glycerophosphate) to 1× final concentration.
5 ml microcentrifuge tubes or equivalent
21-G needles and syringes
Refrigerated microcentrifuge.
Protein assay kit: Pierce BCA Protein Assay Kit.
Laemmli SDS sample loading buffer (6×): 375 mM Tris–HCl pH 6.8, 9% SDS, 50% glycerol, 0.03% bromophenol blue. Add 2-mercaptoethanol (2-ME) at 9% V/V before use.
Protein molecular weight standard: Precision Plus Protein Dual Color Standards.
Heating block.
PAGE Protein gels: precast 4–20% Tris-Glycine gels.
Tris–glycine–SDS running buffer, 10× (Bio-Rad): Dilute stock solution to 1× with water to achieve a working solution of running buffer that is 25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3.
Protein gel electrophoresis system.
0.45 μm low fluorescence polyvinylidene difluoride (PVDF) membrane
Gel transfer system (e.g., Trans-Blot Turbo Transfer System).
Transfer buffer as recommended for the user’s specific transfer system (proprietary for the Trans-Blot Turbo Transfer System).
100% ethanol or methanol for activating PVDF membrane
Blocking buffer (e.g., AdvanBlock).
Primary antibodies: rabbit anti-CD63, rabbit anti-TSG101, mouse anti-β-tubulin, and mouse anti-cytochrome c (Table 1).
Tube rotator or rocker (for 50 ml conical tubes).
1× TBS
1× TBS / 0.1% Tween 20
10% SDS
Secondary antibodies (detection system dependent, e.g., IRDye secondary antibodies 680RD goat anti-mouse, 680RD goat anti-rabbit, 800CW goat anti-mouse, and 800CW goat anti-rabbit).
Blot imaging system (e.g., Odyssey Fc Imaging System).
Table 1.
Table of antibodies used and their concentration
| Name of antibody | Company | Mono/ polyclonal) |
Molecular Weight (kDa) |
Species | Dilution for WB |
Secondary antibody dilution |
|---|---|---|---|---|---|---|
| CD63 | Sigma (Cat #HPA010088) | Poly | 50–60 | Rabbit | 1:1000 | 1:5000 |
| TSG101 | Abcam (Cat #ab125011) | Mono | 50 | Rabbit | 1:1000 | 1:5000 |
| Cytochrome C | ThermoFisher (Cat #66264-1-IG) | Mono | 14 | Mouse | 1:1000 | 1:5000 |
| β-tubulin | Cell Signaling (Cat #86298S) | Mono | 55 | Mouse | 1:1000 | 1:5000 |
2.5. Transmission Electron Microscopy (TEM)
2% Methyl cellulose, viscosity: 25 cP (optional)
Parafilm.
Nickel EM grids (e.g., 10 nm Formvar/1 nm Carbon coated square 300 mesh nickel grids).
EM grid tweezers.
Filter paper (e.g., Whatman).
UranyLess™ EM Stain.
Transmission electron microscope (e.g., JEOL 100CX II TEM 125 kV).
3. Methods
3.1. Cell Culture for Exosome Production
The following is the general protocol for culturing adherent cells to obtain exosomes. In our hands we found that melanoma cell lines can vary up to 2.5-fold in the number of exosomes they secrete. Thus, for each sample of exosomes, the appropriate number of plates to seed in step 2 will need to be determined for each cell line. In order to isolate exosomes from the cells in culture and limit contamination from exosomes present in FBS, the cells must be grown in exosome-depleted medium (see Note 3). To maximize growth, expand the melanoma cell lines in medium containing normal FBS and replace with exosome-depleted medium the day before harvesting the supernatants.
Once cell lines are 70–90% confluent in complete medium, wash the plates/flasks with 1× DPBS and add dissociation reagent until the cells have rounded up and started detaching. Add an equal volume of complete medium and transfer to a conical tube. Count the cells and determine percent viability using trypan blue or another method. Ensure that viability is greater than 95%.
Pellet the cells at 300 × g; resuspend in complete medium and seed cells into each of 3–5 plates at a density that will reach a confluency of 70–85% in 2–4 days. Monitor the plates of cells daily, replacing medium as necessary (see Notes 4 and 5).
When cells reach 70–85% confluency, aspirate off all medium. Wash monolayer by adding 10 ml of plain medium, making sure to tilt plate back and forth and side to side. Aspirate medium completely.
Add 12–16 ml of exosome-depleted medium and culture for 18–24 h. Volume added will depend on the metabolic activity of the cells (see Note 6).
3.2. REIUS Method for Isolating Exosomes
All centrifuge spins are performed at room temperature (RT). In order to maintain sterility, all steps are performed in a biosafety cabinet (see Notes 7 and 8).
Tilt plates back and forth to resuspend andy settled exosomes. While plate is tilted, use a pipette to transfer all supernatant to a 50 ml conical tube. Supernatant from multiple plates can be added to the same tube (see Note 9).
Centrifuge tubes at 300 × g for 5 min to pellet any cells. Pour supernatants into new 50 ml tubes and discard emptied tubes.
Centrifuge tubes at 3000 × g for 15 min to pellet any cell debris. Pour supernatants into new 50 ml tubes and discard emptied tubes.
Using 0.20 μm PES syringe filters, filter the supernatants into new 50 ml tubes. Keep these clarified supernatants at 4 °C until remaining steps can be performed, up to 3 days.
Add 15 ml clarified supernatant to ultrafiltration devices, using one device for each predetermined exosome sample as mentioned in Subheadings 3.1.
Centrifuge at 3000 × g for 15 min. Carefully lift the sample reservoir and aspirate away the filtrate that has passed through the reservoir into the tube. Reinsert the reservoir into the tube.
Apply another 15 ml clarified supernatant to the ultrafiltration unit and centrifuge and aspirate as in step 6, repeating until all clarified supernatant has been filtered (see Note 10).
Measure the volume in the sample reservoir using a micropipette. If the volume is much greater than 100 μl, centrifuge the filter device for another 15 min at 3000 × g (the Amicon devices will not “overspin” or release all liquid×as flowthrough). We routinely measure 115–120 μl of concentrated supernatant even after this extra spin.
Bring the SEC spin columns to room temperature for 15 min, one for each ultrafiltration unit used, keeping the columns upright. Check the integrity of the columns to be sure there are no bubbles, air pockets, or “breaks” in the matrix. If so, do not use that column. Throughout the SEC spin column procedure, take care to avoid introducing bubbles while adding the 1× PBS washes and samples.
After removing the bottom plug, place the SEC spin columns into the provided collection tubes. Remove the cap of each column and immediately screw it back on (see Note 11). Remove the cap again and using a micropipette, discard the 20% ethanol above the frit of the column. To prevent the column matrix from drying, immediately add 250 μl of filtered PBS to the top of the column, replace the cap, and continue with the protocol (see Note 12).
Centrifuge the columns at 50 × g for 10 s. Pour off the flow-through. If PBS still sits above the frit in any of the columns, spin those columns for 5 s at 50 × g, repeating if necessary. Check the integrity of the spin column matrix as in step 9.
Add another 250 μl of filtered PBS, replace the caps, and centrifuge at 50 × g for 10 s. Aspirate or pour off the flow-through. Again, check the integrity of the spin column matrix.
Carefully add a maximum of 100 μl of the ultrafiltered supernatant to each column and replace the cap. This time centrifuge at 50 × g for 1 min. If the cell culture medium contained phenol red, the spin columns will have a pink to orange tint at the top of the matrix depending on how spent the culture medium was.
Transfer the columns to prelabeled and sterile low-binding microcentrifuge tubes.
Add 200 μl of filtered PBS onto the columns. Replace caps and centrifuge at 50 × g for 1 min. The pink to orange color should now be seen throughout the column matrix.
The resultant flow-through in the low-binding microcentrifuge tubes contains concentrated stock exosomes in approximately 200 μl of PBS. Discard the spin columns. Measure and note the final exosome sample volumes. At this point NTA can be used to determine exosome concentration and size. We routinely perform the dilutions for NTA on freshly isolated exosome samples. The stock exosomes are then frozen at −80 °C.
If multiple ultrafiltration units were used for the same cell line undergoing the same treatment in step 5 and there is extra concentrated supernatant still present in those ultrafiltration units after step 13, the excess can be pooled. Adjust the total volume to 100 μl with filtered PBS and isolate the exosomes using a freshly prepped new SEC column (see Note 13).
3.3. NTA by ZetaView
- Prepare the following dilutions of the stock exosomes (dilutions can be kept overnight at 4 °C).
- 1:100 = 990 μl filtered PBS + 10 μl concentrated stock exosomes
- 1:1000 = 900 μl filtered PBS + 100 μl 1:100 dilution
- 1:10,000 = 900 μl filtered PBS + 100 μl 1:1000 dilution (if necessary)
Count exosomes using NTA. We use the following ZetaView settings: 25 °C, 488 nm laser, shutter speed of 400, frame rate of 60, and sensitivity of 89. The instrument is calibrated using 100 nm beads and primed with filtered 1× PBS since the exosomes are isolated in PBS (see Notes 14 and 15).
Exosome samples are injected into the instrument using a 1 cc syringe. Individual particles are visualized in real-time (Fig. 2a). The results are collected from 10 positions and analyzed. Wash with 2–5 ml filtered PBS after each sample. A mean diameter of ~50–60 nm and concentration of approximately 1 × 1011–1 × 1012 exosomes/ml can be expected (Fig. 2b, c).
Fig. 2.
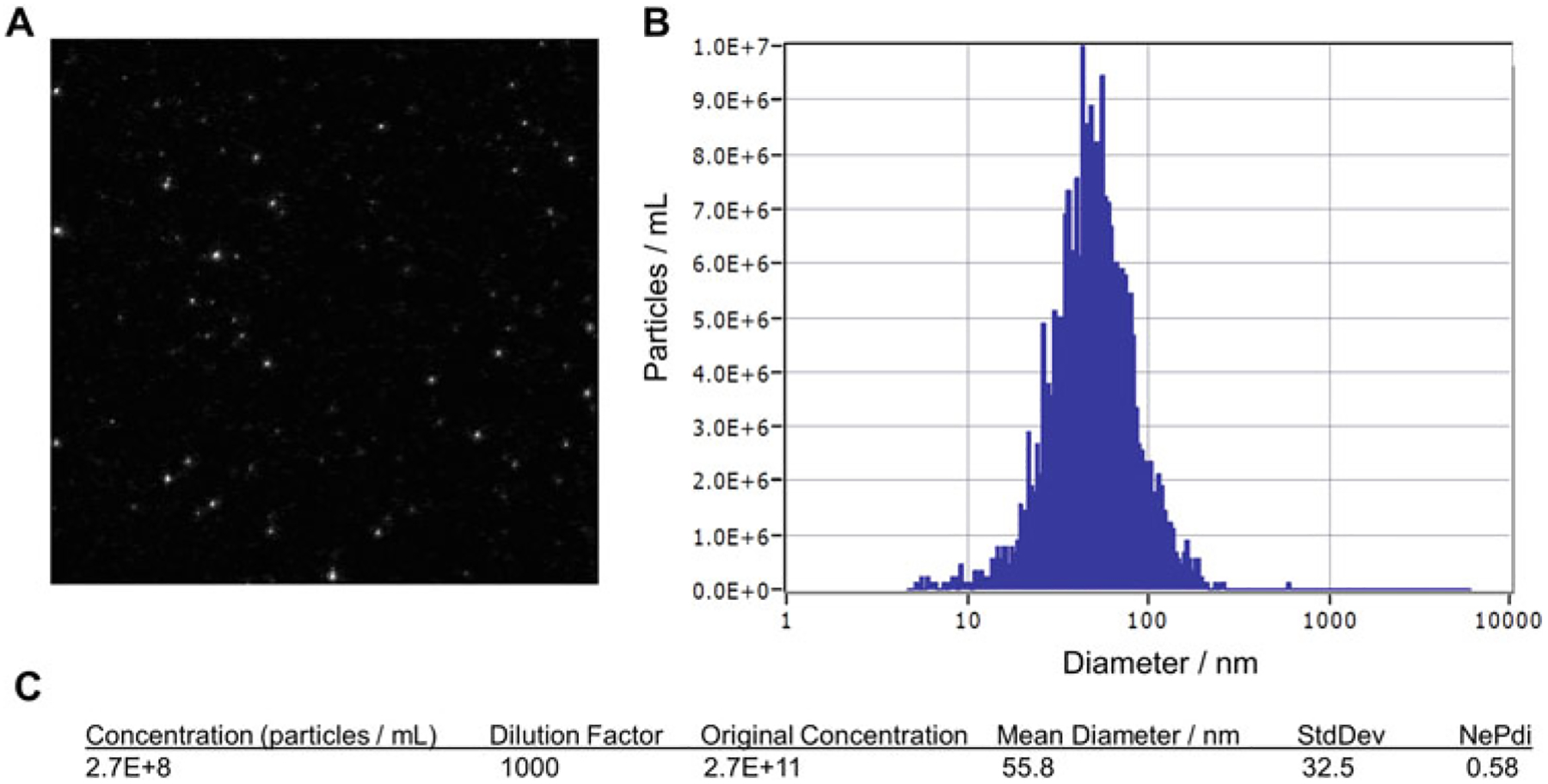
Breakdown of nanoparticle tracking analysis (NTA) report generated by ZetaView. (a) A snapshot of the particles visualized in the particle counting chamber. Particle count should be between 50 and 500 (maximum). (b) Size distribution graph with a single, normally distributed curve (mean, median, and mode are of similar values) indicating a successful exosome isolation. (c) Concentration of particles in the chamber should be range between 1 × 107 and 9.9 × 108. The dilution factor used is multiplied to give the original exosome concentration of the sample. Mean diameter is 50–70 nm depending on the cell line from which the exosomes are derived. The exosome polydispersity index (NePdi) is a ratio of the standard deviation divided by the mean exosome size based on NTA (ZetaView). NePdi higher than 1.0 could indicate that the sample was not passed through a 0.22 μm syringe-filter before ultrafiltration or a that a faulty SEC column was used
3.4. Western Blot Analysis of Exosome Samples
When preparing western blots, include cell lysates to serve as controls for the exosome samples. Samples of enriched exosomes should show expression of CD63 and TSG101 but very little to no expression of β-tubulin (or other cytoplasmic housekeeping genes such as β-actin) and cytochrome C (to demonstrate absence of organelle contamination from mitochondria). Cell lysates should show expression of all four proteins (Fig. 3).
Fig. 3.
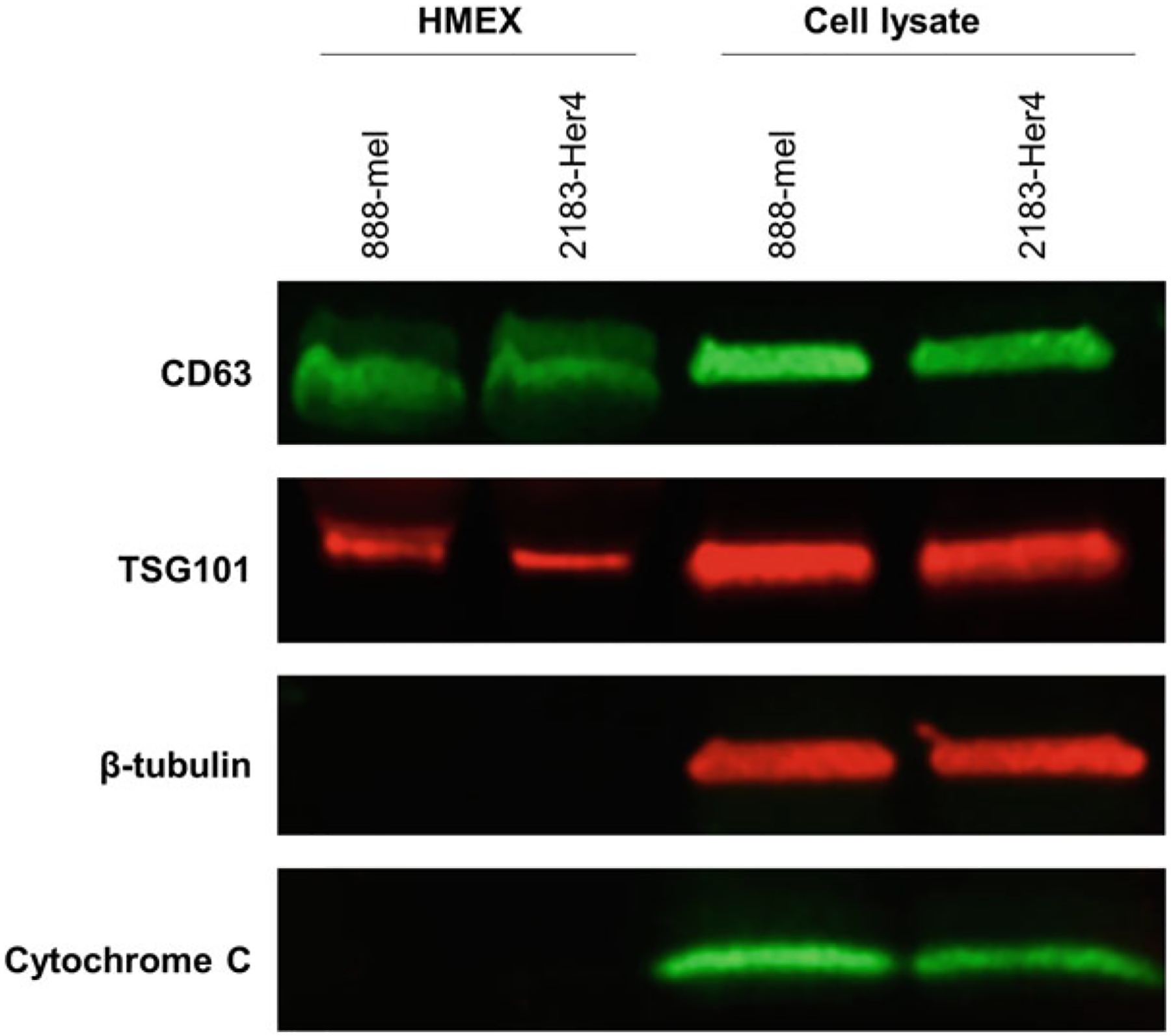
Western blot analysis of exosomes. Cell lysates are included as positive controls for proteins absent in exosomes, such as β-tubulin (cytoplasmic) or cytochrome C (mitochondrial). Most reviewers expect at least two probes for the absence of nonexosomal protein, preferably one cytoplasmic and one organelle-specific protein. Both TSG101 and CD63 must be present for the sample to be defined as an exosome isolate. If only CD63 is present but not TSG101, it is still considered as an EV isolate if β-tubulin and/or cytochrome C is not present
Cell lysates can be prepared directly from monolayers in 150 mm dishes (or T175 flasks), or from cell pellets.
- Prepare lysates from monolayers as follows.
- Place dishes/flasks on ice and wash with ice-cold 1× DPBS.
- Aspirate away DPBS and add 3 ml RIPA buffer with protease/phosphatase inhibitors for 5 min.
- Using a cell scraper and pipette, transfer cell suspension to a 5 ml microcentrifuge tube and incubate for 15 min on ice.
- Pass cell suspension through a 21-G needle 5 times. Incubate another 15 min on ice.
- Split lysate into 1.5 ml microcentrifuge tubes and continue to step 4.
- Prepare lysates from cell pellets as follows.
- Add 1 ml RIPA buffer with protease/phosphatase inhibitors to 5 × 106 cells.
- Resuspend to disperse pellet, transfer to a 1.5 ml microcentrifuge tube, and place on ice for 15 min.
- Pass cell suspension through a 21-G needle 5 times. Incubate another 15 min on ice.
Centrifuge cell lysates in a microcentrifuge at full speed for 10 min at 4 °C. Transfer supernatants to a new microcentrifuge tube. Store at −80 °C.
Measure the protein concentration of both the cell lysates and exosome samples using a protein assay kit. We typically dilute an aliquot of cell lysate 1:10 and 1:25 in RIPA buffer + protease/phosphatase inhibitors for protein analysis. An aliquot of exosome samples is diluted 1:5 in RIPA buffer + protease/phosphatase inhibitors. For the exosome samples, a protein concentration of 1–2 μg/μl can be expected after dilution.
Prepare 20 μg of cell lysate and 20 μg of exosome samples for electrophoresis by adding 6× Laemmli SDS sample loading buffer/2-ME up to a maximum volume of 50 μl (or the maximum volume that can be loaded in the gel wells). Heat samples for 5 min at 95 °C. Centrifuge briefly.
Load samples and molecular weight standard on the gel and run at 200 V for approximately 30 min, or as is appropriate for your electrophoresis system.
Presoak PVDF membrane in 100% ethanol (or methanol) to activate the membrane and then rinse in transfer buffer. Transfer proteins from the gels to the PVDF membrane as directed by your transfer system. For the Trans-Blot Turbo System, we use the preprogrammed protocol “1 Mini-TGX”.
After transfer, rinse the membranes in H2O for several minutes to remove any gel debris. Proceed with antibody incubations (CD63, TSG101, Cytochrome C or β-tubulin) as in step 11 or allow the membrane to air dry for ≥2 h (see Note 16).
If membranes were dried, activate membrane by soaking in ethanol or methanol and rinse twice with H2O.
Place membranes in blocking buffer for 1 h, rocking at room temperature. Dilute primary antibodies to the appropriate concentration in blocking buffer (Table 1). For us, all four antibodies (CD63, TSG101, β-tubulin and cytochrome c) are diluted 1:1000. Incubate blots overnight at 4 °C with rocking, or in 50 ml conical tubes on a rotator.
Rinse blots with 1× TBS/0.1% Tween 20 four times for 5 min each wash. Centrifuge antibodies for 5 min at 10,000 × g to remove any aggregates that may occlude bands during imaging. Dilute secondary antibodies to the appropriate dilution (Table 1) in blocking buffer with 0.01% SDS and incubate blots for 1 h, either on a rocker in dark boxes or in conical tubes on a rotator in the dark (see Notes 17 and 18).
Making sure to minimize light exposure, rinse blots in TBS/0.1% Tween 20 four times for 5 min each wash. Finally, rinse once in 1× TBS for 5 min to remove any residual Tween.
Blots can be imaged immediately or allowed to air-dry for at least 2 h in the dark; they then can be stored in the dark at room temperature for imaging in the future.
3.5. Preparation of Exosomes for TEM
The defining feature of exosomes is inherent in the phospholipid bilayer that keeps the internal region of exosomes discrete from their surroundings. Previously, TEM was used for the morphological characterization of an exosome, primarily based on the presence of the lipid bilayer. While not solely reliant upon TEM for exosome characterization, it is important that isolated exosomes maintain their integrity as demonstrated by an intact bilipid membrane; this can be visualized by combining TEM with negative staining (Fig. 4).
Fig. 4.

TEM image of exosomes. Due to the compartmentalized nature of exosomes, negative staining by UranyLess™ highlights intact bilipid layered vesicles as bright spherical spots relative to the darker, even background stained by UranyLess™. If an exosome is structurally compromised, it can appear as a darkened round spot as the stain is absorbed and retained within the vesicles (black arrow). Darker spots of undefined shapes are concentrations of stain (white asterisks). This phenomenon can be minimized by reducing the duration of time the sample is exposed to UranyLess™ but requires a balance as it would give a lighter background and may impair the contrast provided by the vesicles, which if present, are unstained
As the structure of the exosomes will not be preserved by the chemicals used in this protocol, the resultant EM grid exosome sample should be imaged by TEM as soon as possible. If the exosomes are not immediately imaged after step 6, combine 6 μl of isolated exosomes (in 1× PBS) and 6 μl of 2% methyl cellulose before continuing to step 2.
Place one drop (approximately 10 μl) of exosomes at a concentration of at least 1 × 1011 exosomes/ml onto Parafilm.
Using tweezers, invert an EM grid onto the exosome drop for 1 min. Electrostatic forces and osmosis are sufficient for the exosomes to be uniformly distributed across the grid; this occurs very quickly (1 min). Remove excess liquid by touching the edge of the grid with the edge of filter paper.
Place a drop (10 μl) of UranyLess™ solution onto the Parafilm (see Note 19).
Invert the EM grid containing exosomes onto the UranyLess™ drop for 1 min only to limit aggregation of the stain. Remove excess UranyLess™ by touching the edge of the grid with the edge of the filter paper (see Note 20).
Leave to air dry for at least 30 min and then image by TEM.
4. Notes
The quality of exosome-depleted FBS is critical in regards to its exosome concentration. To verify its quality, prepare an aliquot of medium containing 5% normal FBS and another vial with medium containing 5% exosome-depleted FBS and then analyze by NTA. The medium with exosome-depleted FBS should have approximately 1000-fold less particles. This step assures a more accurate quantification of exosomes after isolation.
ZetaView (Particle Metrix) is well-suited for use as a shared laboratory facility instrument, as it contains an automatic wash-pump system that allows for quick turnaround of sample measurements and eliminates contamination from previous user samples without the need to dismantle the instrument to clean it in preparation for the next user.
Exosome-depleted FBS is not only higher in cost but is inferior at supporting the growth of melanoma cell lines when compared to normal FBS.
We have found that a confluency less than 70% will yield too few exosomes. We typically seed 0.7–1.2 × 106 cells per plate.
Cells produce more exosomes when confluency is between 85% and 95%. When confluency reaches 100%, the yield drops considerably. Therefore, cells should be seeded in a uniform layer by giving a gentle lateral shake (in perpendicular motion, i.e., left to right then front to back) several times before placing in the incubator. Observe under the microscope for uniform confluency. Uneven seeding can cause some areas to be 100% confluent which may cause a reduction in exosome production, yielding an inadequate number for downstream experiments.
Caution is advised when using this small volume of medium in 150 mm plates (or T175 flasks). Check that the incubator shelves do not have a slant that may cause certain areas of the plates to be devoid of medium, which could otherwise lead to cell death and contamination of the supernatant with exosomes released by dead cells.
As NTA is extremely sensitive, any bacterial growth in an isolated exosome sample due to contamination would severely compromise both the NTA and the sample.
The isolated exosomes are not considered sterile as the ultra-filters we use are not sterile and the SEC columns are not manufactured in a GMP facility. However, the SEC column matrix is preserved in 20% ethanol and therefore unlikely to carry contamination. When our exosomes were used for downstream cell culture experiments, no contamination was observed for in vitro experiments that lasted 24 h. We also performed in vivo studies where exosomes were injected into the tail-vein of severe combined immunodeficient (SCID) mice with no observed blood-based complications after 30 days. To limit the potential for contamination, use a biosafety cabinet for all steps.
After supernatants have been collected, the cells can be dissociated from the plates and counted in order to calculate the number of exosomes per cell once the exosome count is determined. Otherwise, discard the monolayers as we have observed that cell proliferation slows in the presence of medium containing exosome-depleted FBS.
Although repeated additions of supernatant from one sample can be added to the ultrafiltration units, it is not recommended to “wash and reuse” the units for a different sample because their membranes can clog with use.
We have found that this removal and replacement of the cap helps to maintain both the packing of the matrix and a consistent flowrate.
The SEC columns are stored in preservative that contains enough alcohol to weaken or damage the integrity of the exosome bilipid layers. Therefore, the PBS wash steps of the columns are critical.
The reuse of SEC columns is not recommended in accordance with manufacturer’s instructions as trace amounts of proteins will continue to be released.
The recommended settings may vary depending on your NTA instrument. Please consult the manufacturer for calibration of the instrument to detect spherical liposomes sized 50–100 nm. For consistency, calibrate the instrument using 100 nm microbeads. S.D. should fall between the bead manufacturer’s QC certification fact sheet specifications, including concentration.
There is no need to filter PBS before use if the specific brand/lot of PBS is checked by NTA for particulates by running a PBS-only sample on the NTA instrument. It should register only background levels of particles (the same as reverse-osmosis filtered distilled water). The PBS should be kept at 4 °C when not in use and procedures followed to ensure sterility is not compromised. If the PBS is kept for an extended period of time, check it by NTA before use. If exosome yield following isolation is unusually high, check by NTA to see if the PBS is contaminated. It is advisable to check for particles whenever a new lot of PBS used.
Drying the PVDF membrane allows it to be used at a future time but can also improve the eventual signal by increasing the retention of the proteins on the membrane. Place the dry membranes in a zip-lock bag and store at 4 °C for up to 2 weeks, −20 °C for up to 2 months or at −70 °C for longer storage [16]. When ready to use, bring membranes to room temperature and activate.
SDS is included in the secondary fluorescent antibody blocking buffer to reduce background fluorescence [17].
We typically use IRDye 800CW secondary antibodies to detect protein targets with low abundance, and IRDye 680RD secondary antibodies to detect more abundant proteins.
In the past, negative staining could only be done using heavy metals such as uranyl acetate, which also had radioactive properties: which also had radioactive properties, making it difficult to procure in many labs. In this TEM staining procedure, UranyLess™ can be used in place of uranyl acetate with similar results.
A wash step following incubation with UranyLess™ is not required. We found that without the wash, the exosome images were more accurate and that most exosomes are spherical and rarely cup-shaped.
Acknowledgments
We would like to thank Dr. Teresa Whiteside and her team for their technical advice. This work was supported by The Katherine Anne Gioia Endowed Chair in Cancer Medicine, Roswell Park Comprehensive Cancer Center; National Cancer Institute (NCI) grants P30CA016056 and 1R50CA211108 (HM) involving the use of Roswell Park Comprehensive Cancer Center’s Flow and Image Cytometry Shared Resources.
References
- 1.Shu S, Yang Y, Allen C et al. (2020) Purity and yield of melanoma exosomes are dependent on isolation method. J Extracell Vesicles 9 (1):1692401. 10.1080/20013078.2019.1692401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig N, Lotze MT (2020) A treatise on endothelial biology and exosomes: homage to Theresa Maria Listowska Whiteside. HNO 68 (2):71–79. 10.1007/s00106-019-00803-1 [DOI] [PubMed] [Google Scholar]
- 3.Whiteside TL (2016) Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem 74:103–141. 10.1016/bs.acc.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalluri R (2016) The biology and function of exosomes in cancer. J Clin Invest 126 (4):1208–1215. 10.1172/JCI81135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valadi H, Ekström K, Bossios A et al. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9 (6):654–659. 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Li X, Zhu R et al. (2016) Lung cancer exosomes initiate global long non-coding RNA changes in mesenchymal stem cells. Int J Oncol 48(2):681–689. 10.3892/ijo.2015.3272 [DOI] [PubMed] [Google Scholar]
- 7.Whiteside TL (2017) Exosomes in cancer: another mechanism of tumor-induced immune suppression. Adv Exp Med Biol 1036:81–89. 10.1007/978-3-319-67577-0_6 [DOI] [PubMed] [Google Scholar]
- 8.Whiteside TL (2015) The potential of tumor-derived exosomes for noninvasive cancer monitoring. Expert Rev Mol Diagn 15 (10):1293–1310. 10.1586/14737159.2015.1071666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu S, Yang Y, Allen C et al. (2018) Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci Rep 8 (1):12905. 10.1038/s41598-018-31323-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahlert C, Kalluri R (2013) Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 91 (4):431–437. 10.1007/s00109-013-1020-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalluri R, LeBleu VS (2020) The biology, function, and biomedical applications of exosomes. Science 367(6478):eaau6977. 10.1126/science.aau6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CH, Chen YC (2019) Clinical significance of exosomes as potential biomarkers in cancer. World J Clin Cases 7(2):171–190. 10.12998/wjcc.v7.i2.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alegre E, Zubiri L, Perez-Gracia JL et al. (2016) Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin Chim Acta 454:28–32. 10.1016/j.cca.2015.12.031 [DOI] [PubMed] [Google Scholar]
- 14.Théry C, Whitwer K, Aikawa E et al. (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7(1):1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altevogt P, Bretz N, Ridinger J et al. (2014) Novel insights into exosome-induced, tumor-associated inflammation and immunomodulation. Semin Cancer Biol 28:51–57. 10.1016/j.semcancer.2014.04.008 [DOI] [PubMed] [Google Scholar]
- 16.EMD-Millipore (2017) Protein blotting handbook: tips and tricks, 6th edn. MilliporeSigma, Billerica, MA. https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/General_Information/1/protein-blotting-handbook.pdf [Google Scholar]
- 17.Li-COR Biosciences (2008) Good westerns gone bad: tips to make your NIR Western blot great. http://biosupport.licor.com