

Summary
Dopaminergic inputs to basal amygdala (BA) instruct learning of motivational salience. This learning depends on intracellular plasticity signals such as cAMP, which is regulated by activation of dopamine receptors. Here, we examine the dynamics of dopamine release and downstream signaling during multiple salient events occurring within tens of seconds. We perform real-time tracking and manipulation of cAMP in BA neurons in vitro and in vivo. Optogenetically-evoked release of dopamine drives proportional increases in cAMP in almost all BA glutamatergic neurons, suggesting widespread actions of dopamine across neurons preferring positive or negative valence. This cAMP response decreases across trials with short inter-trial intervals due to depression of dopamine release. No such depression is evident when photostimulating cAMP production directly. cAMP and protein kinase A (PKA) responses to repeated appetitive or aversive stimuli also exhibit pronounced depression. Thus, history-dependent dynamics of dopamine and cAMP may regulate learning of temporally clustered, salient stimuli.
Graphical Abstract
eTOC
Stimuli associated with appetitive or aversive events acquire motivational salience, invigorating behavior during future stimulus presentations. Lutas et al. find that dopamine-triggered molecular signals in amygdala facilitate learning of the motivational salience of stimuli. Dopamine release weakens when salient events occur in rapid succession, limiting molecular signaling in amygdala neurons.
Introduction
Neural plasticity during associative learning involves neuromodulators such as dopamine that regulate cyclic adenosine monophosphate (cAMP) levels (Bissière et al., 2003; Castro et al., 2013; Johansen et al., 2014; Lee et al., 2021; Tronson et al., 2006). While the timing of dopamine-dependent cAMP signals is important for associative plasticity (Handler et al., 2019; Steinberg et al., 2013; Yagishita et al., 2014), we do not fully understand the spatial extent or the temporal dynamics of cAMP signals evoked by natural release of dopamine.
The basal amygdala (BA) region is critical for learning the valence of initially neutral sensory cues (O’Neill et al., 2018; Wang et al., 2018). Dopamine is released by ventral tegmental area (VTA) axons in the BA during salient events of both positive and negative valence (Lutas et al., 2019). Despite this common dopamine signal, individual BA glutamatergic neurons fall into two equal-sized groups that respond to either appetitive or aversive events (Lutas et al., 2019; O’Neill et al., 2018; Zhang and Li, 2018). It is unknown if dopamine evokes increases in cAMP in all BA neurons during a salient event, or if post-synaptic mechanisms restrict cAMP increases to subsets of neurons.
We address these questions by controlling dopamine release from VTA axons in BA (VTADA→BA) while simultaneously visualizing calcium or cAMP dynamics in BA glutamatergic neurons. Optogenetic stimulation of VTADA→BA axons drove acquisition of stimulus responses in BA neurons across days, similar to the acquisition of responses to cues paired with natural appetitive and aversive outcomes (Lutas et al., 2019). Further, selective disruption of cAMP signals within BA glutamatergic neurons impaired the learning of the motivational salience of a cue. Exogenous dopamine or transient photostimulation of VTADA→BA axons elevated cAMP in most neurons. This widespread increase suggests that the even split in the proportions of positive- and negative-valence BA neurons is unlikely to emerge due to target-neuron-specific dopamine signals.
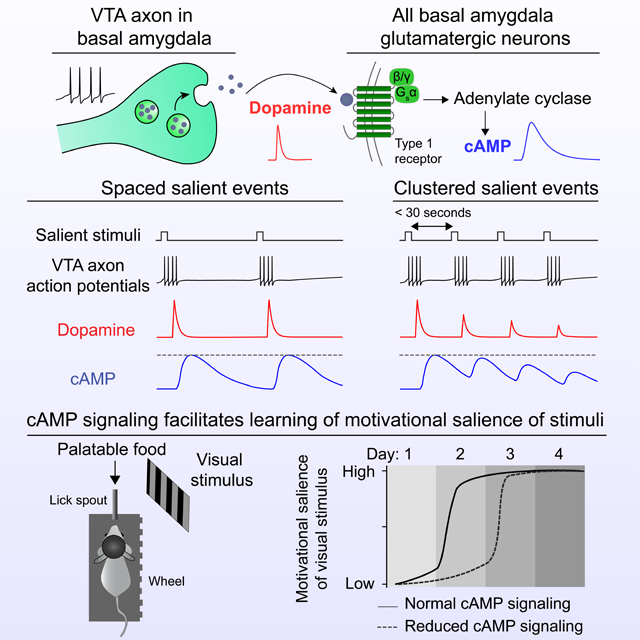
We found that cAMP increases triggered by transient dopamine release or direct photostimulation of cAMP lasted for over 30 seconds. Presynaptic depression at VTADA→BA terminals limited the overall duration of elevated cAMP when multiple dopamine release events were spaced tens (but not hundreds) of seconds apart. This synaptic depression may function as an onset or novelty filter such that the first of a series of sequential salient events would generate the largest magnitude of dopamine-evoked cAMP and, subsequently, the largest effects on neural plasticity. Together, our results suggest that dopamine-evoked increases in cAMP in BA initiate a window for permissive plasticity that is regulated by stimulus history and salience.
Results
Pairing visual cues with optogenetic stimulation of VTADA→BA axons results in cue-evoked calcium responses in BA neurons
BA neurons acquire responses to previously meaningless visual cues when these cues are paired with salient appetitive or aversive outcomes (Lutas et al., 2019; O’Neill et al., 2018; Schoenbaum et al., 1999). To determine whether pairing dopamine release with an arbitrary visual stimulus was sufficient for BA neurons to acquire responses to this stimulus, we combined two-photon calcium imaging of the same BA neurons across several sessions with photostimulation of VTADA→BA axon terminals (Figure S1A–C). Only a small fraction of BA neurons was significantly responsive to the presentation of arbitrary visual stimuli prior to any manipulations (1%; n = 14/1283 neurons from 7 mice; Figure S1D), consistent with prior work (Lutas et al., 2019). We then paired “Cue A” with brief photostimulation of VTADA→BA axon terminals across daily sessions. After three days of pairing, a larger fraction of BA neurons had become responsive to the paired visual stimulus (4%; n = 48/1199 neurons from 6 mice; Figure S1D). Despite the randomized nature of stimulus presentations and the six-second ITI between trials, BA neurons that acquired responses to “Cue A” also acquired responses to the unpaired “Cue B” (Figure S1E,F). Such generalization may relate to sustained effects of transient VTADA→BA axon photostimulation. Nevertheless, responses to “Cue B” were weaker than those to “Cue A” (Figure S1G,H). Therefore, photostimulating VTADA→BA axons immediately following an arbitrary visual cue is sufficient to drive acquisition of cue-evoked responses in a subset of BA glutamatergic neurons.
A cAMP sensor reveals spatially broad dopamine signaling across all BA glutamatergic neurons
To better understand the immediate effects of photostimulating VTADA→BA axons, we expressed a fluorescent biosensor of cAMP, cADDis (Tewson et al., 2016), in BA glutamatergic neurons (Figure 1A,B). In vitro application of the adenylate cyclase activator forskolin (Figure 1C) or dopamine (Figure 1D) generated reliable decreases in overall fluorescence intensity, indicating increases in cAMP concentration (Tewson et al., 2016) (Figure 1C–D).
Figure 1. Endogenous dopamine release elevates cAMP in most basal amygdala glutamatergic neurons.

A) Viral construct and injection strategy for expressing cADDis in basal amygdala (BA) glutamatergic neurons. hSyn: human Synapsin. WPRE: post-transcriptional regulatory element.
B) Schematic and representative images of amygdala brain slice with expression of cADDis in BA neurons. CeA: central amygdala. LA: lateral amygdala.
C) Left: forskolin-mediated activation of adenylate cyclase (AC) drives production of cAMP. Middle: example forskolin-evoked (50 μM) cADDis fluorescence change in BA. Y-axis is inverted for clarity, as increases in cAMP decrease fluorescence. Right: peak response of individual slices. n = 5 slices, 2 mice.
D) Left: Type 1 (D1R) or Type 2 (D2R) dopamine receptors actions on AC. Middle: example dopamine-evoked (20 μM) cADDis fluorescence change in BA. Right: peak response of individual slices. n = 4 slices, 2 mice.
E) Epifluorescence image of cADDis fluorescence and masks for cell somata.
F) Average cumulative distribution of peak responses of individual BA neurons to dopamine and forskolin application. 1 μM dopamine: n = 440 neurons from 3 slices, 1 mouse. 3 μM dopamine: n = 294 neurons, 2 slices, 2 mice. 20 μM dopamine: n = 579 neurons, 4 slices, 2 mice. 50 μM forskolin: n = 597 neurons, 5 slices, 2 mice. n.s., p > 0.05. Kolmogorov-Smirnov test.
G) Schematic of brain slice imaging of cADDis fluorescence and photostimulation-evoked dopamine release.
H) Mean cADDis fluorescence in response to photostimulation of dopamine release (10 s duration; 15.5 Hz). n = 12 slices, 4 mice. Inset: mean cADDis fluorescence with monoexponential fit overlaid (blue line). Fitted decay rate: 20 s.
I) Left: maximum intensity projection of cADDis fluorescence across 15 depths (~10 μm apart). Image color is inverted for clarity. Right: maximum projection of color-coded BA cell masks.
J) Image of normalized difference in cADDis fluorescence between dopamine and baseline conditions in an example slice.
K) Cumulative distribution of peak cADDis responses of individual BA neurons following photostimulation of VTADA→BA axon terminals. n = 1697 neurons from 11 slices, 5 mice.
L-M) Percentage of all neuronal somata (black) or neuropil rings surrounding somata (blue) per slice with significant cADDis responses following photostimulation (L). Peak response of neuronal somata or neuropil rings with significant cADDis responses following photostimulation of VTADA→BA axons (M). n = 11 slices, 5 mice (L,M). All panels: mean ± s.e.m.
As most BA neurons express Type 1 dopamine receptors (D1) (Lutas et al., 2019; Namburi et al., 2015; O’leary et al., 2020), while only few express Type 2 dopamine receptors (D2), dopamine should elevate cAMP in BA neurons. To determine the fraction of BA glutamatergic neurons with increased cAMP in response to dopamine, we extracted signals from the somatic regions of individual neurons (Figure 1E; see Methods). Forskolin, which drives dopamine receptor-independent cAMP production, increased cAMP in all BA glutamatergic neurons. Similarly, application of a high concentration of dopamine (20 μM) potently elevated cAMP in all BA glutamatergic neurons, while lower concentrations drove weaker responses (Figure 1F). This finding demonstrates a near-universal potential for dopamine signaling in BA glutamatergic neurons, and suggests that the diversity of valence preferences of nearby BA glutamatergic neurons (Lutas et al., 2019; O’Neill et al., 2018) may not be due to differences in their inherent ability to increase (or decrease) their cAMP levels in response to dopamine.
Photostimulation of VTADA→BA axons transiently elevates cAMP in almost all BA glutamatergic neurons
While exogenous application of dopamine revealed a widespread increase in cAMP in BA glutamatergic neurons, physiological release of dopamine directly from axon terminals may be more spatially restricted. To evoke endogenous dopamine release, we targeted expression of a red-shifted channelrhodopsin (Chrimson) to VTA dopamine neurons (Bäckman et al., 2006; Klapoetke et al., 2014). To confirm that photostimulating VTADA→BA axons in brain slices reliably evoked dopamine release, we combined widefield photostimulation of Chrimson with two-photon imaging of a green fluorescent biosensor for dopamine, dLight1.1 (Patriarchi et al., 2018) (Figure S2A). Brief trains of optical stimulation (15.5 Hz; 5 s duration) generated transient elevations in dopamine that lasted for several seconds (Figure S2B). Importantly, fluorescence changes in this D1-based dopamine sensor were blocked by application of a D1 antagonist (SCH23390, 300 nM, Figure S2C), confirming that the signals were mediated by dopamine release and not by artifacts related to optical stimulation.
We next tested whether we could visualize changes in cAMP levels in response to photostimulated dopamine release (Figure 1G). Photostimulation of VTADA→BA axon terminals transiently increased cAMP, which peaked later than dopamine and was mediated by D1 dopamine receptors (Figure S2E,F). cAMP remained above baseline for tens of seconds following photostimulation – longer than the duration of elevated dopamine (Figure 1H). The slow decay of cAMP was not due to prolonged activation of dopamine receptors, as similar decay kinetics were observed following direct, transient photostimulation of intracellular cAMP using a blue-light activated adenylate cyclase (biPAC; Figure S3A–C) (Zhang et al., 2021b). This extended elevation in cAMP across tens of seconds may reflect a broad temporal window of plasticity that could result in generalization of plasticity to unpaired cues occurring within that time window (e.g. Figure S1).
We next examined endogenous dopamine-evoked cAMP signals from individual BA neuron somata (Figure 1I,J; see Methods). We detected widespread cAMP responses in over 70% of all BA glutamatergic neuron somata as well as in the surrounding neuropil (largely composed of local dendrites; Figure 1K,L). While this fraction is less than that evoked by near-saturating concentration of bath-applied dopamine, this is likely due to the lower concentration of dopamine released via photostimulation (cf. Figure 1 and Figure S2D). Responses were of similar magnitude in somatic and neuropil compartments, consistent with dopaminergic axon synaptic contacts onto dendrites of these neurons (Muller et al., 2009) (Figure 1M). Thus, most BA glutamatergic neurons express functional D1 receptors and respond to axonal release of dopamine.
Synaptic depression of dopamine release restricts cAMP signaling in BA neurons
We noticed that brief trains of optical stimulation (15.5 Hz; 2, 5, or 10 s duration) generated transient elevations in cAMP that increased sublinearly with pulse train duration (Figure 2A,B). This sublinearity may reflect presynaptic depression of dopamine, as has been observed for dopaminergic inputs to the striatum (Liu and Kaeser, 2019). Indeed, we observed that photostimulating VTADA→BA axons evoked dopamine release that also scaled sublinearly with increasing duration of the pulse train (Figure 2C,D). It is unlikely that cADDis or dLight1.1 sensors are near saturation during responses to photostimulated dopamine release (compare Figure 1F and 1H; see also Figure S2D). Rather, the sublinear scaling of responses with stimulation duration is best explained by a progressive decline in evoked dopamine release.
Figure 2. Synaptic depression of dopamine release restricts postsynaptic cAMP accumulation.
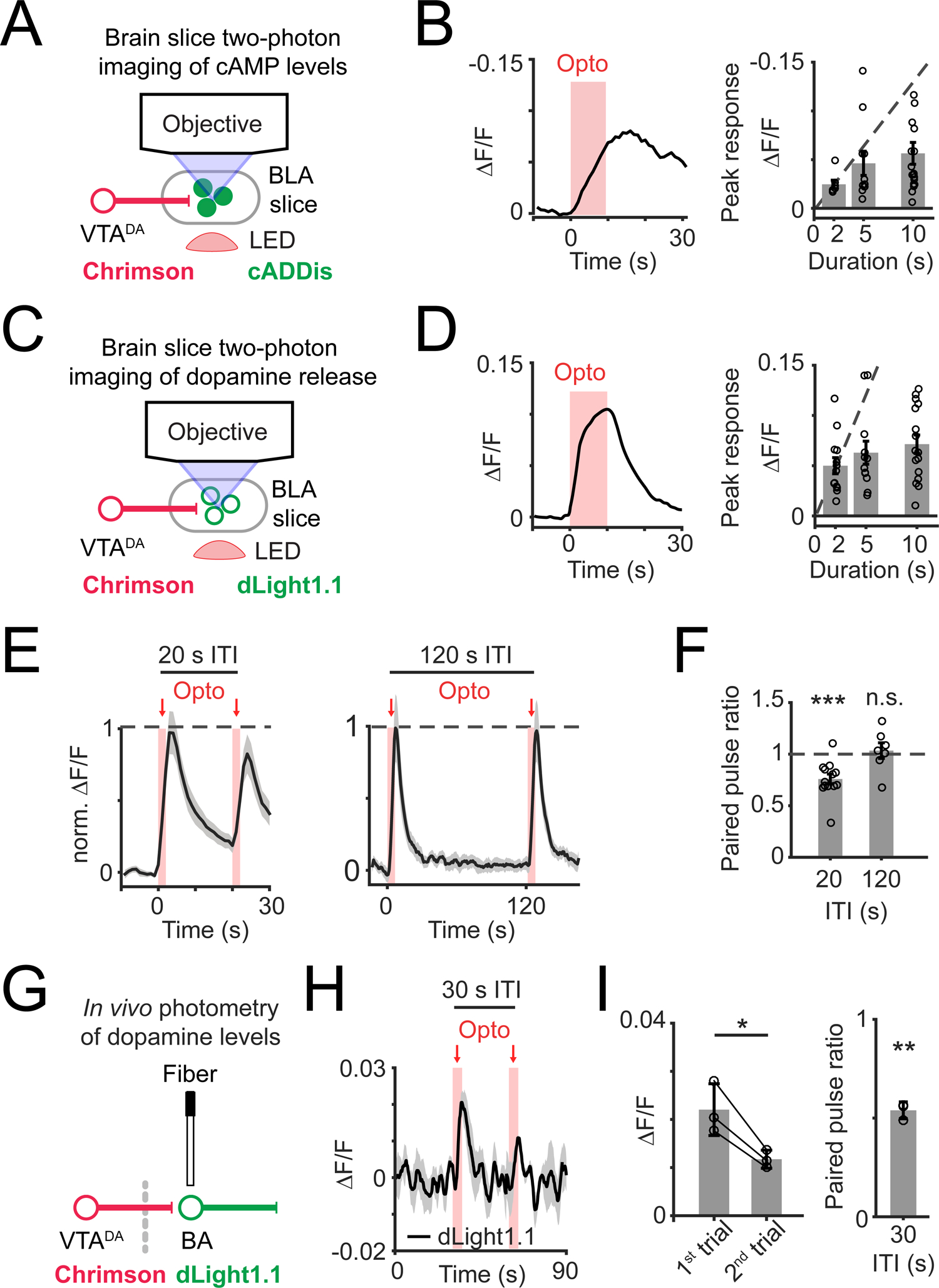
A) Schematic of brain slice imaging of cADDis fluorescence and photostimulation-evoked dopamine release.
B) Left: example recording of cADDis fluorescence in response to a train of red-light pulses (10 s duration; 15.5 Hz). Right: peak response of individual brain slices to photostimulation of dopamine release for 2 s (n = 7 slices, 4 mice), 5 s (n = 11 slices, 5 mice), or 10 s (n = 13 slices, 5 mice). Recordings are normalized to baseline period before the first photostimulation pulse.
C) Schematic of brain slice imaging of dopamine sensor dLight1.1 and photostimulation-evoked dopamine release.
D) Same as in (B) for dLight1.1 imaging. Stimulation duration: 2 s (n = 13 slices, 4 mice), 5 s (n = 12 slices, 4 mice), or 10 s (n = 16 slices, 4 mice).
E) Left: mean time course in response to a pair of photostimulation trains (2 s train duration at 15.5 Hz) with inter-trial intervals (ITIs) of 20 s (left; n = 13 slices, 4 mice) or 120 s (right; n = 7 slices, 2 mice). Recordings are normalized to period before the first photostimulation pulse.
F) Paired pulse ratio of peak response (second pulse train / first pulse train) for ITIs of 20 s (***, p = 0.0005, n = 13 slices, 4 mice) or 120 s (n.s., p = 0.57, n = 7 slices, 2 mice). Two-sided Wilcoxon sign-rank.
G) Schematic of in vivo photometry recordings of dLight1.1 fluorescence and photostimulation of dopamine release.
H) Mean time course of dLight1.1 fluorescence in response to photostimulation of VTADA→BA axons (5 s duration; 20 Hz; 30 s ITI). n = 3 mice.
I) Left: peak response during the first and second photostimulation pulse trains (30 s ITI). Right: paired pulse ratio of peak response (second pulse train / first pulse train). *, p = 0.03; **, p = 0.002, n = 3 mice. Two-sided paired t-test. All panels: mean ± s.e.m.
We also observed a diminished magnitude of dopamine release across repeated stimulation events when inter-trial intervals (ITIs) were 20 seconds long, but not when they were two minutes long (Figure 2E–F).
To examine this depression phenomenon in vivo, we recorded photostimulated dopamine release in BA using fiber photometry (Figure 2G). We previously found that this photostimulation protocol resulted in dopamine release of a similar magnitude as natural (tail shock-evoked) release (Lutas et al., 2019). Here, we found that the magnitude of dopamine release was smaller when a photostimulation event followed a prior event by 30 s, providing direct in vivo evidence for presynaptic depression (Figure 2H,I). This long-lasting depression of dopamine release is consistent with the depression of VTADA→BA axon-evoked glutamate co-released with dopamine (Lutas et al., 2019), and with depression at dopamine projections to the striatum (Adrover et al., 2014; Zhang et al., 2009).
In vivo cAMP dynamics in BA neurons following repeated photostimulation of VTADA→BA axons
We next asked whether cAMP dynamics in BA glutamatergic neurons exhibit similar characteristics in vivo as in our brain slice experiments (Figure 3A). We reliably detected cAMP responses lasting tens of seconds following brief optical stimulation of dopamine release (Figure 3B). Our protocol for in vivo photostimulation of VTADA→BA axons was well below saturating levels, as it drove weaker changes in cAMP than intraperitoneal injection of a high concentration of a D1 receptor agonist (SKF81297; 20 mg/kg; Figure S4A). When we repeatedly photostimulated VTADA→BA axons every 30 s, we found that cAMP transients became profoundly attenuated over the course of several minutes (Figure 3C,D). We did not observe this attenuation when using a 120 s ITI (Figure S4B). This result suggested that the attenuation in cAMP responses could be explained by the presynaptic depression of dopamine release described above.
Figure 3. In vivo fiber photometry recordings of cAMP dynamics following photostimulated dopamine release, tail shock and Ensure delivery.
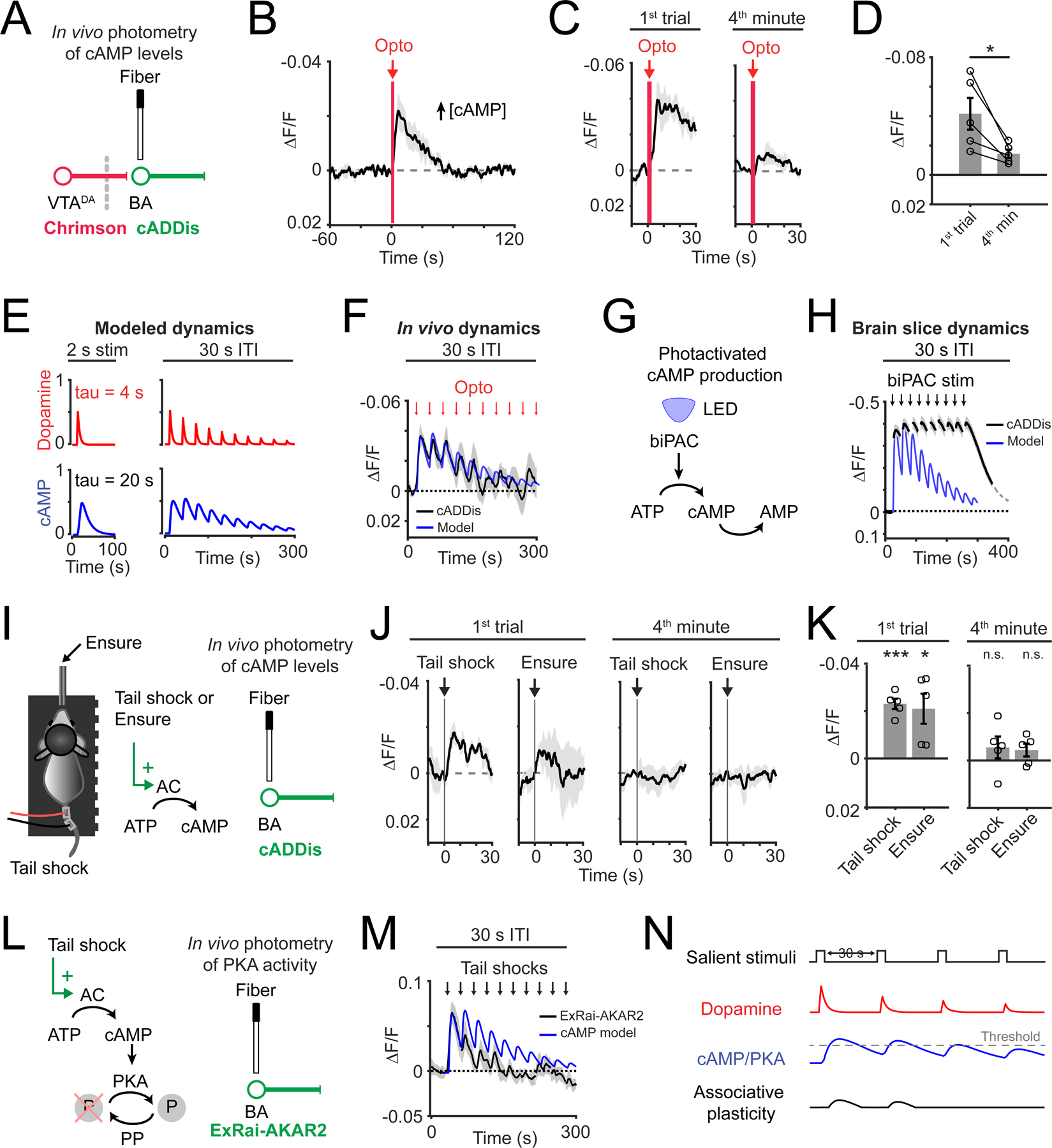
A) Schematic of in vivo photometry recordings of cADDis fluorescence and photostimulation of VTA axon dopamine release in BA.
B) Mean cADDis response to photostimulation (2 s duration; 20 Hz). n = 5 mice.
C) Mean cADDis response to photostimulation (2 s duration; 20 Hz) during first trial (left) or fourth minute of recording (right; average of two 30-s trials). n = 5 mice.
D) Peak response following photostimulation on first trial vs. fourth minute (* p = 0.03, two-tailed paired t-test, n = 5 mice).
E) Simulation of dopamine (top; red) and cAMP (bottom; blue) dynamics for a single event (2 s) (left), or for multiple events (30 s ITI) (right).
F) Mean cADDis response to photostimulation (black line; 2 s duration; 20 Hz; 30 s ITI; n = 5 mice). Simulated cAMP dynamics (blue line) scaled to peak cADDis response.
G) Schematic of cAMP production using biPAC (blue-light activated adenylate cyclase).
H) Mean cADDis response to photostimulation of biPAC in brain slices (black; 2 s duration; 30 s ITI; n = 11 slices, 4 mice). Simulated cAMP dynamics (blue line) scaled to peak cADDis response.
I) Left: schematic of delivery of uncued aversive tail shock or palatable food (Ensure) to an awake, head-fixed mouse. Middle: predicted consequences on cAMP production in BA glutamatergic neurons. Right: schematic of in vivo photometry recordings.
J) Left: mean cADDis response to tail shock delivery (0.3 mA; 50 ms duration; 30 s ITI) or to Ensure delivery (single 5 μL droplet; 30 s ITI) during first trial. Right: same as left, but for the fourth minute of recording. n = 5 mice.
K) Peak response following tail shock or Ensure delivery during the first trial (left) or fourth minute (right). ***, p = 0.0004; *, p = 0.03; n.s., p > 0.05, two-tailed t-test, peak response vs. 0; n = 5 mice.
L) Left: predicted effect of tail shock on protein kinase A (PKA) activity. Dephosphorylation by protein phosphatases (PP) is indicated. Right: schematic of in vivo photometry recordings of ExRai-AKAR2 (biosensor of PKA activity).
M) Mean ExRai-AKAR2 response to tail shock delivery (0.3 mA; 50 ms duration; 30 s ITI; n = 6 mice), normalized to baseline period before first trial. Simulated cAMP dynamics (blue line; based on model in Figure 3E) scaled to peak photometry amplitude.
N) Schematic of observed effect of salient stimuli (30 s ITI) on dopamine and cAMP/PKA responses. The observed depression may attenuate windows of plasticity associated with closely spaced salient stimuli. All panels: mean ± s.e.m.
We explored this possibility by modeling cAMP dynamics in response to dopamine released at variable ITIs (30 s: Figure 3E; 10 s: Figure S4C). We started with the assumption that cAMP dynamics following photostimulation of dopamine axons could be understood as a convolution of the dynamics of extracellular dopamine and of the decay in intracellular cAMP. We fit mono-exponential decay functions to the post-peak response time courses from slice recordings for axon-evoked dopamine release and, separately, for cAMP evoked by direct, transient photostimulation of intracellular cAMP (dopamine: τ = 4 s; cAMP: τ = 20 s; see Methods). We then modeled the attenuation in dopamine release as observed in our measurements of paired-pulse depression (Figure 2E–F; paired pulse ratio: 75%). Our simulation revealed that depression in dopamine release across trials is sufficient to explain a return of evoked cAMP levels to baseline within minutes (Figure 3E; Figure S4C).
We compared this simulation to in vivo cADDis fiber photometry during the same protocol of photostimulation of VTADA→BA axons with 30-s or 10-s ITIs. We found that endogenous cAMP dynamics mirrored those in our simulation (Figure 3F; Figure S4D). To further assess whether a presynaptic rather than postsynaptic mechanism mediated the depression in evoked cAMP responses, we used biPAC to bypass the dopamine receptor and directly stimulate intracellular cAMP production (Figure 3G). When we photostimulated biPAC repeatedly every 30 seconds, cAMP levels remained persistently elevated throughout the recording (Figure 3H). Together, these results demonstrate that transient cAMP elevations such as those that may occur during a rapid sequence of salient events do not accumulate across minutes, likely because of presynaptic depression of dopamine release.
Repeated shocks or rewards evoke progressively weaker cAMP responses in BA neurons
Dopamine is naturally released in the BA during motivationally salient appetitive and aversive events (Lutas et al., 2019). We examined cAMP responses to repeated delivery of appetitive palatable food in hungry mice, and to repeated, aversive tail shocks in sated mice (Figure 3I), both of which drive robust dopamine release in the BA (Lutas et al., 2019). We detected significant responses on the first trial of each sequence (Figure 3J,K). Similar to repeated photostimulation of VTADA→BA axons, repeated food delivery or tail shock attenuated cAMP transients such that, within 5 minutes, we were unable to detect significant evoked increases (Figure 3J–K). The depression of cAMP responses in vivo during tail shocks or food delivered was generally consistent with predictions from our simulated dopamine-evoked cAMP signal, but was more variable than that observed with VTADA→BA axon photostimulation (Figure S4E,F).
We next asked whether the depression of cAMP signals was paralleled by depression of protein kinase A (PKA) activity - a key downstream regulator of synaptic plasticity (Figure 3L). We used fiber photometry and a fluorescent sensor of PKA activity, ExRai-AKAR2 (Zhang et al., 2021a), to track in vivo responses to tail shocks (Figure 3L). We found that tail shocks elicited increases in ExRai-AKAR2 signal that were more robust than responses observed using cADDis (Figure 3M). Consistent with the depression in cADDis responses, we found that ExRai-AKAR2 responses depressed with repeated tail shocks (30 s ITI; Figure 3M). ExRai-AKAR2 reports the ratio of phosphorylation by PKA and dephosphorylation by protein phosphatases. However, given our earlier findings of depressing cAMP responses to repeated dopamine axon photostimulation, it is likely that attenuation in ExRai-AKAR2 responses is due to a weakening in cAMP-mediated phosphorylation by PKA. Our characterization of ex vivo and in vivo cAMP and PKA sensor responses following endogenous dopamine release reveals temporal constraints on sustained cAMP elevation and PKA phosphorylation of targets in BA neurons. These constraints likely increase the relative potency of associative plasticity for novel salient events vs. subsequent events occurring in close succession (Figure 3N).
Enhancing cAMP breakdown leads to impaired learning of motivational salience
To assess whether cAMP signaling in BA glutamatergic neurons was necessary for learning incentive motivational properties of a stimulus, we virally delivered a minimal catalytic domain of phosphodiesterase 4D3 (PDE4D3-cat) to enhance breakdown of cAMP and prevent dopamine-evoked cAMP production (Zhang et al., 2021b) (Figure 4A). cAMP increases following biPAC photostimulation were abolished in neurons expressing PDE4D3-cat (Figure 4A–C). Application of the phosphodiesterase inhibitor IBMX could partly rescue the loss of biPAC-evoked cAMP transients, indicating that neurons expressing PDE4D3-cat were still capable of producing cAMP (Figure 4D).
Figure 4. Enhancing cAMP breakdown in BA glutamatergic neurons disrupts learning of motivational salience.

A) Schematic of phosphodiesterase 4D3 catalytic domain (PDE4D3-cat) enhancing the breakdown of cAMP production following biPAC photostimulation (left) and brain slice two-photon imaging (right).
B) Example images of amygdala brain slice (top left) and cADDis fluorescence in BA glutamatergic neurons (top right). Example image showing neurons expressing cADDis (bottom left) and mKate2-biPAC with mKate2-PDE4D3-cat (bottom right).
C) Left: stimulation of biPAC (2 s duration; 120 s ITI) in control neurons (black; n = 13 slices, 4 mice) or neurons expressing PDE4D3-cat (green; n = 14 slices, 5 mice). Right: same as left for 30 s ITI in control (black; n = 11 slices, 4 mice) or PDE4D3-cat (green; n = 7 slices, 3 mice) neurons.
D) Stimulation of biPAC (2 s) in neurons expressing PDE4D3-cat prior to (green; n = 5 slices, 4 mice) or following application of a phosphodiesterase inhibitor (IBMX, 100 μM; red; n = 5 slices, 4 mice).
E) Top: viral construct for PDE4D3-cat. Bottom: viral delivery of PDE4D3-cat in BA glutamatergic neurons.
F) Left: schematic of visual stimulus and Ensure delivery to an awake, food restricted, head-fixed mouse. Right: training paradigm: habituation following by consecutive days of pairing cue with Ensure (60 trials/day; 30 s ITI).
G) Mean baseline-subtracted lick rate per day throughout Pavlovian conditioning, for control (top; n = 7) or PDE4D3-cat expressing mice (bottom; n = 6).
H) Top: mean baseline-subtracted lick rate during the last second of the 2 s cue, normalized to maximum cue-evoked lick rate across days, for control (n = 7) or PDE4D3-cat expressing mice (n = 6). Bottom: maximum lick rate during each bin across control (black) or PDE4D3-cat expressing (green) mice.
I) Latency to half maximum cue-evoked lick rate. ***, p = 0.0007, two-tailed unpaired t-test. All panels: mean ± s.e.m.
We then tested whether expression of PDE4D3-cat in BA glutamatergic neurons would disrupt learning of motivational salience (Figure 4E). We used a Pavlovian trace conditioning paradigm known to require new protein synthesis in the BA (Kochli et al., 2015). In our paradigm, a visual stimulus was followed by the delivery of Ensure (Figure 4F). Food-restricted control mice displayed cue-evoked anticipatory licks during the visual stimulus on the second day of associative learning, indicating successful cue-outcome learning (Figure 4G). In contrast, mice expressing PDE4D3-cat took significantly longer to develop cue-evoked anticipatory licking, indicating a delay either in associative learning and/or in the expression of incentive salience in the absence of cAMP signaling (Figure 4G–I). These behavioral results support a role for cAMP signaling in BA glutamatergic neurons in the learning of motivational salience.
Discussion
The release of neuromodulators including dopamine in the basal amygdala is critical for cue-outcome association learning (Johansen et al., 2014; Tang et al., 2020; Tye et al., 2010). Dopamine receptor activation and increased cAMP may strengthen synaptic inputs to enhance the salience of stimuli paired with either appetitive or aversive outcomes (Lutas et al., 2019). Here, we show that disruption of transient increases in cAMP impaired learned cue-evoked anticipatory licking. Similar to recent experiments inhibiting PKA activity in downstream neurons in nucleus accumbens (Lee et al., 2021), our results indicate an important role for cAMP dynamics in BA glutamatergic neurons during associative learning
Synaptic depression of dopamine release in BA
We showed that synaptic depression of dopamine release occurs at VTADA→BA axon terminals and explains the weakened cAMP signals in response to repeated VTADA→BA photostimulation. Similar characteristics have been observed in dopamine neuron projections to the dorsal and ventral striatum (Adrover et al., 2014; Michael et al., 1987). Synaptic depression at these mesostriatal dopaminergic synapses is mediated by several factors (Liu and Kaeser, 2019; Zhang et al., 2009). We suspect that similar mechanisms mediate the minutes-long depression we observed in VTADA→BA projections.
The depression of dopamine release may impose temporal constraints on synaptic plasticity when salient events are clustered in time. In this way, the spacing of salient events may affect learning rates and memory consolidation in amygdala-dependent tasks (Jacob and Waddell, 2020; Josselyn et al., 2001). Moreover, as certain medications (e.g. methylphenidate) as well as drugs of abuse (e.g. cocaine) can enhance dopamine synaptic depression (Adrover et al., 2014), these drugs may influence the temporal properties of amygdala plasticity.
What fraction of BA neurons is affected by dopamine release from VTADA→BA axons?
We previously showed that dopamine is released from individual VTADA→BA axons following both appetitive and aversive outcomes (Lutas et al., 2019). Here, we found that dopamine-evoked increases in cAMP in almost all BA glutamatergic neurons. This is likely to be a conservative estimate of affected neurons, given the limited sensitivity of our cAMP sensor (see below). This finding suggests that dopamine release in BA may reflect a permissive signal that gates plasticity in both appetitive- or aversive-preferring glutamatergic neurons (Beyeler et al., 2016; Kim et al., 2016; Lutas et al., 2019; Zhang and Li, 2018; Zhang et al., 2021c). In this way, dopamine could control the rate of learning according to the degree of motivational salience of a given event, by scaling the proportion of BA neurons that undergo a rise in cAMP levels. Meanwhile, learning of the valence of the outcome associated with a cue may be achieved via calcium-dependent processes driven by distinct pathways relaying information about sensory cues and about positive and negative outcomes (Correia and Goosens, 2016).
Limitations of the study
The low affinity of the cAMP sensor we used (Tewson et al., 2016) may explain why our in vivo fiber photometry measurements did not pick up the weaker and/or more sparse increases in cAMP following delivery of palatable food or tail shocks. This sensitivity may have been further impaired by not specifically targeting cADDis to dendritic compartments where dopamine-evoked cAMP increases may be concentrated (Pinard et al., 2008). We partially overcame this sensitivity issue by using a high signal-to-noise biosensor of PKA activity (Zhang et al., 2021a), which revealed depressing responses to trains of tail shocks. Future studies using higher affinity cAMP sensors (Massengill et al., 2021) or fluorescence lifetime optimized biosensors (Lee et al., 2019, 2021; Massengill et al., 2021) as well as targeting of sensors to dendritic spines should enhance real-time interrogation of biochemical signaling in response to endogenous salient events.
Conclusions and future directions
We established an all-optical platform using reporters and actuators of intracellular cAMP to interrogate dopamine-dependent cAMP signals in BA glutamatergic neurons in vitro and in vivo. Future studies can employ this approach to study cAMP dynamics in BA excitatory neurons as well as parvalbumin-positive interneurons (Chu et al., 2012; Pinard et al., 2008), in both basic and pre-clinical settings such as following exposure to addictive substances, or to stressful, traumatic events. In addition, these tools can be used to understand other neuromodulatory signals in the BA (e.g., norepinephrine, PACAP, and serotonin) that act via influences on cAMP. While most in vivo studies of cellular plasticity have focused on changes in electrical and calcium activity, continued optimization of tools to detect molecular signaling cascades will expand our understanding of the underlying biochemical signals that control neural circuit plasticity (Lee et al., 2019, 2021).
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mark Andermann (manderma@bidmc.harvard.edu).
Materials availability
For inquiries regarding cADDis plasmid and virus generated during this study, please contact Montana Molecular.
Plasmids for mKate2-biPAC and mKate2-PDE4D3-cat were previously generated (Zhang et al., 2021b) and deposited to Addgene [catalog # 169127 & 169128].
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mouse care and experimental procedures were approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center. Mice were housed in a 12-hour-light/12-hour-darkness environment with standard mouse chow and water provided ad libitum, unless specified otherwise. Food restricted mice were singly housed. For both in vitro and in vivo experiments, adult male and female mice between the ages of 9 to 20 weeks were used for this study. The EMX1-Cre and SLC6A3-IRES-Cre (DAT-IRES-Cre) strains used in this study were maintained on a C57Bl/6 background.
METHOD DETAILS
Stereotaxic surgeries
Viral injections, fiber implantations, and GRIN lens implantations were performed as described in Lutas et al., 2019, with the following specifications and modifications.
For targeting expression into BA glutamatergic neurons, mice (8 – 12 weeks of age) expressing Cre recombinase driven by the Emx1 promoter (EMX1-Cre) were injected at (relative to Bregma): anteroposterior (AP): − 1.6 mm; dorsoventral (DV): − 4.8 mm; mediolateral (ML): ± 3.3 mm. For targeting expression into VTA dopamine neurons, DAT-Cre mice were injected at AP: − 3.2 mm; DV: − 4.5 mm; ML: ± 0.4 mm.
For in vivo experiments using the cAMP sensor cADDis, we unilaterally injected AAV1-hSyn-DIO-GreenDownward-cADDis (Children’s Hospital Vector Core; 300 nL). For cADDis or dLight1.1 (AAV1-hSyn-dLight1.1; Children’s Hospital Vector Core; 300 nL) brain slice imaging experiments, injections were targeted bilaterally to increase the number of useable slices. For experiments using biPAC (AAV1-EF1a-DIO-mKate2-biPAC; Children’s Hospital Vector Core) or PDE4D3-cat (AAV1-EF1a-DIO-mKate2-PDE4D3-cat; Children’s Hospital Vector Core) together with cADDis imaging, viruses were mixed 1:1 and a total volume of 300 nL was injected bilaterally. For in vivo calcium imaging experiments, we unilaterally injected AAV1-hSyn-FLEX-GCaMP6s (Addgene; 300 nL). For in vivo experiments using the PKA activity sensor ExRai-AKAR2, we unilaterally injected AAV9-hSyn-FLEX-ExRai-AKAR2 (300 nL) (Zhang et al., 2021a).
For fiber photometry experiments, optic fibers (400 μm diameter core; multimode; NA 0.48; 5.0 mm length; Doric Fibers) were implanted over BA at (relative to Bregma): AP: −1.6 mm; DV: −4.7 mm; ML: 3.3 mm.
For in vivo BA cell body imaging of GCaMP6s, mice were implanted with a singlet gradient index (GRIN) lens (GRINtech, NEM-100-25-10-860-S-1.0p; 1.0 mm diameter; 9 mm length; 250 μm focal distance on brain side at 860 nm, (NA 0.5); 100 μm focal distance on air side (NA 0.5); non-coated). GRIN lens implantation coordinates for cell body imaging of BA neurons in EMX1-Cre transgenic mice (relative to Bregma): AP: −1.6 mm, ML: 3.2 mm, DV: −4.8 mm. In order to ensure a snug fit for the lens to reduce brain motion and to increase surgical survival rate, we pre-set the insertion tracks by lowering a syringe needle with a slightly narrower diameter (20-gauge, 0.9 mm) to a depth of 0.1 mm above the final depth of the lens.
For in vivo PDE4D3-cat experiments, we bilaterally injected AAV1-EF1a-DIO-mKate2-PDE4D3-cat (Children’s Hospital Vector Core; 300 nL) and cemented titanium headposts similar to GRIN lens implantation surgeries.
Widefield epifluorescence imaging of acute brain slices
Following 3 – 5 weeks of expression, acute slices were prepared as described in Lutas et al., 2019, and widefield fluorescence imaging was performed on an upright microscope (Axioskop 2 plus; Zeiss) equipped with an sCMOS II camera (Prime, Photometrics). Fluorescence excitation for imaging was achieved using a 470 nm LED (Thorlabs). Image acquisition was performed using ImageJ Micro-manager (Edelstein et al., 2014). Image acquisition frame rate was 2 Hz for cADDis fluorescence imaging. A 10x (Olympus) or 20x (Zeiss) objective was used for all imaging experiments. During imaging, slices were continuously superfused (flow rate: 2–5 ml/min) with oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) at room temperature. To prevent oxidation of dopamine, 50 μM Na-metabisulfite was included in all ACSF solutions during dopamine application experiments.
General two-photon imaging methods
Two-photon imaging was performed using a two-photon resonant-galvo scanning microscope (NeuroLabWare) at 15.5 frames/second and 796 × 512 pixels/frame as described previously (Lutas et al., 2019). An InSight X3 laser (Spectra-Physics) was used to excite the fluorophores (920–1050 nm), and the emission light was filtered (green: 510/84 nm; red: 607/70 nm; Semrock) before collection with photomultiplier tubes (H11706-40; Hamamatsu). The XY scanning was performed using resonant/galvo mirrors and the Z scanning was achieved with an electrically-tunable lens (Optotune).
Two-photon imaging of acute brain slices
For two-photon imaging of acute brain slices, slices were prepared as described in Lutas et al. 2019, and transferred to a recording chamber perfused with ACSF (oxygenated with 95% O2 and 5% CO2; flow rate: 2–5 mL/min) at either room temperature or 32° C as indicated. Imaging was performed with a 16× 0.8 NA water-immersion objective (Nikon). The excitation wavelength used was 920 nm.
In optogenetic experiments involving Chrimson, stimulation was triggered by the onset of image acquisition frames (15.5 frames/second) for 2-, 5-, or 10-second duration. The gating property of the PMT (H11706-40; Hamamatsu) was triggered at the onset of the frame to protect the PMT from optogenetic stimulation light and lasted for 10 ms. Thus, the top ~16% of each frame during the stimulation was blank, but much of the frame (~84%) provided near-simultaneous imaging of biosensor signals during the photostimulation. A 620 nm LED (1 mW/mm2, Luxeon Star LEDs) driven by an Arduino-controlled driver (Luxeon Star LEDs) was used for photostimulation.
In optogenetic experiments involving biPAC, the PMT was powered off during the photostimulation (2-s; 470 nm LED; 1 mW/mm2, Luxeon Star LEDs), which was driven by an Arduino-controlled driver (Luxeon Star LEDs).
To block phosphodiesterase activity, IBMX (100 μM in DMSO; Tocris R&D systems) was applied to the brain slice for at least 10 minutes. Recordings of cADDis following biPAC stimulation were performed before and after application of IBMX. To block dLight1.1 (dLight1.1 is engineered from the D1 receptor) or cADDis signals, we applied an antagonist of the D1 receptor (SCH23390; 300 nM; Tocris R&D systems) while recording signals evoked by photostimulation of dopamine release from VTADA→BA axons in brain slices.
In vivo two-photon imaging via implanted lens
Two-photon imaging via implanted lenses was performed as previously described (Lutas et al., 2019) with the following adjustments. A 10× 0.5 NA air objective was used (TL10X-2P; ThorLabs). For optogenetic stimulation of Chrimson via the implanted lens, a 617 nm LED (M617L3; ThorLabs) controlled by an LED driver (T-Cube; ThorLabs) was used (5 – 10 mW at the objective face). One of the stimuli (“Cue A”) was paired with photostimulation of VTADA→BA axon terminals, which occurred 200 ms after the visual stimulus offset, and the second (“Cue B”) was not paired with any outcome (Figure S1C).
Fiber photometry recording
Fiber photometry recordings were performed as described in Lutas et al., 2019, using head-fixed mice that were free to run on a circular treadmill. Fiber optic cables (1 m long; 400 μm core; 0.48 NA; Doric Lenses) were coupled to implanted optic fibers with zirconia sleeves (Precision Fiber Products). Excitation and emission light was passed through a four-port fluorescence mini-cube (FMC4_E(460–490)_F(500–550)_O(580–650)_S, Doric Lenses), which allowed for collection of GFP fluorescence and excitation of red-shifted channelrhodopsins. For biosensor photometry recordings, the excitation light (~ 100 μW at the face of the patch cord) was provided by a 465 nm LED (Plexon LED and driver). For optogenetic stimulation, the excitation light (~ 5 mW at the face of the patch cord) was provided by a 620 nm LED (Plexon LED and driver) which was controlled by an Arduino Uno. Emission light was collected by a femtowatt photoreceiver (Newport 2151), demodulated using a lock-in amplifier (SR830; Stanford Instruments) and digitized at 1 kHz sample rate (PCIe-6321; National Instruments). Data acquisition was controlled using a custom script in MATLAB (MathWorks). In a subset of experiments the D1 receptor agonist (SKF81297; 20 mg/kg in saline; Tocris R&D systems), was injected intraperitoneally while recording cADDis photometry signals. In these experiments, endogenous dopamine release was achieved with Chrimson photostimulation of VTADA→BA terminals and we compared peak cAMP responses following photostimulation and injection of SKF81297. Delivery of uncued appetitive (Ensure) or aversive (tail shock; AMPI Iso-Flex stimulus isolator; 0.3 mA, 50 ms) outcomes was performed as previously described (Lutas et al., 2019).
Pavlovian conditioning behavior
Following three weeks of recovery from surgery and expression of the viral construct, mice were chronically food restricted to maintain body weight at 85% of free-feeding body weight(Lutas et al., 2019). Mice were habituated for two days (30 minutes per day) to head fixation and trained to lick at a spout to receive Ensure delivery. Afterwards, mice were trained on a Pavlovian conditioning task in which a full-screen square wave drifting grating (0°; 2 Hz, 0.04 cycles/degree; 80% contrast) was presented for 2 seconds followed by the delivery of a drop of Ensure (~5 μL). Each training day consisted of a single 30 minute session during which a total of 60 visual stimuli were presented with the inter-trial interval of 30 seconds. All mice were trained on the Pavlovian conditioning task for four consecutive days. All behavioral training was conducted using MonkeyLogic (Asaad and Eskandar, 2008; Lutas et al., 2019) and licks were detected by a capacitance-sensing lickspout (3D printed with conductive filament connected to a capacitance sensor, MPR121; Adafruit).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Statistical details are found in the figure legends. The numbers of samples in each group were based on those in previously published studies. Experiments were conducted by an investigator with knowledge of the animal genotype and treatment. All virus expression, optic fiber implants, and GRIN lens placements were verified by post hoc histology. Mice in which either the virus expression or optic fiber was not appropriately located (< 10 % of the time) were excluded from analysis. All data presented as bar and line graphs indicate mean ± s.e.m. with individual data points also plotted. Statistical analyses were performed in MATLAB. Significance levels are indicated as follows unless otherwise specified: *p < 0.05; **p < 0.01; ***p < 0.001.
Data analysis
All data analyses were performed using MATLAB (Mathworks) and ImageJ (NIH).
Fiber photometry analysis
Photometry signals were sampled at 1 kHz, low pass filtered below 100 Hz, and downsampled to 50 Hz. We calculated ΔF/F = (F − F0)/F0, where F0 the mean of baseline window prior to the event of interest. For analysis of the responses to an individual event, all trials containing presentations of that event during a run were averaged to obtain a mean timecourse, and then the peak response during the event window was obtained. We also did not use the sliding-window method for baseline normalized to avoid distorting the slow dynamics of extracellular dopamine and intracellular cAMP signals.
Widefield fluorescence brain slice imaging analysis
Movies were initially corrected for x-y motion using identical methods (efficient subpixel registration to averaged reference image) as used for two-photon imaging analyses above. For cADDis cAMP sensor imaging, as cells were very bright at baseline (as evoked cAMP leads to a decrease in fluorescence of this sensor) and showed slower dynamic changes in fluorescence upon dopamine application, regions-of-interests were automatically segmented by using morphological filters to identify bright spherical regions (Liang et al., 2018). Briefly, a mean projection through the movie was first applied to obtain a single mean image. Local normalization was then applied by subtracting the local mean (Gaussian kernel with sigma = 8 pixels) and dividing by local variance across pixels (Gaussian kernel with sigma = 150 pixels). Basic MATLAB functions were then used to remove small unconnected structures and fill in gaps in larger structures. The image was then binarized and regions of interest were segmented by applying a Euclidian distance transform followed by a watershed transform (a common strategy used to segment spherical objects). We estimated neuropil signals by taking circular annuli around the region of interest, as described above for two-photon image analysis.
Two-photon brain slice and in vivo imaging analysis
Image registration for brain slice and in vivo two-photon calcium imaging of BA cell bodies was performed as previously described (Lutas et al., 2019). Briefly, to correct for x-y motion, each frame from an imaging session was registered to a reference image (average of 1000 frames within a session) using efficient subpixel registration methods (Bonin et al., 2011). For extraction of signal from cell body regions of interest (ROIs) from volumetric brain slice imaging (15 depths; ~ 10 μm apart), we used CellPose (Stringer et al., 2020), which optimally identified ROIs from bright cADDis expressing cell bodies. For in vivo calcium imaging analysis, we used PCA/ICA to extract masks of pixels with correlated activity, corresponding to individual axons or cell bodies (Mukamel et al., 2009). Timecourses were extracted by averaging each of the pixels within each binarized mask. We calculated neuropil activity as the median value of an annulus surrounding each ROI (inner radius: 15 pixels; outer radius: 50 pixels; pixels belonging to any other ROI were excluded from these annulus masks). This timecourse of neuropil activity was then subtracted from the activity timecourse of the associated ROI to create a fluorescence timecourse, F(t), where t is time of each imaging frame. The change in fluorescence was calculated by subtracting a running estimate of baseline fluorescence (F0(t)) from F(t), then dividing by F0(t): ΔF/F(t) = (F(t) − F0(t))/F0(t), where F0(t) is a running estimate of baseline fluorescence calculated as the 10th percentile of F(t) in the previous 32-second sliding window (Petreanu et al., 2012).
Criteria for determining responsivity to cues
To determine if a cell was responsive to each cue in our in vivo calcium imaging experiments, we used previously established, conservative criteria (Lutas et al., 2019), which are described here. We performed a Wilcoxon sign-rank test for each frame post-stimulus onset against the 1-s baseline period prior to stimulus onset, with Bonferroni correction for multiple comparisons across frames (p < 0.01). If three consecutive frames were significantly different than the baseline period, a cell was considered responsive to that cue. For all cells with significant responses to at least one cue, a preferred cue was determined as the cue evoking the largest response during the cue period. For estimation of a cell’s mean cue-evoked response magnitude, and for estimation of a cell’s response bias to a given cue, we averaged all trials containing presentations of that cue during the run to obtain a mean timecourse for that cell, and then the maximum response during the 2-s duration of the cue presentation was used as that cell’s response magnitude.
Modeling dopamine-evoked cAMP dynamics
The long-lasting cADDis response we recorded likely reflects true long-lasting cAMP elevations as the dissociation constant of the sensor is expected to be less than a second. While the off kinetics were not measured in the original publication of cADDis (Tewson et al., 2016), the biosensor is constructed from the protein Exchange Factor directly Activated by cAMP (EPAC), which exhibits fast off kinetics (Kraemer et al., 2001). The dissociation constant (koff) of EPAC was measured to be ~ 20 s−1 (Kraemer et al., 2001). In a single-ligand system, the half-life of dissociation is calculable as the natural log of 2 divided by koff [ln(2) ÷ koff]. Based on this calculation, the half-life of EPAC is around 35 ms.
To model the dynamics of dopamine and cAMP, we first fit monoexponential functions to averaged traces of either dLight1.1 or cADDis recordings. We used the decay rate from the dLight transient to first model dopamine dynamics in response to 2 s square inputs (30 s intervals between each input), which exponentially depressed such that the second input was 25% weaker than the first pulse (as observed in our measurements of dopamine synaptic depression). We convolved this input kernel with an exponential (4 s time constant; step size of 0.1 s). We then used this convolved waveform, which models dopamine release with synaptic depression across trials as an input kernel, to estimate cAMP dynamics (20 s time constant, step size of 0.1 s). For qualitative comparison with in vivo recordings, we normalized the modeled cAMP dynamics to the peak of the first evoked response.
Cue-evoked licking during Pavlovian conditioning
The licking behavior was sampled at 1 kHz (PCIe-6321; National Instruments) and, for analysis, was binned every 500 ms and converted to frequency (licks / s). For each trial, licking frequency was normalized to the 2 second period before the cue presentation. All mice developed cue-evoked licking that was significantly greater than baseline licking. As a measure of the rate of learning as determined by cue-evoked licking, we first normalized each mouse’s cue-evoked licking to the maximum lick rate across all four days. We then quantified the latency to reach 50 % of the maximum cue-evoked lick rate.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Virus strains | ||
| AAV2/1.hSyn.FLEX.GCaMP6s.WPRE | Addgene | CAT# 100845 |
| AAV2/1.hSyn.DIO.GreenDownward-cADDis.WPRE | Boston Children’s Hospital Viral Core | Zhang et al., 2021b |
| AAV2/1.hSyn-dLight1.1.WPRE | Boston Children’s Hospital Viral Core | Lutas et al., 2019 |
| AAV2/1.EF1a.DIO.mKate2-biPAC.WPRE | Boston Children’s Hospital Viral Core | Zhang et al., 2021b |
| AAV2/1.EF1a.DIO.mKate2-PDE4D3-cat.WPRE | Boston Children’s Hospital Viral Core | Zhang et al., 2021b |
| AAV2/9.hSyn.FLEX.ExRai-AKAR2-CW3SL | Richard Huganir | Zhang et al., 2021a |
| AAV2/1.hSyn.FLEX.Chrimson-tdTomato | UNC Vector Core | |
| Chemicals, peptides, and recombinant proteins | ||
| SCH23390 hydrochloride | Tocris | Cat# 0925 |
| SKF81297 hydrobromide | Tocris | Cat# 1447 |
| Dopamine hydrochloride | Tocris | Cat# 3548 |
| Forskolin | Tocris | Cat# 1099 |
| IBMX | Tocris | Cat# 2845 |
| Experimental models: Organisms/strains | ||
| Mouse: EMX1-cre; C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Mouse: DAT-cre; C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:020080 |
| Software and algorithms | ||
| MATLAB R2019 | Mathworks | https://www.mathworks.com/products/matlab.html; RRID: SCR_001622 |
| ImageJ 1.49t | NIH | https://imagej.nih.gov/ij/; RRID: SCR_003070 |
| Scanbox | Neurolabware | https://scanbox.org/ |
| Other | ||
| 10x, 0.5 NA objective | ThorLabs | CAT# TL10X-2P |
| 4x, 0.28 NA objective | Olympus | XLFUOR |
| 16x, 0.8 NA objective | Nikon | CFI75 |
| Two-photon microscope | Neurolabware | http://neurolabware.com/ |
| InSight X3 laser | Spectra-Physics | InSight X3 |
Highlights.
Basal amygdala cAMP signaling facilitates learning of motivational salience of cues
Axonal dopamine release increases cAMP in most basal amygdala glutamatergic neurons
Presynaptic depression of dopamine release limits postsynaptic cAMP accumulation
cAMP responses depress following clustered appetitive or aversive stimuli
Acknowledgements
We thank the Andermann lab including K. Evans, R. Essner, N. Nguyen and Drs. K. McGuire, H. Kucukdereli, O. Amsalem, J.S. Alvarado for feedback. V. Flores-Maldonado and P. Prasad aided with mouse care. Dr. J. Madara aided with brain slice preparation. Boston Children’s Hospital Viral Core provided viral packaging. Drs. I. Hong and R. Huganir shared ExRai-AKAR2 virus. This work was supported by NIH F32 DK112589, a Davis Family Foundation Award, and a BNORC pilot grant (P30DK046200; A.L.), a Lefler Fellowship (S.X.Z.), and NIH R01 DK109930, DP1 AT010971, DP1 AT010971-02S1, R01 MH12343, McKnight Foundation, Klarman Family Foundation, and Harvard Brain Science Initiative Bipolar Disorder Seed Grant, supported by Kent and Liz Dauten (M.L.A).
Footnotes
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adrover MF, Shin JH, and Alvarez VA (2014). Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J. Neurosci. 34, 3183–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaad WF, and Eskandar EN (2008). A flexible software tool for temporally-precise behavioral control in Matlab. J. Neurosci. Methods 174, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. [DOI] [PubMed] [Google Scholar]
- Beyeler A, Namburi P, Glober GF, Simonnet C, Calhoon GG, Conyers GF, Luck R, Wildes CP, and Tye KM (2016). Divergent routing of positive and negative information from the amygdala during memory retrieval. Neuron 90, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissière S, Humeau Y, and Lüthi A (2003). Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 6, 587–592. [DOI] [PubMed] [Google Scholar]
- Bonin V, Histed MH, Yurgenson S, and Reid RC (2011). Local diversity and fine-scale organization of receptive fields in mouse visual cortex. J. Neurosci. 31, 18506–18521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LRV, Brito M, Guiot E, Polito M, Korn CW, Hervé D, Girault JA, Paupardin-Tritsch D, and Vincent P (2013). Striatal neurones have a specific ability to respond to phasic dopamine release. J. Physiol. 591, 3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H-Y, Ito W, Li J, and Morozov A (2012). Target-specific suppression of GABA release from parvalbumin interneurons in the basolateral amygdala by dopamine. J. Neurosci. 32, 14815–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SS, and Goosens KA (2016). Input-specific contributions to valence processing in the amygdala. Learn. Mem. 23, 534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler A, Graham TGW, Cohn R, Morantte I, Siliciano AF, Zeng J, Li Y, and Ruta V (2019). Distinct Dopamine Receptor Pathways Underlie the Temporal Sensitivity of Associative Learning. Cell 178, 60–75.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob PF, and Waddell S (2020). Spaced Training Forms Complementary Long-Term Memories of Opposite Valence in Drosophila. Neuron 106, 977–991.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Diaz-Mataix L, Hamanaka H, Ozawa T, Ycu E, Koivumaa J, Kumar A, Hou M, Deisseroth K, Boyden ES, et al. (2014). Hebbian and neuromodulatory mechanisms interact to trigger associative memory formation. Proc. Natl. Acad. Sci. U. S. A. 111, E5584–E5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Neve RL, Nestler EJ, and Davis M (2001). Long-Term Memory Is Facilitated by cAMP Response Element-Binding Protein Overexpression in the Amygdala. J. Neurosci. 21, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Pignatelli M, Xu S, Itohara S, and Tonegawa S (2016). Antagonistic negative and positive neurons of the basolateral amygdala. Nat. Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochli DE, Thompson EC, Fricke EA, Postle AF, and Quinn JJ (2015). The amygdala is critical for trace, delay, and contextual fear conditioning. Learn. Mem. 22, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer A, Rehmann HR, Cool RH, Theiss C, De Rooij J, Bos JL, and Wittinghofer A (2001). Dynamic interaction of cAMP with the rap guanine-nucleotide exchange factor epac1. J. Mol. Biol. 306, 1167–1177. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Chen Y, Lodder B, and Sabatini BL (2019). Monitoring behaviorally induced biochemical changes using fluorescence lifetime photometry. Front. Genet. 10, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lodder B, Chen Y, Patriarchi T, Tian L, and Sabatini BL (2021). Cell-type-specific asynchronous modulation of PKA by dopamine in learning. Nature 590, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Fratzl A, Goldey G, Ramesh RN, Sugden AU, Morgan JL, Chen C, and Andermann ML (2018). A Fine-Scale Functional Logic to Convergence from Retina to Thalamus. Cell 173, 1343–1355.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, and Kaeser PS (2019). Mechanisms and regulation of dopamine release. Curr. Opin. Neurobiol. 57, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutas A, Kucukdereli H, Alturkistani O, Carty C, Sugden AU, Fernando K, Diaz V, Flores-Maldonado V, and Andermann ML (2019). State-specific gating of salient cues by midbrain dopaminergic input to basal amygdala. Nat. Neurosci. 22, 1820–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massengill CI, Bayless-Edwards L, Ceballos CC, Cebul ER, Qin M, Whorton MR, Ye B, Mao T, and Zhong H (2021). Highly sensitive genetically-encoded sensors for population and subcellular imaging of cAMP in vivo. BioRxiv 2021.08.27.457999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AC, Ikeda M, and Justice JB (1987). Mechanisms contributing to the recovery of striatal releasable dopamine following MFB stimulation. Brain Res. 421, 325–335. [DOI] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, and McDonald AJ (2009). Dopaminergic innervation of pyramidal cells in the rat basolateral amygdala. Brain Struct. Funct. 213, 275–288. [DOI] [PubMed] [Google Scholar]
- Namburi P, Beyeler A, Yorozu S, Calhoon GG, Halbert SA, Wichmann R, Holden SS, Mertens KL, Anahtar M, Felix-Ortiz AC, et al. (2015). A circuit mechanism for differentiating positive and negative associations. Nature 520, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’leary TP, Sullivan KE, Wang L, Clements J, Lemire AL, and Cembrowski MS (2020). Extensive and spatially variable withincell-type heterogeneity across the basolateral amygdala. Elife 9, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P-K, Gore F, and Salzman CD (2018). Basolateral amygdala circuitry in positive and negative valence. Curr. Opin. Neurobiol. 49, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong W-H, Folk RW, Broussard GJ, Liang R, Jang MJ, et al. (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science (80-. ). 360, eaat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Gutnisky DA, Huber D, Xu N, O’Connor DH, Tian L, Looger L, and Svoboda K (2012). Activity in motor-sensory projections reveals distributed coding in somatosensation. Nature 489, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard CR, Muller JF, Mascagni F, and McDonald AJ (2008). Dopaminergic innervation of interneurons in the rat basolateral amygdala. Neuroscience 157, 850–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, and Gallagher M (1999). Neural encoding in orbitofrontal cortex and basolateral amygdala during olfactory discrimination learning. J. Neurosci. 19, 1876–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, and Janak PH (2013). A causal link between prediction errors, dopamine neurons and learning. Nat. Neurosci. 16, 966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, and Pachitariu M (2020). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 2020 181 18, 100–106. [DOI] [PubMed] [Google Scholar]
- Tang W, Kochubey O, Kintscher M, and Schneggenburger R (2020). A VTA to basal amygdala dopamine projection contributes to signal salient somatosensory events during fear learning. J. Neurosci. 40, 3969–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewson PH, Martinka S, Shaner NC, Hughes TE, and Quinn AM (2016). New DAG and cAMP Sensors Optimized for Live-Cell Assays in Automated Laboratories. J. Biomol. Screen. 21, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, and Taylor JR (2006). Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat. Neurosci. 9, 167–169. [DOI] [PubMed] [Google Scholar]
- Tye KM, Tye LD, Cone JJ, Hekkelman EF, Janak PH, and Bonci A (2010). Methylphenidate facilitates learning-induced amygdala plasticity. Nat. Neurosci. 13, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gillis-Smith S, Peng Y, Zhang J, Chen X, Salzman CD, Ryba NJP, and Zuker CS (2018). The coding of valence and identity in the mammalian taste system. Nature 558, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita S, Hayashi-Takagi A, Ellis-Davies GCR, Urakubo H, Ishii S, and Kasai H (2014). A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science (80-. ). 345, 1616–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Li B (2018). Population coding of valence in the basolateral amygdala. Nat. Commun. 9, 5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JF, Liu B, Hong I, Mo A, Roth RH, Tenner B, Lin W, Zhang JZ, Molina RS, Drobizhev M, et al. (2021a). An ultrasensitive biosensor for high-resolution kinase activity imaging in awake mice. Nat. Chem. Biol. 17, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PEM, and Dani JA (2009). Controls of Tonic and Phasic Dopamine Transmission in the Dorsal and Ventral Striatum. Mol. Pharmacol. 76, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Lutas A, Yang S, Diaz A, Fluhr H, Nagel G, Gao S, and Andermann ML (2021b). Hypothalamic dopamine neurons motivate mating through persistent cAMP signalling. Nat 2021 5977875 597, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Guan W, Yang T, Furlan A, Xiao X, Yu K, An X, Galbavy W, Ramakrishnan C, Deisseroth K, et al. (2021c). Genetically identified amygdala–striatal circuits for valence-specific behaviors. Nat. Neurosci. 2021 2411 24, 1586–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.