
Fig. 1: The major UPR pathways initiated from the ER.
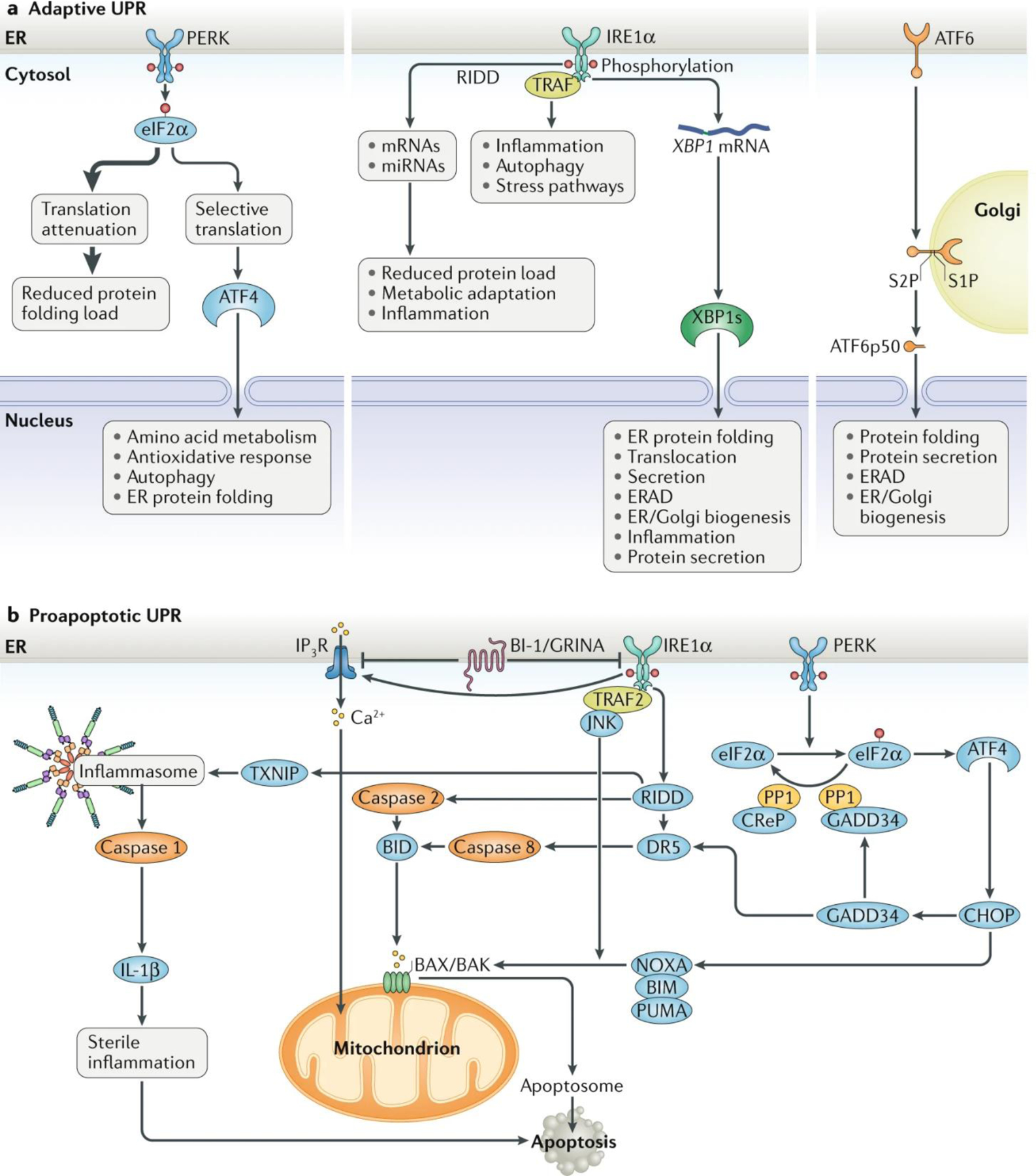
a | Adaptive unfolded protein response (UPR). Under endoplasmic reticulum (ER) stress, three major UPR branches are activated: (1) PERK phosphorylates eukaryotic translation initiation factor 2 subunit-α (eIF2α), reducing the overall frequency of mRNA translation initiation. However, selective mRNAs, such as ATF4 mRNA, are preferentially translated in the presence of phosphorylated eIF2α. ATF4 activates the transcription of UPR target genes encoding factors involved in amino acid biosynthesis, the antioxidative response, autophagy and apoptosis. (2) IRE1α RNase splices XBP1 mRNA, which encodes a potent transcription factor that activates expression of UPR target genes involved in ER proteostasis and cell pathophysiology. IRE1α RNase can also cleave ER-associated mRNAs or non-coding functional RNAs, leading to their degradation through regulated IRE1-dependent decay (RIDD), which modulates the protein folding load, cell metabolism, inflammation and inflammasome signalling pathways. The IRE1α cytosolic domain may also serve as a scaffold to recruit adaptor proteins, for example tumour necrosis factor receptor-associated factor (TRAF) family members, thereby activating inflammatory responses under non-canonical ER stress conditions. (3) ATF6 transits from the ER to the Golgi apparatus, where it is cleaved by site-1 protease (S1P) and site-2 protease (S2P), yielding an active cytosolic ATF6 fragment (ATF6p50). This fragment migrates to the nucleus, activating transcription of the UPR target genes involved in ER protein folding homeostasis and cell physiology. Additionally, unfolded or misfolded proteins accumulated in the ER lumen may be degraded through the proteasome-based ER-associated protein degradation (ERAD) machinery that is regulated by the ATF6-mediated and/or IRE1α–X-box-binding protein 1 (XBP1)-mediated UPR branches. b | Proapoptotic UPR. Under ER stress, the PERK–eIF2α UPR branch induces translation of ATF4, which can activate expression of the proapoptotic factor CCAAT/enhancer-binding protein homologous protein (CHOP) and GADD34. GADD34 targets protein phosphatase 1 (PP1) to dephosphorylate eIF2α and thereby restore mRNA translation. Constitutive repressor of eIF2α phosphorylation (CReP) also serves as a cofactor to provide PP1 specificity for phosphorylated eIF2α under ER stress. CHOP promotes ER stress-induced apoptosis by modulating GADD34, death receptor 5 (DR5) and the members of the BCL-2 or BH3-only family, including NOXA, BIM and PUMA, to stimulate protein synthesis and exacerbating protein folding defect. Furthermore, the IRE1α UPR branch is involved in caspase 2-dependent, caspase 8-dependent or BAX/BAK-dependent apoptosis through RIDD or activation of TRAF2–JUN N-terminal kinase (JNK) signalling. The IRE1α-mediated RIDD also regulates thioredoxin-interacting protein (TXNIP) to activate inflammasome-dependent and caspase 1–IL-1β-dependent sterile inflammation, leading to apoptosis. In addition, Ca2+ release from the ER via inositol 1,4,5-trisphosphate receptor (IP3R), which interacts with the ER-located antiapoptotic proteins BAX inhibitor 1 (BI-1) and GRINA, contributes to mitochondrial reactive oxygen species release and the activation of the BAX/BAK-dependent apoptosome. miRNA, microRNA.