

Abstract
Compared with traditional de novo drug discovery, drug repurposing has become an attractive drug discovery strategy due to its low-cost and high efficiency. Through a comprehensive analysis of the candidates that have been identified with drug repositioning potentials, it is found that although some drugs do not show obvious advantages in the original indications, they may exert more obvious effects in other diseases. In addition, some drugs have a synergistic effect to exert better clinical efficacy if used in combination. Particularly, it has been confirmed that drug repositioning has benefits and values on the current public health emergency such as the COVID-19 pandemic, which proved the great potential of drug repositioning. In this review, we systematically reviewed a series of representative drugs that have been repositioned for different diseases and illustrated successful cases in each disease. Especially, the mechanism of action for the representative drugs in new indications were explicitly explored for each disease, we hope this review can provide important insights for follow-up research.
Keywords: Drug repositioning, Drug discovery, Neurological diseases, Cancer, Antimicrobial, Antiviral
Graphical abstract

1. Introduction
Drug discovery and development is an expensive, time-consuming, and risky business. The average cost of bringing a new drug to the market is estimated at $ 1.24 billion [1]. As shown in Fig. 1 , traditional drug development strategies are roughly divided into four stages: drug discovery, preclinical studies, clinical trials, and post-marketing safety monitoring. Due to regulatory requirements regarding safety, efficacy, and quality in animal research and clinical trials, the time required to develop a new drug is approximately 12–17 years [2]. However, drug repositioning process significantly saves the development time. It only costs researchers approximately 3–12 years to develop an old drug for new treatment (Fig. 1). Furthermore, the cost of drug repositioning is much lower than traditional drug development. This strategy provides many research institutions with opportunities to develop drugs with less investment. Therefore, in order to speed up the drug development process, lower the risk of failure and reduce costs, pharmaceutical companies have adopted drug repositioning as an effective alternative.
Fig. 1.

Comparison of traditional drug development and drug repositioning process.
In recent years, it can be seen from Fig. 2 that the investment in drug research and development is increasing, however, the number of approved drugs is still limited. Thence, people are increasingly interested in drug repositioning due to a variety of factors. The purpose of drug repositioning is to determine new indications for old drugs, which can be divided into the following categories: (i) Drugs that have been successfully marketed. (ii) Drugs with no specific indications and safety problems in clinical trials. (iii) Drugs that are terminated due to budget and other reasons. Compared with traditional drug discovery, drug repositioning may allow compounds to skip to Phase II, thereby reducing time to market and lowering investment costs. In addition, since clinical data are available at the beginning of a development project, the risks associated with further development are greatly reduced [3]. Another great advantage of drug repositioning is that many cases have been proven to be sufficiently safe in preclinical trials. Besides, for an approved drug, not only preclinical information but also clinical profiles are also available, which accelerates the development of drug discovery [4]. A crucial purpose of drug repositioning is to find new drug-disease relationships. Multiple methods have been developed to achieve this goal, including computational methods, biological experimental methods, and the combination of both [5]. With the open-source of many drug-related databases, this advancement has accelerated the development of a computational approach. At the same time, the mixed approach that combining biology and computational method greatly improves the efficiency of drug repositioning while reducing costs. Drug repurposing strategy should avoid hasty proof-of-concept. Instead, a detailed and comprehensive plan should be proposed, and the information obtained in the previous development cycle should be used to appropriately simplify the work. When conducting preclinical research, drug pharmacology and dose intensity need to be considered. Early development must not bypass scientific inspections [6]. Besides, drug repositioning faces some major challenges. For example, if the initial clinical trials do not meet current regulatory requirements, new Phase I trials may be needed to complete the data. Most successful cases are derived from an understanding of pharmaceutical pharmacology or clinical efficacy following a retrospective analysis of the original indications of the drug. These repositioners have weaker intellectual property protections that will reduce companies’ return on investments. A majority of drug repositioning applications are based on positive clinical or epidemiological observations or discovering efficacy in vivo. With the rapid development of computing methods and data explosion, the mixed advantages of drug repositioning will become more prominent.
Fig. 2.

The number of approved drugs and investment by the FDA from 1998 to 2018.
In this perspective, we systematically summarized several applications and notable strategies of drug repositioning in different types of diseases, including neurological diseases, cancer, microbial, and virus infections. As for neurological disease, we reviewed 9 potential candidates that can be repurposed in the treatment of Alzheimer's disease. These drugs, including Carmustine, Bexarotene, Tamibarotene, Imatinib, Valsartan, Nitrendipine, Liraglutide, Acitretin, Minocycline, had been approved with specific indications. Although they are old drugs, one important consideration is that the safety depends on the indication. Any change of dosage or route of administration will need a re-examination of the drugs. For oncological diseases, all reviewed drugs except ciglitazone and clofibric acid are approved drugs. These two are experimental compound that require further study than old drugs. Due to lack of safety and toxicity data makes the late stages of development for a repositioned drug considerably important. For antimicrobial repurposing sections, all reviewed drugs are approved drugs. It should be noted that, flupenthixol and thioridazine are withdrawn drugs. Flupenthixol caused a relapse in all patients and observed a significant weight decrease in clinical [7]. Thioridazine was withdrawn worldwide in 2005 due to its association with cardiac arrythmias. Even if the adverse effects of such drugs lead to the eventual withdrawal from the market, this does not prevent their research in drug repurposing. In the antivirus repurposing review, emricasan, Suramin, emetine, CVL218, PJ-34, and Beclabuvir are categories in the investigational group. These compounds required complete evaluations of mechanism, dosage, formulation, and safety. We also focused on the current major challenges of drug repositioning and provide some suggestions for accelerating the application of drug repositioning.
2. Neurological diseases
In recent years, identifying relevant drugs for neurological disorders is still an important task in drug development. Compared with de novo drug discovery, drug repositioning is more cost-effective and time efficient. This approach has achieved significant success in neurological diseases. Alzheimer's disease (AD) is the most common disease that belongs to neurodegenerative disease. It is a collective term for loss of memory and other cognitive abilities that seriously affect daily life. It is predicted that the number of patients suffering with AD will reach 74.7 million by 2030 and 131.5 million by 2050 [8]. Reducing the risk of dementia is a priority task in the work of the World Health Organization (WHO).
AD was first described by the German psychiatrist Aloysius Alzheimer in 1907 [9]. When conducting a pathological study of the brain tissue of patients with dementia, it was found that there are two types of lesions in the brain of AD patients, senile plaque (SP) and neurofibrillary tangles (NFTs) [10]. SP is composed of β-amyloid, and NFTs are caused by abnormal phosphorylation of tau protein [11]. Amyloid precursor protein (APP) exists on the surface of neurons. Under normal circumstances, APP is broken down by enzymes on the surface of neurons and releases β-amyloid protein. Many β-amyloid proteins are assembled into insoluble fibers to eventually produce SP [12]. When neurons communicate, signals are transmitted from the soma to the synapse. These signals are transmitted through microtubules, which are stabilized by normal tau proteins. The tau protein appears abnormal during AD, destroying the normal structure of the microtubules, so the neuron's skeleton can no longer be maintained and falls apart [13]. The abnormal tau protein aggregates in neurons to form filaments. Because there is no backbone support, neurons degenerate, connections between neurons are lost, tau protein is abnormally accumulated, and eventually neurons die. As shown in Fig. 3 , the coexistence of these two lesions is necessary for the occurrence of AD, but which lesion occurs first is still being studied. In fact, reducing SP is not enough to eradicate AD, and the relationship between β-amyloid and tau protein is not clear [13]. Therefore, the exact molecular mechanism leading to dementia remain the focus, and the relative importance of different pathological entities as drug substrates remains controversial [14]. Even though there are currently four approved drugs for treating AD, we still need to establish an understanding of the pathogenesis of the disease to develop more candidates.
Fig. 3.

Potential pathological process of Alzheimer's disease [15].
Studies have shown that there is a significant relationship between hypertension and Alzheimer's disease [16,17], but the relationship between the two is much complicated. At present, because the pathological mechanism of AD is still unclear and only stays in the theoretical stage of mechanism research [18], there is no specific method for the treatment of AD. Thus, the studies suggest that only some cognitive improvement drugs can treat AD. Anti-cancer drugs showed a great therapeutic effect on AD. Clinical statistics show that breast cancer patients undergoing chemotherapy have a reduced risk of developing AD in old age [19]. The reason for this phenomenon perhaps is shared signaling pathways, thus providing credibility for the role of anticancer drugs in AD [20].
2.1. Drugs repositioned on Alzheimer's disease
Carmustine (Table 1) was approved by the FDA for the treatment of brain cancer and was found to be effective for AD. As an alkylating agent, carmustine induces cross-links bases pair in DNA [21]. It inhibits cancer cell replication by hindering the normal replication process of DNA [22]. Drug repositioning study reveals that carmustine mainly regulate APP to reduce amyloid-β(Aβ) aggregation [23]. Compared with the control group in the neural experiment, carmustine significantly reduced the normalized level of Aβ in the cells [23]. The experimental conclusion summarizes that long-term use of carmustine at a non-toxic dose may reduce the production of Aβ to achieve the purpose of treatment for AD. In addition, carmustine interacts with secretase independently, which can avoid side effects caused by off-target. This increases the safety of the drug in the treatment of AD. Carmustine acts on different targets demonstrating that the drug repurposing strategy is on the basis of polypharmacology. Bexarotene (BEXA) (Table 1) is a Retinoid X Receptor (RXR) agonist that increases apolipoprotein E (ApoE) expression and microglial phagocytosis. It was approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL). The mechanisms of anticancer are inhibition of cell cycle progression, inhibition of angiogenesis and metastasis [24]. In 2016, Krishna and colleagues found that BEXA can restore cognitive function by reducing cholesterol and Aβ [25], this result indicates that BEXA become an important drug for the treatment of AD. The primary mechanism of action is that RXR participates in the formation of ApoE-related lipoprotein particles and promotes the degradation of Aβ. Agonists of these receptors also promote phagocytosis by microglia and macrophages. The research indicates that BEXA acts on specific targets but repurposes different diseases settings. Tamibarotene ( Table 1 ) is a retinoic acid receptor (RAR) agonist, and it was approved in Japan for the treatment of acute promyelocytic leukemia (APL) [26]. Since Tamibarotene is a synthetic retinoid, it can regulate the immune system and reduce inflammatory cytokines and chemokines, which control the excess stimulation of astrocytes and microglia around Ab plaques. Tamibarotene is reported to have fewer side effects than other retinoids, so it is considered a promising drug candidate for the treatment of AD. Reported results strongly implied that tamibarotene could result in impaired β-secretase cleaving and lowering Aβ production [27]. Tamibarotene controls multiple target genes involved in the pathology of AD. Mechanisms of action for the drug revealed that the repurposing strategy is related to polypharmacology. Acitretin decreases the expression of STAT1 and STAT3-dependent signaling which interferes with the JAK pathway [28]. In a transgenic mouse model of AD, retinoids led to increased ADAM10 expression. Therefore, the systematic retinoids especially acitretin present a valuable therapeutic strategy for the treatment of AD. It should be noted that, compared with the placebo group, acitretin is generally safe and well-tolerated in clinical trials [29]. This result preliminarily proves that the acitretin seems to be an ideal candidate drug for the treatment of AD. Since acitretin acts on multiple targets pertaining to multiple disease pathways, the repurposing strategy is on the basis of polypharmacology. Imatinib ( Table 1 ) is a selective tyrosine kinase inhibitor. It is also the first-line drug for the treatment of chronic myeloid leukemia [30] and gastrointestinal stromal tumors [31]. Studies have shown that Imatinib can be used to treat Alzheimer's disease [32] due to interferes c-Abl/p73 signaling pathway and decreasing the tau protein phosphorylation in hippocampal neurons. The repurposing strategy is also related to the polypharmacology of the drug. However, Imatinib has been proven to effectively reduce the production of Aβ in vitro and in animal models, it is not obviously in humans, so further research is needed.
Table 1.
Drug repurposing examples and their mechanism of action in Neurological Diseases.
| Target | Structure | Drug | Drug Class | Proposed Mechanism | Original Indications |
|---|---|---|---|---|---|
| DNA/RNA | 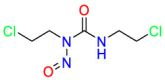 |
Carmustine | Anti-cancer | Cultured cells overexpressing APP | brain tumors, multiple myeloma, Hodgkin's disease and non-Hodgkin's lymphomas. |
| Retinoic acid receptor RXR-alpha/beta/gamma | 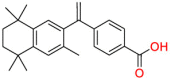 |
Bexarotene | Anti-cancer | Stimulate the expression of apolipoprotein E | cutaneous T-cell lymphoma (CTCL) |
| Retinoic acid receptor alpha/beta | 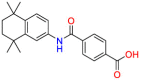 |
Tamibarotene | Synthetic retinoid | Reduce significantly the insoluble Aβ levels | Investigated for treatment in leukemia |
| Tyrosine-protein kinase ABL1 | Imatinib | Anti-cancer | Inhibit GIST cell proliferation | malignant gastrointestinal stromal tumors | |
| Angiotensin receptor blockers (ARBs) | 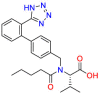 |
Valsartan | Anti-hypertensive | Block AT1 or processing of angiotensin II | hypertension to reduce the risk of fatal and nonfatal cardiovascular events |
| Calcium channel blockers | 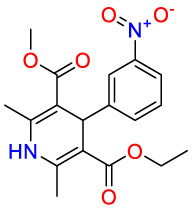 |
Nitrendipine | Anti-hypertensive | Calcium antagonists | mild to moderate hypertension |
| GLP1 analogs |  |
Liraglutide | Type II diabetes | GLP-1 analogue; Activating GLP-1 receptor insulin secretion | diabetes and intended for weight loss |
| Retinoid therapy |  |
Acitretin | Severe psoriasis and dyskeratosis | Induce apoptosis of primary human keratinocytes | severe psoriasis in adults |
| Tetracycline antibiotics |  |
Minocycline | Broad-spectrum antibiotics | Bind to a position of ribosome 30S subunit and inhibiting protein synthesis | inflammatory lesions of acne vulgaris |
Antihypertensive drugs can significantly reduce the accumulation of amyloid-β. Attempting to choose some antihypertensive drugs as neuroprotective agents can achieve the purpose of treating AD because the neuroprotective and cognitive functions of AD may be mediated through related mechanisms of angiotensin receptor blockers (ARB) and calcium channel blockers (CCB) associated with hypertension. Valsartan ( Table 1 ), an angiotensin receptor blocker, blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II in hypertension treatment. A drug-related study found that Valsartan significantly reduces the accumulation of AD β-amyloid in a mouse model. This drug received attention in AD treatment because Valsartan alleviates the deterioration of Aβ-mediated cognitive ability even if it is administered less than two times a dose for the treatment of hypertension [33]. In 2014, Yang et al. found that Valsartan could ameliorate cognitive decline by regulating the expression and activity of AChE, and reduce oxidative damage by decreasing the level of MDA in cortex and hippocampus [34]. This finding further supports the potential strategy of Valsartan in the treatment of AD based on polypharmacology. Hypertension is one of the risk factors for dementia, while antihypertensive drugs with different mechanisms have different effects on dementia [35,36]. Previous studies have found that calcium channel blockers seem to be most effective drugs. Nitrendipine ( Table 1 ), a second-generation calcium channel blocker, is used for the treatment of hypertension by inhibiting the influx of extracellular calcium across the myocardial and vascular smooth muscle cell membranes. The possible explanation for nitrendipine in AD treatment is to block the uncontrolled calcium influx into the neurons. This repurposing case indicates that nitrendipine exerts a specific mechanism of action in different diseases settings. It has also been shown to be particularly beneficial and safe in hypertensive patients with a high risk of dementia [37]. The relationship between hypertension and dementia is not yet known, so further research is needed.
Liraglutide (Table 1) is a glucagon-like peptide-1 (GLP-1) receptor agonist in type Ⅱ diabetes treatment. Given that the connection between AD and central insulin signaling is gradually recognized, Liraglutide has also been proven to have a neuroprotective mechanism [38]. Liraglutide interferes Phosphoinositide 3-kinase/mitogen associated protein kinase (PI3K/MAPK) dependent pathways by binding to glucagon like peptide-1 receptor (GLP-1R) to increase the presence of Aβ transporters in cerebrospinal fluid [39]. Because Liraglutide is currently approved for the treatment of type 2 diabetes, so it acts like a shortcut in the treatment of AD. Minocycline ( Table 1 ), an anti-inflammatory tetracycline compound, binds to the bacterial 30S ribosomal and inhibits protein synthesis. Reports have established the presence of inflammatory markers in AD patients’ brains. Minocycline could exert anti-inflammatory effects through the blood-brain barrier and has neuroprotective effects. Studies have shown that Minocycline prevents the deposition of Aβ and the death of neurons [40], and protects hippocampal neurogenesis in the presence of Aβ [41]. The repurposing strategy is on the basis of specific mechanism of action but repurpose for different diseases settings. Unfortunately, clinical trials have shown that Minocycline has different efficacy at different doses compared with placebo. The results of clinical trials show that high doses have no obvious benefit. Besides, the cognitive function of mild patients was not improved after accepting Minocycline treatment. Therefore, Minocycline needs further research in drug repositioning.
Although some of the drugs discussed here appear to be good candidates for further research, it is uncertain whether any of them will succeed as successful therapies for Alzheimer's disease and a more systematic approach is needed to screen approved drugs and analyze epidemiological biological and clinical data to identify new potential candidates for repurposing in Alzheimer's disease. It is also important to consider the optimal stage of the disease for treatment based on the mechanism of action of new indications and thus to determine whether the trial should target early-clinical Alzheimer's patients [42].
2.2. Representative cases of drug repositioning in other neurological diseases
One of the most representative cases of drug repositioning in neurological disease comes from galantamine. Galantamine is a phenanthridine alkaloid isolated from galanthus caucasicus in 1947 [43]. After a few years, its structural characteristics were obtained and used for surgery and anesthesia. Galantamine was mainly used as a cholinesterase inhibitor in the 1960s to treat diseases such as polio and muscle atrophy. Because its activity is better than pyridostigmine but weaker than neostigmine, it has not attracted widespread attention [44]. Another reason that hinders the development of galantamine is its extremely low synthesis efficiency as galantamine is more difficult to synthesize than other analogs. Research in the 1990s [45] found that galantamine can improve memory dysfunction in mice, and it is speculated that it may be effective in treating AD. Later, it was confirmed that galantamine can specifically bind to the active site of acetylcholinesterase, thereby improving the function transmission of cholinergic nerves, improving the cholinergic energy in the brain of AD patients, and improving cognitive ability [46]. In 2001, the FDA approved the registration of galantamine for the treatment of mild to moderate AD [47]. Galantamine does not improve the daily activities of untreated patients with severe AD. The dual-mode action of galantamine (AChE inhibition and allosteric nicotinic receptor modulation) develops new therapeutic strategies. The dual mechanism is shown in Fig. 4 a. This case is also considered a polypharmacological strategy due to its drug repurposing target inconsistent with the original target.
Fig. 4.

a. Galantamine targets two sites of action: Nicotinic receptors and Acetylcholinesterase. b. Amantadine stimulates dopamine release and prevents dopamine reuptake. COMT = Catechol-O-transferase; DA = dopamine; MAO = monoamine oxidase (Source-Medscape) [48,51].
Galantamine binds to presynaptic and postsynaptic nicotinic acetylcholine receptor (nAChR) on cholinergic neurons but different with acetylcholine (ACh) [48]. The response of nAChR to ACh will be enhanced, when galantamine and ACh bind to their respective binding sites at the same time. Activation of nAChR promotes the release of neurotransmitters that are important for memory [49]. Therefore, regulating nAChR has important clinical benefits in AD, including improving memory function. In addition, galantamine inhibits the utilization of ACh by inhibiting AChE. Compared with butyrylcholinesterase, galantamine is 50 times more selective for AChE. This high selectivity mat improves the tolerability. Galantamine has been proven efficient in the symptomatic treatment of Alzheimer's disease patients [50].
Amantadine is an antiviral drug synthesized in the 1950s and approved to prevent and treat the symptoms of influenza A infection. Subsequently, amantadine was used to develop treatments for neurological disorders [52]. The drug repositioning of amantadine was accidently discovered during the case observation of a patient in 1968 [53]. Experimental data linked its mechanism of action to the antagonist effect of the N-methyl-d-aspartate (NMDA) subtype of glutamate receptors [54].
Parkinson's disease (PD) is performed by the loss of dopaminergic neurons causing a decrease in dopamine levels [55]. Amantadine is a non-competitive antagonist of the NMDA receptor, which stimulates dopamine release and prevents dopamine reuptake (Fig. 4b). Subsequently, it was discovered that amantadine modulates the activity of glutamatergic corticosteroid and subthalamic-pallidal pathways, which is the core mechanism for the development of dyskinesia in PD [56]. In 1973, amantadine was the first drug approved by the FDA for the treatment of dyskinesia in PD. In 2017, the extended-release formulation was approved for use in the treatment of levodopa-induced dyskinesia [57]. The drug is currently only used to treat Parkinson's disease in the United States. This successful case provides a new treatment strategy and provides novel polypharmacology directions.
3. Oncological diseases
In the past decades, significant work has been done in search of novel methods for the treatment of cancer. However, cancer is still one of the leading causes of death worldwide. The consideration method in drug development does not only include de novo design but also search approved known molecules used in a non-oncological situation [58]. The explosive growth of a great number of cancer genome data and the gradual disclosure of internal high-throughput screening data by pharmaceutical companies have made in silico drug repositioning more convenient. More than 200 non-cancer drugs have been shown to have anticancer activity. A typical example of this is propranolol, which is often used as a blood pressure lowering drug and has since been proven to be effective in relieving tumors in combination with other cancer treatment strategies. The scientists of the Dana-Farber team used the PRISM molecular barcode method to select 4518 drugs from the Drug Repurposing Center for screening. The screening targets covered 578 cell lines of 24 tumor types. Surprisingly, a great number of non-oncology drugs can selectively inhibit cancer cell line subsets [59]. Some of these drugs even discovered cancer targets that were not previously involved [59]. Researchers found that the mechanism of action of most anti-cancer drugs is mainly through the replacement of proteins. This study found that some drugs do not only block proteins but instead activate or stabilize protein-protein interactions [59].
3.1. Colorectal cancer (CRC)
Colorectal cancer is the second leading cause of cancer death worldwide, mutations in the TP53 gene are the leading cause of colorectal cancer [60]. The p53 protein is encoded by the TP53 gene [61]. p53 can not only promote the repair and survival of damaged cells but also promote the permanent removal of irreparable damaged cells through apoptosis or autophagy [62]. Therefore, the complex regulation of p53 can prevent tumor incidence and progression [62].With the development of genomics and screening of existing drugs, it has been discovered that non-oncology drugs have shown anticancer activity [63,64]. Chlorpromazine (CPZ) ( Table 2 ) is mainly used to treat psychiatric diseases by blocking postsynaptic dopamine receptors in cortical and limbic areas of brain. Studies have found that the apoptosis of tumor cells induced by CPZ depends on the expression of p53 [61]. Therefore, CPZ offers potential for the treatment of CRC and provides effective research ideas for drug repositioning due to polypharmacology. Dehydroepiandrosterone (DHEA) ( Table 2 ) is a major C19 steroid produced by the adrenal. It has been reported that DHEA can significantly inhibit 1,2-dimethylhydrazine-induced colon tumors in BALB/c mice [65] in 1984. The mechanism of action for DHEA is to reduce the number of abnormal crypt foci (ACF) induced by azoxymethane (AOM) to achieve the purpose of treatment. The results of acting on different targets should make a wary of side effects. Orlistat ( Table 2 ) is a fatty acid synthase (FASN) inhibitor for obesity management, which works by inhibiting the absorption of dietary fat. In 2011, a study by Zhuang et al. found that orlistat can inhibit the growth of colorectal cancer by arresting the cell cycle in the G1 cycle and activating the apoptosis of caspase-3 [66]. Additionally, orlistat inhibited inflammation, hyperplasia, and tumor progression through NF-kB activation [67]. Since fatty acid synthesis plays an important role in the survival of cancer cells, fatty acid synthase may be a potential target for cancer treatment. The polypharmacological properties of orlistat provide new ideas for drug repurposing. Pantoprazole ( Table 2 ) is an irreversible proton pump inhibitor (PPI), and it was used for the treatment of gastroesophageal reflux disease. Since PPIs are commonly effective in cancer patients, studies investigating drug repositioning are particularly important for therapy. Zeng et al. found that pantoprazole can effectively inhibit TOPK, highly expressed in multi types of cancers, especially in colorectal cancer cells [68] via virtual screening. Thus the polypharmacological properties of pantoprazole provide novel directions to different diseases therapy. Dichloroacetate (DCA) ( Table 2 ) is a specific inhibitor of pyruvate dehydrogenase kinase (PDK). It increases pyruvate flux into the mitochondria by inhibiting the PDK, promoting glucose oxidation. As early as 1956, Otto Warburg observed that cancer cells inherently depend on glycolysis to produce chemical energy [69]. PDK inhibitors can attenuate the effect of glycolysis to promote the oxidative phosphorylation of mitochondria and inhibit the growth of colorectal cancer cells [70]. This case indicates that repurposing strategy is related to a similar mechanism of action but applies in different diseases settings. Non-hormonal compounds are binding to estrogen receptors that could produce beneficial effects on the skeletal system without adverse effects on other tissues. These compounds were called selective estrogen receptor modulators (SERM). Raloxifene ( Table 2 ) is a SERM that the FDA approves to treat and prevent postmenopausal osteoporosis in women. Through research on the prevention of different types of colon cancer, it was discovered that raloxifene may act as an ER-β antagonist to achieve the purpose of preventing colon cancer [71]. The death of patients due to tumor recurrence is also a major obstacle in cancer treatment. A reasonable explanation is that the cells continue to exist and enter the cell cycle after the induced stimulation during the dormant period. Comparison of different tumor cells disclosed that dormant colorectal cancer (CRC) cells are chemo-resistant [72]. Therefore, it is hoped that drugs can interfere with the dormant state to kill cancer cells to prevent cancer recurrence completely. The repositioning strategy is related to mechanisms of action in different diseases settings. Itraconazole participates in compound synthesis and detoxification via hydroxylation reaction and inhibits cytochrome P450 (CYP) enzymes [73]. CYP enzymes are important for the biosynthesis of sterols, which are vital components of fungal cell membranes. In 2018, Simon et al. reported that the broad-spectrum antifungal drug itraconazole ( Table 2 ) could switch dormant cells into global senescence. This evidence supports the therapeutic potential of itraconazole in CRC [74]. Pre-clinical validation studies indicate that itraconazole interference Wnt signaling pathway inhibition and tumor growth arrest [75]. The drug repurposing strategy is related to itraconazole polypharmacology. Tanshinone ⅡA (Tan IIA) ( Table 2 ), as a member of the major lipophilic active constituent isolated from the root of Chinese medicinal herb, can mediate the apoptosis of colorectal cancer cells. Tan IIA is the most abundant diterpene quinone in Salviae miltiorrhizae and used to treat cardiovascular disease. Tan IIA protects cardiomyocytes from ischemia and hypoxia injury by inhibiting the expression of miR-1 through the p38-MAPK signaling pathway [76]. Sayilaxi et al. proved that Tan IIA reduces the viability of CRCs by promoting the lysis of mitochondria [77]. Furthermore, Researches reveal that Tan IIA inhibits the growth of cancer cells by stimulating autophagy in MEK-ERK-mTOP pathway [78]. The repurposing strategy indicates that Tan IIA acts on multiple targets pertaining to multiple diseases pathways. p53-MDM2 protein-protein interaction plays a key role in almost all tumor cells [79]. The identification of inhibitors of p53-MDM2 from the approved drugs library could increase the successful rate of anticancer drug discovery. The antipsychotic drug fluspirilene ( Table 2 ) as a virtual screening drug has been shown to be an effective p53-MDM2 small molecule inhibitor [80]. Experimental results show that fluspirilene can significantly inhibit the growth of cancer cells including CRCs. Fluspirilene exerts a specific mechanism of action in different diseases settings, providing novel insight for drug repurposing.
Table 2.
Drug repurposing examples and their mechanism of action in Colorectal Cancer.
| Target | Structure | Drug | Drug Class | Proposed Mechanism | Original Indications |
|---|---|---|---|---|---|
| D2 dopamine receptor | 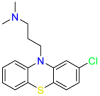 |
Chlorpromazine (CPZ) | Antipsychotic agent | Tumor cell apoptosis induced by p53 expression | For the treatment of schizophreniato control nausea and vomiting; |
| Estrogen receptor | 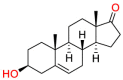 |
Dehidroepiandrosterona (DHEA) | Over-the-counter dietary supplements | DHEA has a chemo-preventive effect on colon cancer precursors | Indicated for the treatment of lupus, depression, adrenal insufficiency, cervical cancer |
| Fatty acid synthase |  |
Orlistat | Treat obesity | Inhibit pancreatic lipase | Indicated for obesity management including weight loss and weight maintenance |
| (H+, K+)-ATPase enzyme |  |
Pantoprazole | Gastric protection | Prevent the final step in gastric acid production | Treatment of gastroesophageal reflux disease associated with a history of erosive esophagitis |
| Pyruvate dehydrogenase kinase | 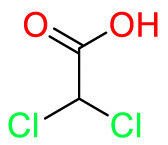 |
Dichloroacetate (DCA) | Treat brain cancer | Inhibit the enzyme pyruvate dehydrogenase kinase | Used for the treatment of warts and cauterization |
| Estrogen receptor | 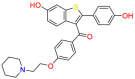 |
Raloxifene | Reduce the risk of breast cancer | Mediate anti-estrogen effects on breast and uterine tissues | Indicated for the prevention and treatment of osteoporosis |
| Cytochrome P-450-dependent enzymes | 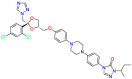 |
Itraconazole | Against Histoplasmosis; Blastomycosis; Cryptococcal meningitis; Aspergillosis | Lead to impaired ergosterol synthesis | For the treatment of the following fungal infections in immunocompromised and non-immunocompromised patients |
| VEGF/VEGFR2 | 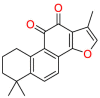 |
Tanshinone IIA | Treatment of coronary heart disease; angina; myocardial infarction | Interfere with the tumor cell cycle, promote tumor cell apoptosis, and inhibit tumor cell invasion and metastasis. | Used to treat patients suffering from myocardial infarction (MI), angina pectoris, stroke, diabetes, sepsis |
| 5-Hydroxytryptamineptamine receptor 2A | 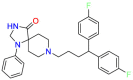 |
Fluspirilene | Chronic schizophrenia | Guanine nucleotide binding to G protein triggers signal transduction and regulates the activity of downstream effectors. | Used for schizophrenia |
3.2. Ovarian cancer
Malignant tumors of the ovary are one of the common malignant tumors of female reproductive organs [81]. Epithelial cancer is the most common among malignant ovarian tumors. The incidence of ovarian malignant tumors ranks third in gynecological tumors, but the mortality rate is the first, so it poses a serious threat to women's lives. For the treatment of ovarian cancer, platinum-based combined chemotherapy is currently used in clinical practice. Niraparib, Rucaparib, and Olaparib are targeted drugs for ovarian cancer.
Peroxisome proliferators-activated receptors (PPARs) are ligand-activated receptors in the nuclear hormone receptor family, and its three subtypes have been discovered including PPARα, PPAR β/δ, and PPARγ [82]. PPARα plays an important role in regulating inflammation and tumorigenesis. The activation of PPAR-β/δ can enhance fatty acid metabolism, while the activation of PPAR-γ can cause insulin sensitization and enhance glucose metabolism [83]. Previous studies have shown that [84,85] ligands that activate PPARγ can be used as effective therapeutic strategies to inhibit ovarian cancer. Anti-tumor drug repositioning mainly starts from the research of mechanism. It is hoped that the method of drug repositioning can not only achieve the effect of treating ovarian cancer, but also obtain a certain preventive effect.
Ciglitazone decreases differentiation and angiogenesis in human umbilical vein endothelial cells to exert an anti-hyperglycemic effect. Ciglitazone ( Table 3 ) is a highly selective agonist of PPARγ and improves insulin sensitivity. In vivo studies have found that ciglitazone can inhibited glucose uptake in ovarian cancer cells and decreased expression of glucose transporter-1 (GLUT-1) [86]. Based on mechanistic studies, the polypharmacology of ciglitazone is beneficial for drug repurposing. Clofibric Acid (CA) ( Table 3 ) is a ligand of PPARα, and used for the treatment of hyperlipidemia. In 2007, a study revealed that CA can be used to inhibit the growth of human ovarian cancer [87]. Studies have found that the main mechanism is that CA can induce the expression of carbonyl reductase (CR) in ovarian cancer cells, thereby reducing the level of PGE2 and inhibiting the expression of VEGF, leading to the inhibition of angiogenesis and ultimately inducing tumor cell apoptosis [87]. Another study found that compared with the control group, the group that received CA can effectively treat peritoneal cancer caused by ovarian cancer without damaging the organs [88]. The action of CA on multiple targets associated with multiple disease pathways demonstrates that its drug repurposing strategy is based on polypharmacological properties. Ritonavir ( Table 3 ), an HIV protease inhibitor, cleaves both structural and functional proteins to inhibit the formation of infectious virions to produce the antiviral effect. When the body's innate immune system is weakened, it increases the risk of solid tumors. Researchers found that ritonavir can inhibit the apoptosis of a variety of tumor cells [89,90]. Ritonavir inhibits cancer cells from exerting anti-tumor effects by depleting G1 cyclin and mediating G1 cell cycle arrest of ovarian cancer cells [91]. Ritonavir synergistically inhibits AKT to increase the efficacy of cell apoptosis. Therefore, it is recommended that ritonavir be used as an enhancement drug for the treatment of ovarian cancer. The drug repurposing strategy is based on specific mechanism of action for different diseases settings. Inhibiting mevalonate pathway has an inhibitory effect on the growth of ovarian cancer cells, so it is also a novel strategy to consider drugs targeting this pathway for the treatment of ovarian cancer [92]. Alendronate possibly inhibits the mevalonate pathway to inhibit bone resorption for the treatment of osteoporosis. Alendronate ( Table 3 ) showed a concentration-dependent inhibitory effect on SKOV3 cells. Studies have found that alendronate attenuates the activation of Rho by inhibiting the mevalonate pathway, thereby inhibiting the migration of ovarian cancer cells [93]. Either in vivo or in vitro evaluations have shown the inhibitory effect of alendronate on the growth of ovarian cancer cells [94]. The drug acting on single target but repurpose for different diseases settings. Statins are usually used as drugs that inhibit the mevalonate pathway to treat hypercholesterolemia [95]. Also,it can cause specific apoptosis of tumor cells [96], and it has been shown that the combination of statins and traditional chemotherapy strategies can improve the efficacy of targeted tumors [[97], [98], [99]]. Statins used to treat cardiovascular disease through inhibit the 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA). It has been proposed and proved that inhibition of the HMG-CoA may against cancer. Lovastatin is an HMG-CoA reductase inhibitor, and it works by reducing the production of cholesterol. Lovastatin ( Table 3 ) may lead to cell cycle progression and cell proliferation by affecting the cholesterol biosynthesis pathway, thereby mediating antineoplastic effects [100]. The specific mechanism of action for lovastatin provides drug repurposing strategy. Ivermectin ( Table 3 ) may inhibit LPS-induced production of inflammatory cytokines for the treatment of inflammation [101]. Ivermectin inhibits the proliferation of cancer cells by regulating a variety of signal pathways. It mainly blocks cell cycle progression and promotes apoptosis by blocking KPNB1 [102]. Besides, ivermectin can also inhibit the growth of ovarian cancer by targeting molecules in the energy metabolism pathway [103]. This evidence is sufficient to prove that ivermectin has great potential for the treatment of ovarian cancer. The case also proved that ivermectin acts on multiple targets pertaining to multiple diseases pathways.
Table 3.
Drug repurposing examples and their mechanism of action in Ovarian Cancer.
| Target | Structure | Drug | Drug Class | Proposed Mechanism | Original Indications |
|---|---|---|---|---|---|
| PPARγ | 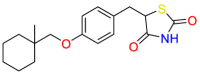 |
Ciglitazone | Never used as a medication | Inhibit ovarian cancer cell growth | Ciglitazone may potentially be used in ovarian hyperstimulation syndrome |
| PPARα | 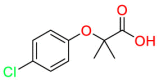 |
Clofibric Acid | Regulate blood lipids | Inhibit angiogenesis and induce apoptosis | Clofibric acid is a biologically active metabolite of the lipid-lowering drugs |
| HIV protease | 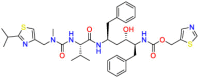 |
Ritonavir | HIV infection | Inhibit cell cycle and induce apoptosis | Ritonavir is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. |
| Farnesyl pyrophosphate synthase | 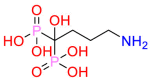 |
Alendronate | Anti-osteoporotic drugs | Inhibit mevalonate pathway | Indicated for the treatment and prevention of osteoporosis |
| Hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase | 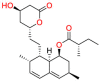 |
Lovastatin | Anti-dyslipidemia drugs | Inhibition of mevalonate pathway | Indicated to reduce the risk of myocardial infarction, unstable angina in individuals without symptomatic cardiovascular disease |
| Yes-associated protein 1 (YAP1) | 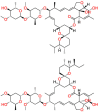 |
Ivermectin | Anti-parasitic agent | Inhibit cell proliferation | Ivermectin cream is indicated for the treatment of inflammatory lesions associated with rosacea |
| 5′-AMP-activated protein kinase subunit beta-1 | 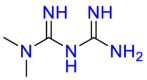 |
Metformin | Diabetes | Lead to a decrease in insulin resistance and a clinically significant decrease in plasma fasting insulin levels | Indicated as an adjunct to diet and exercise to increase glycemic control |
Metformin (Table 3) is the first choice for treating diabetes in clinics by lowering hepatic glucose output, and the discovery that metformin can inhibit the growth of cancer cells in vivo and in vitro has aroused great interest from scientists. Studies have shown that metformin can reduce cancer mortality [[104], [105], [106]]. Many different studies on the mechanism of action of metformin were performed. When cisplatin was used in combination with metformin, as the concentration of metformin increased, the half-maximal inhibitory concentration of cisplatin on the ovarian cancer CP70 cell line decreased significantly. In addition, in an apoptosis experiment of ovarian cancer epithelial cells, it was found that metformin can induce the apoptosis of OVCAR-3 and OVCAR-4 cell lines that depend on adenosine phosphate active protease (AMPK) and inhibit cancer cells. Although there are many studies on the anticancer mechanism of metformin, it is still in the experimental stage, and its safety and effectiveness are still to be explored. Mechanism studies proved that the repurpose strategy is based on polypharmacology.
3.3. Melanoma
Melanoma is a malignant tumor that develops from melanocytes that originate in the nerve ridge. Melanoma is a very deadly skin cancer that starts in melanocytes. Ultraviolet (UV) light radiation from sunlight and the number of melanocytic nevi is the main environmental risk factors for melanoma skin cancer development [107]. In the past, the main treatment strategies for malignant melanoma were surgery, radiotherapy, and chemotherapy. Nowadays, there are currently some drugs on the market that can be used to treat melanoma, but it is still difficult to treat melanoma because of drug resistance and high metastasis. Through the study of melanoma signaling pathways, it has been found that STAT3 plays an important role, so it is proposed to use STAT3 inhibitors as potential anti-melanoma compounds [108]. Besides, some drugs have anticancer activity against a variety of solid tumors, including melanoma. James et al. [109] developed a method based on knowledge graphs to find high-quality melanoma drug candidates. This method uses a more systematic biological graph probabilistic analysis to improve the quality of drug screening, and finally screened 25 candidate drugs for the treatment of metastatic melanoma. Riedel et al. [110] obtained haloprogin by screening the FDA-approved drug library. The synergistic effect of this drug and the organometallic drug RAPTA-T can effectively kill cancer cells. Currently, it is mainly used in the development of melanoma drugs.
Niclosamide (Table 4) is a drug approved by the FDA to treat parasitic infections [111]. The mechanism of action for eradicate tapeworm infections via diminishing the potential of the inner mitochondrial membrane to inhibit oxidative phosphorylation to Ref. [112]. [113]. Besides, the molecular mechanism investigation indicates that niclosamide exhibits anticancer activity by suppressing STAT3 phosphorylation [114]. Mahdi Hatamipour et al. encapsulated niclosamide in nanoliposomes and showed anti-tumor activity against melanoma in vivo [115]. Due to the poor solubility and low bioavailability of niclosamide hindered its clinical application [116], it is hoped that its application in cancer can be ameliorated through preparation. Niclosamide, which acts on multiple targets about multiple disease pathways, provides a drug repurposing strategy. FDA approves auranofin for the treatment of rheumatism. Studies speculated that it induces heme oxygenase 1 (HO-1) mRNA, an inducible heme-degrading enzyme with anti-inflammatory properties. Because protein kinase C (PKC) plays a key role in the function of melanoma cells, the PKC inhibitor auranofin is considered as a potential treatment for melanoma. Experiments revealed that auranofin inhibits melanin synthesis by reducing the activity of intracellular tyrosinase [117]. The drugrepurposing strategy is on the basis of polypharamcoloy. FDA approved disulfiram (DSF) ( Table 4 ) in 1951 for the treatment of alcohol addiction via inhibiting all the identified aldehyde dehydrogenase isoforms. DSF showed metal-dependent anti-tumor activity in the 1970s [118]. The report showed [119] that DSF inhibited cell line proliferation and induced apoptosis of melanoma cells in the presence of free copper, so it was identified as one of the effective compounds that preferentially reduce the proliferation of melanoma subtypes. DSF induces apoptosis by reducing the mitochondrial membrane potential [120]. In this case, the drug repurposing strategy of DSF is related to polypharmacology. Drug resistance is a vital issue in the treatment of tumor drugs. When DSF is combined with copper, it shows that it can inhibit drug resistance-related targets, so DSF is considered to have great development potential. Riluzole (GRM1 blocker) ( Table 4 ) is used to treat amyotrophic lateral sclerosis (ALS) [121]. Early studies have found that ectopic expression of glutamate receptor 1 (GRM1) in melanocytes can transform into melanoma. Therefore, riluzole is proposed as an effective drug for the treatment of melanoma [122]. It inhibits the metabolic activity of melanoma by blocking the signal transduction of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathways. Although riluzole is well tolerated [123], further clinical test results are needed. Drug repurposing strategy of riluzole is related to polypharmacology. Leflunomide ( Table 4 ) was approved by the FDA in 1998 for the treatment of rheumatoid arthritis (RA). Leflunomide is a dihydroorotate dehydrogenase (DHODH) inhibitor [[124], [125], [126]]. Besides, reported studies reveal that leflunomide exerts the effects via inhibiting the transcriptional elongation of genes that are required for melanoma growth [127]. Since leflunomide is effective against a variety of cancer cells and required different concentrations. It is necessary to continue to investigate the safest dose for the treatment of melanoma. The repurposing strategy is related to the polypharmacology properties. Through the screening of FDA-approved drugs, it was identified that haloprogin ( Table 4 ) interacts with RAPTA-T to synergistically kill cancer cells [128]. Haloprogin possibly treats Tinea infections via inhibiting oxygen uptake and disrupting yeast membrane structure and function. On the other hand, combination treatment can be administered in very low concentrations and efficiently inhibit melanoma growth. In the mouse model, haloprogin shows superior to standard drugs for the treatment of melanoma. However, whether the combination drugs can inhibit the spread of melanoma needs further research. Although the mechanism of action is unknown, the drug repurposing strategy is suspected to be related to polypharmacology due to different targets.
Table 4.
Drug repurposing examples and their mechanism of action in Melanoma.
| Target | Structure | Drug | Drug Class | Proposed Mechanism | Original Indications |
|---|---|---|---|---|---|
| STAT3 | 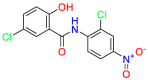 |
Niclosamide | Anti-helminthic drug | Induce the apoptosis of melanoma cells | For the treatment of tapeworm and intestinal fluke infections |
| AKT |  |
Auranofin | Rheumatoid Arthritis | Increase reactive oxygen species (ROS) levels and apoptosis | Used in the treatment of active, progressive or destructive forms of inflammatory arthritis |
| Aldehyde dehydrogenase | 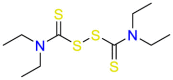 |
Disulfiram | Inhibit the enzyme acetaldehyde dehydrogenase | Affect the intermediate metabolism of alcohol | For the treatment and management of chronic alcoholism |
| Glutamate |  |
Riluzole | Lou Gehrig's disease | Anticonvulsant | For the treatment of amyotrophic lateral sclerosis |
| Mitochondrial enzyme dihydroorotate dehydrogenase | 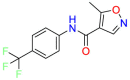 |
Leflunomide | Rheumatoid Arthritis | Reduce the cell viability of melanoma cells | For the management of the signs and symptoms of active rheumatoid arthritis (RA) |
| Unknown | 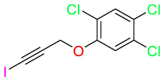 |
Haloprogin | Antifungal agent | Inhibit the absorption of oxygen and destroy the structure and function of the yeast membrane | Used to treat fungal (Tinea) skin infections |
3.4. Represented cases of drug repositioning in cancer
During the 1950s–1960s, thalidomide was widely used as an antiemetic for pregnant women, and it caused tens of thousands of malformed babies. However, thalidomide combined with dexamethasone can treat multiple myeloma (MM). MM is an incurable malignancy of terminally differentiated B-cells accounting for approximately 1–2% of all human cancers [129]. Thalidomide, a synthetic derivative of glutamic acid, is an oral immunomodulatory therapeutic drug. Its activity as a single agent in refractory or relapsed MM was first reported in 1999 [130]. According to reported researches, the anti-angiogenesis effect is mediated by inhibition of VEGF and β fibroblast growth factor (FGF) [131] (Fig. 5 ). Besides, thalidomide inhibits production of MM growth factors including IL-6, TNF-α, and vascular endothelial growth factor(VEGF) [129]. It can also block the G1 growth of MM cells. Although the exact mechanism of thalidomide in the treatment of multiple myeloma is not completely understood and its anti-angiogenic effect is only part of the means of its anti-myeloma activity, the treatment results for multiple myeloma are particularly impressive.
Fig. 5.

The pharmacological mechanisms of thalidomide. Thalidomide arrests multiple myeloma (MM) cells in the G1 phase by inhibiting adhesion of MM cells to bone-marrow stromal cells [132].
4. Antimicrobial repositioning
The current speed of discovery of antibiotics is much lower than the peak period of 1940–1960 [133]. The main reason is that antimicrobial resistance has weakened the speed of antimicrobial development. Antimicrobial resistance is a serious global problem, new drugs, and novel treatments are urgently needed to improve this situation [134]. As with other drugs, de novo antibacterial discovery is a long and arduous process, which not only requires time and cost but also has very high safety and toxicity risk [135]. Due to its effectiveness, drug repositioning has made substantial progress in the development of alternative drugs for the treatment of bacterial and fungal infections over the past decade. And it has become one of the major strategies for the drug development of pharmaceutical companies. Recent research has found that bithionol, a drug previously used to treat parasitic infections in horses, is being reused to kill antibiotic-resistant bacteria, including Methicillin-resistant Staphylococcus aureus (MRSA), a common hospital-acquired infection. This is a successful case in which scientists are addressing resistance by exploring drug repositioning.
S. aureus infection can cause osteomyelitis. The current clinical challenge is mainly its relapse and drug resistance [136]. Existing drugs cannot eradicate small-colony variants (SCV), so drug repositioning is needed to find new drugs to treat chronic infections. Through high-throughput screening from FDA-approved drug libraries, four drugs were finally identified as having bactericidal activity against SCV [137], with the most obvious effect of sitafloxacin. In addition, many non-antibacterial drugs have been reused for bacteria and fungi. For example, the antidepressant azathioprine has anti-biofilm activity against E. coli [138]. The activity of anthelmintics against Gram-positive and Gram-negative bacteria and fungi has been reported [139,140]. Mazumdar et al. [141] found that cardiovascular drug amlodipine (AML) ( Table 5 ), if combined with existing antibacterial drugs, may overcome the resistance of existing drugs. AML exerts antihypertensive and antibacterial effects by inhibiting voltage-gated calcium channels and β-lactamase [142], respectively. Therefore, drug repurposing strategy is related to the polypharmacology of drugs.
Table 5.
Drug repurposing examples and their mechanism of action in antimicrobial applications.
| Drug | Structure | Drug Class | Repurposed Indication | Initial Mechanism | Original Indications |
|---|---|---|---|---|---|
| Amlodipine | 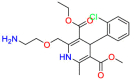 |
Cardiovascular | Antibacterial | Act on vascular smooth muscle to lead to a reduction in peripheral vascular resistance | Hypertension. Coronary artery disease. Angina |
| Pentamidine | Antiprotozoal | Antibacterial | Inhibition of protein and RNA synthesis and antagonism of receptors | For the treatment of pneumonia | |
| Sertraline | 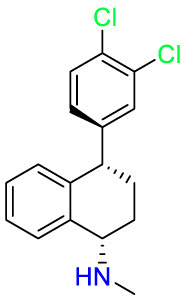 |
Antidepressant | Antifungal | Inhibition of CNS neuronal uptake of serotonin (5HT) | Used for the treatment of major depressive disorder (MDD) |
| Aspirin | 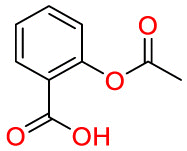 |
NSAIDs | Methicillin-resistant Staphylococcus aureus (MRSA) | Target cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) | Pain, fever, and inflammation |
| Flupenthixol | 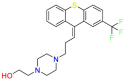 |
Antipsychotics | Antibacterial | Antagonizing dopamine actions | Used for maintenance therapy of chronic schizophrenic patients |
| Thioridazine | 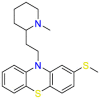 |
Antipsychotic | Methicillin-resistant Staphylococcus (S.) aureus | Block dopamine (DA) receptors | For the treatment of schizophrenia and generalized anxiety disorder |
| Rapamycin (TOR) | 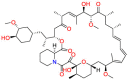 |
Potent immunosuppressant | Antifungal; Antineoplastic | Selectively block the transcriptional activation of cytokines thereby inhibiting cytokine production | Used to treat lymphangioleiomyomatosis |
| Prochlorperazine | 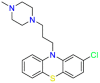 |
Antipsychotics | Antibacterial | Block D2 dopamine receptors in the brain | Indicated for the symptomatic treatment of severe nausea and vomiting |
| Lacidipine | 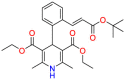 |
Cardiovascular drug | Gram-positive and Gram-negative | Dilating peripheral and coronary arteries, reducing peripheral vascular resistance | Indicated for the treatment of hypertension either alone or in combination with other ntihypertension agents |
| Nifedipine | 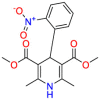 |
Cardiovascular drug | Gram-positive and Gram-negative | Interfere with cell biosynthesis | Indicated to treat vasospastic angina and chronic stable angina |
| Zidovudine | 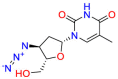 |
Antiretroviral drug | Synergistic effects with polymyxin | Prevent the formation of phosphodiester linkages | Used in combination with other antiretroviral agents for the treatment of human immunovirus (HIV) infections. |
| Tamoxifen | 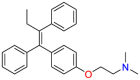 |
Breast cancer | Antifungal | Estrogen receptor antagonist | Indicated to treat estrogen receptor-positive metastatic breast cancer in adults |
| Clioquinol |  |
Antiseptics | Antifungal | Act as a zinc and copper chelator | Used for the treatment of various intestinal diseases |
The drug repurposing strategy has obvious advantages in the combined treatment of antimicrobial drugs. Pentamidine ( Table 5 ) is suspected to interference of critical functions in DNA, RNA and protein synthesis [143]. Through screening, it was found that pentamidine can be combined with antibacterial drugs to treat multidrug-resistant Gram-negative infections. The mechanism of action studies reveal that Pentamidine altered the integrity of the outer membrane of E. coli, providing a physical basis for this synergy [144]. Different mechanisms of action demonstrate that the drug repurposing strategy of pentamidine is related to polypharmacology. However, whether the laboratory results can be successfully converted into clinical results needs further research. Sertraline ( Table 5 ) is an antidepressant which belongs to selective serotonin reuptake inhibitors (SSRIs). The combination of sertraline and amphotericin B has a synergistic effect on Alternaria alternata. Zhai et al. reported [145] that sertraline exhibits a unique effect on cryptococcosis by interfering with translational inhibition of protein synthesis. Costa Silva et al. proved that SSRIs promote apoptosis and reduce the biological activity of C. albicans by changing the integrity of the mitochondrial membrane [146]. The antifungal activity on a variety of fungi makes sertraline promising as an antifungal drug. Due to the lack of antibiotics that can be used to treat methicillin-resistant S. aureus (MRSA), an alternative therapy continues to be found. It proved that sertraline acts as SSRI but repurposed for different diseases settings. Aspirin relieves pain and reduces fever mainly due to decreased prostaglandins and TXA2. However, aspirin (Table 5) plays an anti-staphylococcal effect by inhibiting the expression of alpha-toxin and matrix adhesion genes [147]. The differences of targets demonstrated that the repurposing strategy is related to polypharmacology. Others proved that although aspirin is less effective than ordinary antibiotics, it can exert a synergistic effect as an adjuvant for MRSA [148]. Flupenthixol is a powerful antagonist of both D1 and D2 dopamine receptors [149]. Recently, it is found that flupenthixol reduces the proton pump activity of S. aureus by decreasing the transmembrane potential. Different targets and mechanisms of action indicate that the repurposing strategy is related to polypharmacology. In view of the increasing number of pathogenic organisms with multidrug resistance, combination therapy may prove to be the best option.
Thioridazine (Table 5) was a typical antipsychotic drug act on Dopamine D2 and D1 receptor in its early period, and it was stopped using in 2005 due to adverse cardiac reactions [150]. Thioridazine proved to be sensitive to methicillin-sensitive S. aureus (MSSA), Enterococcus, and M. tuberculosis, the mechanism of action is suspect the inhibition of membrane-bound enzymes [151]. Although thioridazine has lost its role in the treatment of psychosis, it still has great potential in antifungals. Different mechanisms of action in different targets indicate that drug repurposing may provide a strategy for developing antimicrobial drugs. Signaling pathways that activate mitogen-activated protein kinase (MAPKs) elicit many of the responses due to changes in environmental conditions and other stimuli. These pathways were first elucidated in the unicellular eukaryote Saccharomyces cerevisiae. The MAPK pathways of yeast among the understood signal transduction have greatly improved our knowledge about the functions and regulation of these pathways. Also, these studies provide guidelines for drug repurposing [152]. Subsequently, Immunosuppressants often lead to reduced immune function in the treatment of autoimmune diseases, accompanied by fungal infections. Therefore, research on the antifungal activity of immunosuppressants has been increased. Rapamycin (Table 5), a serine/threonine protein kinase inhibitor, directly regulates protein synthesis, cellular proliferation and lipid metabolism. On the other hand, rapamycin has a synergistic effect with amphotericin B in a limited dose [153]. The drug exerts its antibacterial effect mainly by retarding cell-cycle progression at the G1/S transition. Although rapamycin cannot be used as an antibacterial drug in clinical trials, it is still a potential antibacterial drug due to its target. The advantage of this repurposing strategy is combination therapy due to act on different targets. Prochlorperazine mainly works on D2 dopamine receptors and is used for the treatment of anxiety and schizophrenia. In vitro screening shows that prochlorperazine has strong antibacterial activity in Bacillus and Enterobacter [154]. Especially, it was found to reduce the proton motive force of S.aureus by reduction in the transmembrane potential [155]. Prochlorperazine biased towards adjuvants for combined use against multiple fungi in the subsequent development process. Lacidipine (Table 5) is a powerful third-generation calcium channel blocker used to treat hypertension [156]. In 2007, it was found through in vitro screening that the drug has essentially antibacterial effects on S. aureus and V. cholerae [157]. The primary mechanism may be the combination of lacidipine and conventional antibacterial drugs to cause structural modification or enhance its antibacterial effect [157]. The mechanism of antibacterial action of lacidipine needs further study. Nifedipine ( Table 5 ) is L-type calcium channel blocker. In the experiment, it showed promising antibacterial activity against Cryptococcus, Candida and yeast. The compounds screened to have antibacterial effect belong to dihydropyridines, so it is believed that the antibacterial effect is mainly related to its structure. Experimental results show that the antifungal effect of the calcium channel blocker (CCB) involves the regulation of Cch1-Mid calcium channels [158]. Although nifedipine belongs to non-dihydropyridine CCB, it belongs to L-type CCB [159]. Mechanism studies suggest that nifedipine repurposes in different disease settings. Zidovudine ( Table 5 ) is used to treat HIV due to its antiretroviral activity [160]. K. pneumoniae lacks effective treatment drugs due to multi-drug resistance. It has recently been proposed that the combination of zidovudine and polymyxin B shows excellent antibacterial efficacy and minimizes the resistance to polymyxin [161]. Zidovudine was activated in bacteria by thymidine kinase (TK), and interference in DNA resulted in DNA chain termination. The repurposing strategy of zidovudine has been demonstrated in different diseases settings [162].
FDA-approved small-molecule compound library was screened for ESKAPE pathogens, of which ebselen (EB) and 5-fluoro-2′-deoxyuridine (FdUrd) have improved mouse survival rate in lethal models of septicemic MRSA infection. The results indicate the potential of these drugs to treat resistant bacterial infections [163]. In addition, Breger et al. identified 15 drugs with antifungal activity [164]. In recent years, several different methods have also been derived to find new indications for the rapid location of old drugs. Arielle et al. [165] developed a high-throughput assay that ultimately identified 75 drugs by screening generic drug libraries. Tamoxifen (Table 5) initially targets estrogen receptor that is prevalent in receptor-positive breast cancers. But the drug also affects neutrophils and produces neutrophil extracellular traps (NETs), a mesh of proteins that kill bacteria. The ‘off-target effects’ could have critical clinical implications. Another method is to screen based on the specific effective structure of antibacterial drugs to find potential antibacterial drugs. Ejim et al. [166] proposed screening from approved drug libraries, and obtained six non-antibacterial drugs, which enhanced the activity of Mexicycline against a reference Pseudomonas aeruginosa strain. Bacterial resistance is accompanied by a gradual decrease in treatment options, and multiple aspects need to be taken to address the issue of global antimicrobial resistance. In short, the combination of drug repositioning of antimicrobials and the rapid development of new drug mechanisms can reduce the abuse of antimicrobials or reduce the constant threat of antimicrobial resistance. It is through different approaches that humans can cope with the challenges posed by antimicrobial resistance.
4.1. Represented cases of drug repositioning in antimicrobial repositioning
Fungicides are broad-spectrum chemical agents that inactivate microorganisms. When bactericides are used on living tissues, they are often referred to as antiseptics [167]. Clioquinol was produced as an antiseptic and sold as an oral intestinal acaricide in 1934 for the treatment of various intestinal diseases [168]. In the early 1970s, it was withdrawn from the market due to its association with subacute myeloid optic neuropathy (SMON) [168]. Alsterholm M et al. investigated antibacterial activity of a combination of clioquinol and corticosteroids against C.albicans, E.coli, S.aureus, S.epidermidis and Streptococcus pyogenes [169]. Among all topical skin drugs, 3% clioquinol has the strongest antifungal activity. Based on reported researches, it is believed that clioquinol can be a promising formulation in topical epidermal mycosis [167]. It can also be used in combination with systemic antifungal agents to reduce the occurrence of drug resistance and enhance efficacy. It has a broad antifungal spectrum and has moderate to strong activity among common pathogenic fungi. In addition, studies have shown that clioquinol does not exhibit an antibacterial effect by directly binding ergosterol to exhibit plasma membrane instability. Through in-depth study of the mechanism, it is shown that clioquinol can act on the cell wall more effectively than other derivatives to exert antibacterial effects [170]. Therefore, reasonable structural modification can speed up the development of antifungals and enhance the application.
5. Antivirus repositioning
Influenza virus infection has become a serious worldwide health problem due to its infectivity. The virus was first discovered in tobacco plants. Researchers have discovered that diseases in tobacco plants are transmitted through a vector called tobacco mosaic virus. Over the past few decades, scientists have successfully developed dozens of antiviral drugs against viral proteins or host factors. Antiviral drugs could be divided into three main categories, including anti-DNA virus drugs, anti-RNA virus drugs, and anti-retroviruses drugs. The virus needs to enter the cell before replicating, eventually causing the virus to become infected. Although there are both vaccines and antiviral drugs, some genes of viruses constantly change and antiviral resistance emerges. Due to this, it is urgent to develop efficient antivirals.
Although Zika virus infection shows mild or even asymptomatic symptoms, there is currently no effective treatment. For similar public health events, traditional drug development processes are complex and time-consuming. Protein-targeting drugs may have limitations, hope to find new indications for existing drugs [171]. Barrows et al. [172] identified 24 potential anti-ZIKA viruses’ drugs through screening. Xu et al. [173] also systematically identified emricasan, an oral caspase inhibitor, as a highly effective anti-ZIKA drug. Although the FDA-approved drugs show good safety and pharmacokinetics, they have been proven to affect the fetus, which means clinical trials are essential [172]. Drug targets network analysis can be used to identify important genes in the infection pathway in the screening process.
In 2014, the large-scale ZIKA virus (ZIKV) outbreak in Brazil caused global health problems. Mechanism studies have found that ZIKV infection leads to increased caspase-3 activation, which in turn leads to cell death [174]. Emricasan ( Table 6 ) is the first caspase inhibitor tested in human which is used to protect liver cells from excessive apoptosis. Emricasan exerts antiviral effects by reducing neuronal cell death and inhibiting the activity of caspase-3, which is induced by ZIKV. The repurposing strategy is based on the same target but repurposing for different diseases settings. PhA-690509 ( Table 6 ) as an investigational drug for cell cycle-dependent kinase (CDK), inhibits all three ZIKV strains in a dose-dependent manner. As one of the first drugs to be screened and proven to have antiviral activity against ZIKV, PhA-690509 as well as niclosamide inhibits viral RNA replication and finally achieves the purpose of inhibiting ZIKV infection [173]. Due to both DNA and RNA viruses modifying CDK function to favor viral replication, it plays a prominent role in viral infections. The study demonstrates that the repurposing strategy is related to the exact mechanism in different diseases settings. Mycophenolic acid (MPA) ( Table 6 ) was used as an immunosuppressant in the early stage to prevent dengue infection [175]. MPA depletes guanosine nucleotides and inhibits their proliferation, thereby suppressing cell-mediated immune responses and antibody formation [176]. Through the screening of cells that are susceptible to the ZIKA virus, it is found that MPA shows the most effective antiviral effect on ZIKV [177]. The mechanism of the drug's effect may be due to targeting the ZIKV replication process [177]. In addition, the safety of MPA makes it a potential drug for the treatment of the ZIKA virus. Research studies indicate that the drug repurposing strategy of MPA is related to specific mechanism of action but repurpose for different disease settings. Mefloquine inhibits merozoite invasion and proteins interactions involved with nutrient uptake to exert an antimalarial effect. Mefloquine ( Table 6 ) was identified that has an effective anti-dengue virus and anti-ZIKA virus activity by virtual screening [178]. Different from the wet experimental screening method, mefloquine was obtained by calculating the electrostatic potential map of the compound through molecular modeling, and the antiviral activity was tested by experiment [178]. Since this drug has shown adverse events of anxiety or hallucinations in clinical trials, mefloquine needs to be tested in vivo to further evaluate its safety. Also, its detailed mechanism of action remains to be further studied. Suramin ( Table 6 ) is an anti-parasitic drug initially, and it was more recently shown inhibition on multiple viruses. After the outbreak of ZIKV, it is believed that it can be used to treat ZIKV infection. Suramin inhibits the early stage of the replication and affects the entry of the virus, thereby reducing the proportion of cells infected by ZIKV. The intervention of suramin will not only affect the synthesis of viral RNA but also reduce the infectivity [179]. The results verify that the repurposing strategy is on the basis of different diseases settings. The FDA approved sofosbuvir (Table 6) in 2013 to treat chronic hepatitis C virus (HCV), and it is also the first drug that treats certain HCV without the need for interferon therapy [180]. Like the mechanism that initially achieves therapeutic effects by sofosbuvir, it mainly reduces the number of viral infections by restraining viral replication [181]. The drug repurposing strategy with specific mechanism but repurpose for different diseases settings. Sofosbuvir is well tolerated and safe in its original pharmacological effects, and these advantages can support its development as an anti-ZIKV drug. Favipiravir (T-705) ( Table 6 ) is an RNA polymerase inhibitor that works on influenza. Although favipiravir is well tolerated in the Ebola virus disease, it does not show strong antiviral activity. Favipiravir acts as the primary mechanism of anti-influenza or antiviral drugs through selective and potent inhibition of RNA-dependent RNA polymerase (RdRp) of RNA viruses [182]. Experimental studies have shown that the infectivity of the virus gradually decreases with increasing doses [183]. Although Favipiravir exhibits a dose-dependence, the Pharmacokinetics (PK) and tolerability of relevant doses need to be studied to determine the best dosing regimen of favipiravir. Toremifene ( Table 6 ), selective estrogen receptor (ER) modulators, is used to treat certain breast cancers. Toremifene-Mefloquine-Posaconazole is a group of drugs that can effectively block the entry of Ebola [184]. The main mechanism of action may be direct binding to the internal fusion loop region of Ebola virus glycoprotein (GP). These three drugs achieve a synergistic effect by inhibiting Niemann-Pick C1 (NPC1) protein, Acid sphingomyelinase (ASM) activity, and Nicotinic acid adenine dinucleotide phosphate (NAADP)-stimulated lysosomal calcium release [184]. Drugs used to treat EBOV lost potency due to low activity. The combined method can not only prevent the development of drug resistance but also reduce the dosage of each drug. Polypharmacology of toremifene provides the potential for drug repurposing. Pregnant women and people with weakened immune systems are often threatened by human cytomegalovirus. Cardiac glycoside (GC) ( Table 6 ) is a type of glycoside compound that can enhance myocardial contractility. It inhibits the Na+-K+-ATPase on tissues to increase intracellular Ca2+ concentrations to promote inotropy and bradycardia [185]. GC has recently been shown to have anti-cancer and antiviral activities [186]. As a typical cardiac glycoside compound, digoxigenin can effectively inhibit HCMV at a concentration of nanomolar level [187]. Studies have found that cardiac glycosides may inhibit HCMV replication by regulating transcription and translation. Therefore, it is recommended to explore its significance on the combination treatment in subsequent development. Different mechanisms of action on different targets lead to drug repurposing strategies. Emetine (Table 6), an FDA approved drug for the treatment of amoebiasis, blocks protein synthesis in both mammalian and parasitic cells. The combination of emetine and Ganciclovir synergistically inhibits murine cytomegalovirus (MCMV) in a mouse model, but its anti-HCMV activity significantly reduced in vitro. Unlike emetine inhibiting RNA and DNA virus replication, another experiment indicates that emetine disrupts the interaction between MDM2-p53 and MDM2-IE2 induced by HCMV and exerts antiviral effects. The case of drug repurposing for emetine is related to polypharmacology. Studies have found that the dose of emetine used to treat antiviral is far lower than the standard dose, and it does not show toxicity [188]. The advantage of emetine as a potential candidate drug for anti-HCMV is that it adopts a new type of host-dependent antiviral mechanism and has higher safety, whether it is a single drug or a combination drug. Nitazoxanide (Table 6) initially acts on pyruvate-flavodoxin oxidoreductase for the treatment of diseases caused by other protozoa or helminths. It was later proved that it has effective activity against a variety of RNA and DNA viruses, including influenza and a variety of viruses [189]. Nitazoxanide has been regarded as a broad-spectrum antiviral drug due to inhibit the replication of several RNA and DNA viruses [190]. The drug repurposing strategy is on the basis of specific mechanism but repurpose for different diseases settings. Manidipine ( Table 6 ) is used as a calcium channel blocker for the treatment of hypertension. Human cytomegalovirus (HCMV) is a widespread herpes virus, and congenital HCMV infection is one of the causes of fetal death [191]. The IE2 protein of HCMV is an important regulator of viral and host cell gene expression [192]. Studies have shown that manidipine explicitly inhibits the replication of HCMV by interfering with the IE2-dependent activation of the viral E gene [193]. Mechanisms studies demonstrated that the repurposing strategy for manidipine is related to polypharmacology. Ferroquine (FQ) ( Table 6 ) is used for treating malaria may due to inhibit β-hematin formation. Recently, it is investigated as a novel HCV inhibitor. The study of its antiviral mechanism found that the entry stage of HCV is an attractive stage for virus intervention [194]. Ferroquine not only inhibits the binding of HCV into target cells, but also inhibits RNA replication, and finally exhibits antiviral activity independent of genotype. In addition to single-agent antiviral studies, the combination of FQ and interferon (IFN) or HCV NS3/4A protease inhibitor also showed a synergistic antiviral effect. The different mechanism of action for FQ provide drug repurposing strategy. Ayami Sato et al. found that suberoylanilide hydroxamic acid (SAHA) ( Table 6 ), an HDAC inhibitor for T-cell lymphoma, was used as a promising anti-HCV drug. SAHA inhibits the replication of HCV in OR6 cells without cytotoxicity [195]. SAHA is an effective drug for the treatment of HBV-negative hepatocellular carcinoma (HCC) patients, but SAHA should be used with caution in HBV-positive HCC patients [196]. SAHA is effective in drug repositioning strategy due to their pharmacological activities in multiple targets.
Table 6.
Drug repurposing examples and their mechanism of action in antivirus applications.
| Drug | Structure | Drug Class | Repurposed Indication | Mechanisms | Original Indications |
|---|---|---|---|---|---|
| Emricasan |  |
Nonalcoholic steatohepatitis (NASH) | ZIKV infection | Anti-cell-death | Investigated for treatment in hepatitis (viral, C), liver disease |
| PhA-690509 |  |
Cyclin-dependent kinase A inhibitor | ZIKV infection | Inhibit caspase-3 activity in SNB-19 cells | Anti-cancer |
| Mycophenolic acid | 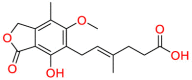 |
Immunosuppressant | ZIKV infection | Inhibit an enzyme needed for the growth of T cells and B cells | For the prophylaxis of organ rejection in patients receiving allogeneic renal transplants |
| Mefloquine | 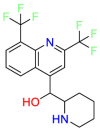 |
Chloroquine resistant or multidrug resistant falciparum malaria | ZIKV infection | Asexual red blood cell type killing Plasmodium falciparum; Plasmodium vivax (killing blood proliferator) | Against chloroquine-resistant forms of Plasmodium falciparum |
| Suramin | 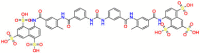 |
African trypanosomiasis and onchocerciasis | ZIKV infection | Block the replication of a variety of viruses | For treatment of onchocerciasis |
| Sofosbuvir | 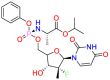 |
Chronic hepatitis C | Inhibit ZIKV RNA polymerase | Against hepatitis C virus (HCV). | Used in combination therapy with other antiviral medications to treat HCV infected patients |
| Favipiravir | 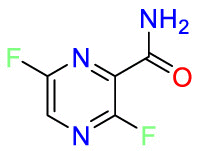 |
Influenza | Against Ebola Virus | Inhibit RNA dependent RNA polymerase (RdRp) activity of influenza virus | Used to treat cases of influenza that were unresponsive to conventional treatment |
| Toremifene | 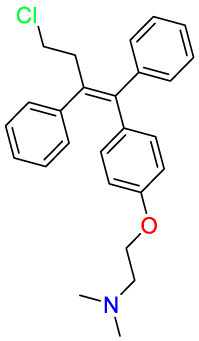 |
Metastatic breast cancer | Against Ebola Virus | Overexpression of NPC1 inhibits virus invasion | For the treatment of metastatic breast cancer |
| Cardiac glycosides | 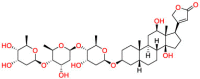 |
Chronic cardiac insufficiency | Anti-HCMV | Drugs interfere with host pathways where viruses are carried away | manifest and chronic cardiac insufficiency |
| Emetine |  |
Anti-parasitic drugs | Anti-HCMV | Direct killing of amoeba trophozoites | severe invasive amoebiasis |
| Nitazoxanide | 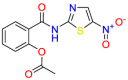 |
Anti-parasitic drugs | Anti-HCMV; Anti-Influenza | Inhibit RNA and DNA virus replication | For the treatment of diarrhea in adults and children caused by the protozoa Giardia lamblia |
| Manidipine | 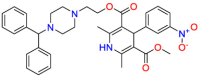 |
Anti-hypertensive drug | Anti-HCMV | Block the entrance of extracellular calcium into the cell | For the treatment of hypertension |
| Ferroquine |  |
Antimalarial | Anti-HCV | Inhibit the formation of β-hematin | Antimalarial drug candidate |
| Suberoylanilide hydroxamic acid |  |
Anticancer | Anti-HCV | Catalyze the removal of the acetyl moiety from the lysine residues of proteins | For the treatment of cutaneous T-cell lymphoma |
The coronavirus SARS-CoV-2 is the pathogen of COVID-19. Coronavirus belongs to the Coronaviridae family. COVID-19 is currently the seventh known coronavirus that can infect human, the remaining six are HcoV-229E, HcoV-OC43, HcoV-NL63, HcoV-HKU1, SARS-CoV, and MERS-CoV [197]. Due to its high replication rate, it has rapidly spread around the world [198]. With the rising number of confirmed cases, the World Health Organization announced that COVID-19 has become a public emergency of international concern. The main clinical symptoms of confirmed cases are fever, cough, dyspnea, and vomiting [199]. The receptors of SARS-CoV-2 and SARS-CoV are both angiotensin-converting enzyme 2 (ACE2), and mucosal cells contain many ACE2 receptors. The virus could continue infecting new hosts by generating numerous copies of its genome and packaging them into virions. Finally, the capsid and RNA are composed to generate new virus particles and released out of the cell to infect new cells [197]. COVID-19 is a serious respiratory disease. Since there are no drugs to treat the virus, drug repurposing is an existing effective response strategy.
Rameez et al. [200] identified raltegravir, paritaprevir, bictegravir, and dolutegravir as anti-SARS-CoV-2 by targeting 3C-like protease (3Clpro) and 2′-O-ribose methyltransferase (2′-O-Mtase) of potentially therapeutic drugs. Chen et al. [201] applied the development of SARS-CoV enzyme-specific inhibitors to SARS-CoV-2 and obtained 16 candidate drugs through virtual screening. Among them, the antiviral drugs Epclusa and Harvoni have inhibitory effects on HCV. Zhou et al. [202] used web-based drug repositioning methods to quantify the interaction between drugs and targets, and identified 16 potential anti-human coronaviruses (SARS-CoV and SARS-CoV-2) drugs. Table 7 summarizes drugs screened by different methods or for different target proteins as potential drugs for further research.
Table 7.
Drug repurposing examples and their mechanism of action in COVID-19 applications.
| Drug | Structure | Drug Class | Repurposed Indication | Initial Mechanisms | Original Indications |
|---|---|---|---|---|---|
| Remdesivir | 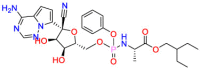 |
Antiretroviral | COVID-19 | Inhibit action of RNA polymerase | Used to treat RNA virus infections |
| Raltegravir | 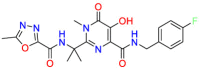 |
Anti-HIV | COVID-19 | Act on HIV integrase and prevent HIV replication | For the treatment of HIV-1 infection in conjunction with other antiretrovirals. |
| CVL218 | 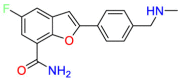 |
PARP1 inhibitor | COVID-19 | Against SARS-CoV-2 replication. Anti-inflammatory effect | Broad spectrum anticancer agent |
| PJ-34 | 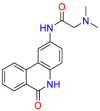 |
PARP1 inhibitor | COVID-19 | Reduce RNA binding, which prevents viral replication | Anticancer agent |
| Beclabuvir | 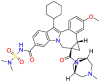 |
Anti-HCV | COVID-19 | Bind to thumb site 1 of the hepatitis C virus (HCV) NS5B RNA-dependent RNA polymerase | Antiviral drug for the treatment of HCV infection |
| Saquinavir | 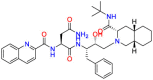 |
HIV protease inhibitor | COVID-19 | Bind to active sites of HIV-1 and 2 proteases | For the treatment of HIV-1 infection |
| Pemirolast | 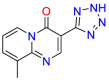 |
Bronchial asthma | COVID-19 | Inhibit the antigen-induced release of inflammatory mediators | For the prevention of itching of the eyes |
| Isoniazid Pyruvate | 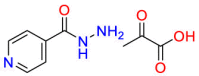 |
Tuberculosis | COVID-19 | Not clear | Isoniazid pyruvate is a metabolite of isoniazid |
| Nitrofurantoin | 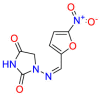 |
Urinary tract infection | COVID-19 | Nitrofurantoin is reduced by bacterial flavoproteins to reactive intermediates | Used to treat acute uncomplicated urinary tract infections |
| Eriodictyol | 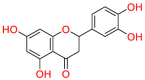 |
Antioxidant | COVID-19 | Active the expression of critical transcription factors | Used for antiallergenic, antimicrobial, anticancer, and antioxidant |
| Ribavirin | 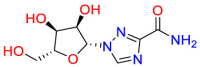 |
Antiretroviral drug | COVID-19 | Inhibition of influenza virus RNA polymerase | Indicated for the treatment of chronic Hepatitis C virus |
| Teicoplanin |  |
Glycopeptide antibiotic | COVID-19 | Stop genomic viral RNA release and viral replication cycle | For the treatment of bacterial infections caused by susceptible microorganism |
| Bortezomib | 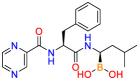 |
Treat multiple myeloma and mantle cell lymphoma | COVID-19 | Reversible inhibitors of chymotrypsin like activity of 26S proteasome in mammalian cells | Used for the treatment of adult patients with multiple myeloma or mantle cell lymphoma |
| Flurazepam | 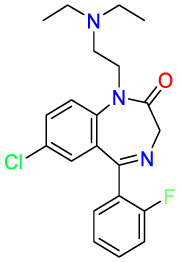 |
Anticonvulsant | COVID-19 | Inhibition of limbic system on the control of brainstem reticular structure | For short-term and intermittent use in patients with recurring insomnia and poor sleeping habits |
Remdesivir is a nucleoside analog with antiviral activity and the first drug approved by the FDA for the treatment of COVID-19. Previous reports have shown that remdesivir has a variety of antiviral activities in vitro [[203], [204], [205]], including SARS-CoV [206] and MERS-CoV [207]. Remdesivir is a diastereomeric monophosphate phosphoramidate prodrug that inhibits viral RNA polymerase. Its active analog enters and accumulates in the cell. Consistent with other antiviral mechanisms of action, it exerts anti-SARS-CoV-2 activity by inhibiting RNA-dependent RNA polymerase. However, the WHO recommended publishing that remdesivir did not significantly improve the survival rate and hospitalization of patients. Therefore, its clinical effectiveness and safety need to be further evaluated. Raltegravir is an antiretroviral drug used in the treatment of HIV-1 infection. It impedes the insertion of HIV-1 DNA into the host cell genome. Subsequently, raltegravir was identified as a potentially effective drug against the main protease (Mpro) of SARS-CoV-2 based on a computational method [200]. Since Mpro is essential to the life cycle of the virus, it is recognized as the best drug discovery target [208]. Raltegravir was screened against Mpro and RdRp. The drug exerts an antiviral mechanism of action that is consistent but repurposed in different disease settings. Although the computational approach can speed up the process of drug repositioning, it still needs further evaluation in vitro and in vivo. The method based on machine learning predicts that poly ADP-ribose polymerase 1 (PARP1) inhibitor CVL218 is an effective drug against coronavirus and has been experimentally verified in vitro [209]. Researchers speculated that CVL218 might target the N-terminal domain of SARS-CoV-2 to impede viral replication. They also emphasized that PARP inhibitors may inhibit the replication of coronavirus. Clinical studies of non-cancerous diseases have proved that PARP1 inhibitors treat acute lung diseases caused by SARS-CoV-2 infection [210]. PJ-34 is the first-generation PARP1 inhibitor. Past studies have shown that PARP1 inhibitors play a central role in inflammatory injury and related diseases, especially in inflammatory diseases of the lung [211]. In particular, PJ-34 can target the N-terminal domain of the coronavirus nucleocapsid (N) protein and reduce the RNA binding ability, thereby inhibiting virus replication [209]. This is also the basic research that PJ-34 is considered to be an effective antiviral drug [212]. The method of describing the interaction between drugs and viral targets based on the knowledge map also confirmed that PJ-34 might have anti-SARS-CoV-2 activity. But further clinical verification is needed. The repurposing strategy of CVL218 and PJ34 is based on specific mechanisms of action but repurposes for different diseases settings.
Beclabuvir is a non-nucleoside NS5B polymerase inhibitor of the RdRp of the hepatitis C virus (HCV) [213]. Both SARS-CoV-2 and HCV are + ssRNA viruses. Thus, an attempt was made to identify effective SARS-CoV-2 inhibitors by screening RdRp inhibitors. Beclabuvir was identified as a potential drug for the treatment of SARS-CoV-2 through virtual screening of approved drugs [213]. It had been proved that beclabuvir can efficiently bind to RdRp of the SARS-CoV-2. In this case, drug repurposing strategy is based on specific mechanism of action but apply in different diseases. Molecular docking predicted saquinavir is an anti-SARS-CoV-2 drug, and its binding mode is highly similar to the Mpro ligand N3 [214]. Saquinavir binds to the active site of the HIV-1 and HIV-2 proteases to prevent the cleavage of viral polyproteins. In silico study demonstrated that saquinavir inhibited 3CLPro activity in SARS-CoV-2 [215]. In addition, in vitro studies have shown that low-dose saquinavir can alleviate the replication of SARS-CoV-2 [216]. Although the calculation method still has certain limitations and required experimental verification, this method speeds up drug discovery and provides a guiding role for the experiment [217]. In the early period of the COVID-19 outbreak, in order to find effective treatment as soon as possible, Micholas Dean Smith used to screen compounds at the interface between S protein and human ACE2 receptor by virtual screening. Results show that pemirolast has the strongest binding affinity [218]. Besides, the higher-ranking compounds include isoniazid pyruvate, nitrofurantoin, and eriodictyol. Among them, pemirolast is used to treat chronic asthma through inhibiting the antigen-induced release of inflammatory mediators [219]; isoniazid pyruvate is a metabolite of isoniazid [220]; nitrofurantoin is used to treat urinary tract infections via reduced bacterial flavoproteins to reactive intermediates, and eriodictyol is used to treat asthma via activates the expression of transcription factors in a dose-response manner [221]. Although a preliminary ranking of small molecules can be performed through large-scale virtual screening, the compounds obtained based on calculations need to be experimentally verified. The specific mechanism of action should be studied for the development of fully effective and safe COVID-19 drugs. Ribavirin is a guanine analog. The antiviral activity not only interferes with polymerase, but also directly inhibits the production of natural guanosine by inhibiting inosine monophosphate dehydrogenase directly, thereby reducing the viability of the virus [222]. Studies have reported [223] that the current three-drug combination (interferon beta-1b + lopinavir-ritonavir + ribavirin) is the safest and effective, but more clinical trial data are necessary. Drug repurposing mainly aims to enhance drug efficacy through combination therapy. There are interactions between ribavirin and multiple domains of SARS-CoV-2 infection. The reduction of TMPRSS2 expression by ribavirin can be explained as the mechanism of action against SARS-CoV-2. The repurposing strategy is related to specific mechanism of action but repurpose in different type of virus. Teicoplanin is an antibiotic used to treat staphylococcal infections. It exerts antibacterial activity by binding to lipid substrate, resulting in inhibition of bacterial peptidoglycan synthesis [224]. This drug has shown activity against a variety of viruses [225,226]. Teicoplanin blocks the virus replication by blocking the protease that mediates the cleavage of the viral S protein [227]. A recent retrospective report showed that the mortality rate of the teicoplanin group was more reduced than the control group [228], but there was no statistically significant difference. In this case, the drug repurposing strategy is based on polypharmacology properties. After evaluating the antiviral activity of several HIV-1 protease inhibitors against SARS-CoV-2 in vitro, researchers found that nelfinavir showed the lowest EC50 value and the highest selectivity index (SI). Thus, it is considered as a candidate for the treatment of COVID-19 [229]. Li et al. reported that nelfinavir is active against SARS-CoV-2 in Vero E6 cells, and it displayed multi targets properties through docking results [230]. The drug repurposing strategy of nelfinavir is based on the virtual screening of antiviral drugs. The specific mechanism of action needs further study. Researchers from the Tokyo National Institute of Infectious Diseases subsequently reported that the combined use of nelfinavir and cepharanthine showed satisfactory results [231]. But the research is currently only in vitro testing, and further research is needed.
5.1. Represented cases of drug repositioning in antivirus applications
One of the representative cases of drug repositioning in antiviral is the use of Chloroquine (CQ) to treat ZIKA. CQ is a commonly used anti-malarial drug, which has important clinical significance. Besides, CQ is also used to treat rheumatoid arthritis and other autoimmune diseases. It has been proven that CQ can reduce ZIKV-related morbidity and mortality [232]. One of the main challenges of ZIKV is congenital fetal malformations caused by an infection in pregnant women. CQ has a long and successful history, and there is no clear evidence that it can cause fetal harm when used to prevent malaria [233]. Therefore, CQ is recommended for the prevention and treatment of ZIKV. CQ hinders the fusion of flavivirus envelope protein and endosomal membrane by affecting endosomal acidification. Cell proteases are essential for the lysis of flavivirus precursor membrane (prM) protein during virus excretion. This transition is pH-dependent, and changes in intracellular pH may cause the release of fewer infectious virus particles [234].
Furthermore, studies have shown that CQ can inhibit SARS CoV-2 replication in Vitro, but it has not shown obvious advantages in vivo. It is currently proposed that the antiviral mode of action may intervene life cycle of the virus. Another suggested mechanism is to inhibit quinone reductase 2 [235], which is necessary for sialic acid, while coronaviruses are abundant in sialic acid [236]. Recent antiviral studies have shown that CQ interferes with the glycosylation process of the cellular receptor ACE2 to inhibit SARS CoV-2 from entering cells [237]. It is necessary to pay attention to the G6PD activity in the blood of patients in clinical trials. The use of CQ in patients with G6PD deficiency may bring high-risk adverse reactions [238].
6. Conclusion
Drug repositioning has become a successful drug discovery strategy due to reduced costs and faster approval procedures. Many successful examples have also shown the great potential of this method in practice. Drug repositioning has multiple advantages, including safety, money-saving market potential advantages, and return on investment potential. It is known that the safety of existing drugs reduces the risk of drug discovery, and this advantage fetches up shortcomings of drug development due to safety failures. As the cost of repositioning is much lower than the de novo drug development, it provides a new strategy for non-profit organizations and academic centers. The repositioned drug Thalomid and its derivatives have brought more than 2.8 billion U.S. dollars in revenue (https://ir.celgene.com/investors/default.aspx), this result also proves the return-on-investment potential of drug repositioning. Although drug repositioning significantly reduces the time and cost associated with the drug development process, the limitations of its application should not be underestimated. A majority of the cases only stay in experiments in vivo and in vitro. Thus, drug repositioning could be improved in many different ways. For example, computational approaches can predict new indications for old drugs and learning their intrinsic features simultaneously, which can help us to identify potential candidates and greatly improve screening efficacy by computational methods.
The rapid growth of disease and drug-related data sets in recent years has also brought rapid development to computational drug repositioning, especially structure-based drug repositioning. Strategies like machine learning and knowledge-based mapping such as network pharmacology can more accurately analyze the internal relationship between drugs and targets, thereby obtaining more meaningful information. Considering the sudden global major public health event such as COVID-19, computational drug repositioning is one of the most feasible strategies. To better understand the role of different methods in drug repositioning, and provide methodological guidance for future public health emergencies, we recommend combining experimental and computational approaches to improve the efficiency of drug repositioning. In this article, we reviewed new mechanisms of action for drugs on common diseases including neurological diseases, cancer, microbial, and virus infections. The opportunities for drug repositioning are of great potential. Although different drugs adopt different identification approaches for repositioning, most of them stay before phase II clinical trial, which is still far away from markets. A better way is to combine multiple approaches both computationally and experimentally for different situations and exploring specific mechanisms, providing novel insights and helping identify candidates for new treatment.
Notes
The authors declare no competing financial interest.
CRediT authorship contribution statement
Yi Hua: Writing – original draft. Xiaowen Dai: Data curation. Yuan Xu: Data curation. Guomeng Xing: Data curation. Haichun Liu: Data curation. Tao Lu: Writing – review & editing. Yadong Chen: Writing – review & editing. Yanmin Zhang: Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 81973182, No. 81803370), State Key Laboratory Innovation Research and Cultivation Fund (No. SKLNMZZ202018), and “Double World-classes” Construction Program of China Pharmaceutical University (No. CPU2018GF02).
Abbreviations
- AD
Alzheimer's Disease
- WHO
World Health Organization
- SP
Senile Plaque
- NFTs
Neurofibrillary Tangles
- APP
Amyloid precursor protein
- BEXA
Bexarotene
- RXR
Retinoid X Receptor
- CTCL
T-cell lymphoma
- DHEA
Dehydroepiandrosterone
- RAR
Retinoic Acid Receptor
- ApoE
Apolipoprotein E
- ARB
Angiotensin Receptor Blockers
- CCB
Calcium Channel Blockers
- SERM
Selective Estrogen Receptor Modulators
- GLP-1
Glucagon-Like Peptide-1
- nAChR
Nicotinic Acetylcholine Receptor
- ACh
Acetylcholine
- NMDA
N-methyl-d-aspartate
- PPI
Proton Pump Inhibitor
- PD
Parkinson's Disease
- ACF
Abnormal Crypt Foci
- AOM
Azoxymethane
- FASN
Fatty Acid Synthase
- DCA
Dichloroacetate
- PDK
Pyruvate Dehydrogenase Kinase
- CRC
Colorectal Cancer
- Tan IIA
Tanshinone IIA
- PPARs
Peroxisome Proliferators-Activated Receptors
- CA
Clofibric Acid
- CR
Carbonyl Reductase
- YAP1
Yes-Associated Protein 1
- AMPK
Adenosine Phosphate Active Protease
- PKC
Protein Kinase C
- DSF
Disulfiram
- ALS
Amyotrophic Lateral Sclerosis
- MAPK
Mitogen-Activated Protein Kinase
- PI3K
Phosphatidylinositol 3-Kinase
- DHODH
Dihydroorotate Dehydrogenase
- MM
Multiple Myeloma
- NPC1
Niemann-Pick C1
- NAADP
Nicotinic Acid Adenine Dinucleotide Phosphate
- ASM
Acid Sphingomyelinase
- FGF
Fibroblast Growth Factor
- VEGF
Vascular Endothelial Growth Factor
- SCV
Small-Colony Variants
- AML
Amlodipine
- SSRIs
Selective Serotonin Reuptake Inhibitors
- MRSA
Methicillin-Resistant S. aureus
- CCB
Calcium Channel Blocker
- SMON
Subacute Myeloid Optic Neuropathy
- ZIKV
ZIKA Virus
- CDK
Cycle-Dependent Kinase
- MPA
Mycophenolic Acid
- HCV
Hepatitis C Virus
- PK
Pharmacokinetics
- FQ
Ferroquine
- SAHA
Suberoylanilide Hydroxamic acid
- HCC
Hepatocellular Carcinoma
- ACE2
Angiotensin-Converting Enzyme 2
- 3Clpro
3C-Like Protease
- PARP1
Poly ADP-ribose Polymerase 1
- RdRp
RNA-dependent RNA polymerase
- prM
precursor Membrane
- CQ
Chloroquine
- G6PD
Glucose-6-Phosphate Dehydrogenase
References
- 1.Kaitin K.I. Deconstructing the drug development process: the new face of innovation. Clin. Pharmacol. Therapeut. 2010;87:356–361. doi: 10.1038/clpt.2009.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tobinick E. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009;22:53. doi: 10.1358/dnp.2009.22.2.1303818. [DOI] [PubMed] [Google Scholar]
- 3.Sleigh S.H., Barton C.L. Repurposing strategies for therapeutics. Pharmaceut. Med. 2010;24:151–159. [Google Scholar]
- 4.Novac Natalia. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013;34:267–272. doi: 10.1016/j.tips.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Xue H., Li J., Xie H., Wang Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018;14:1232–1244. doi: 10.7150/ijbs.24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clout A.E., Della Pasqua O., Hanna M.G., Orlu M., Pitceathly R.D.S. Drug repurposing in neurological diseases: an integrated approach to reduce trial and error. J. Neurol. Neurosurg. Psychiatry. 2019;90:1270. doi: 10.1136/jnnp-2019-320879. [DOI] [PubMed] [Google Scholar]
- 7.Wistedt B. A controlled study of the clinical effects of the withdrawal of depot fluphenazine decanoate and depot flupenthixol decanoate in chronic schizophrenic patients. Vol. 78. 1982. A depot neuroleptic withdrawal study; pp. 301–304. [DOI] [PubMed] [Google Scholar]
- 8.Prince M., Wimo A., Guerchet M., Ali G.-C., Wu Y.-T., Prina M. 2015. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends. [Google Scholar]
- 9.Drouin E., Drouin G. The first report of Alzheimer's disease. Lancet Neurol. 2017;16:687. doi: 10.1016/S1474-4422(17)30258-2. [DOI] [PubMed] [Google Scholar]
- 10.Swerdlow R.H. Pathogenesis of Alzheimer's disease. Clin. Interv. Aging. 2007;2:347–359. [PMC free article] [PubMed] [Google Scholar]
- 11.O'Brien R.J., Wong P.C. Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Congdon E.E., Sigurdsson E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018;14:399–415. doi: 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teich Andrew F., Arancio O. Is the Amyloid Hypothesis of Alzheimer's disease therapeutically relevant? Biochem. J. 2012;446:165–177. doi: 10.1042/BJ20120653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karran E., Mercken M., Strooper B.D., Karran E., Mercken M., De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. Nat Rev Drug Discov 10: 698-712. [DOI] [PubMed] [Google Scholar]
- 15.Dong Y., Li X., Cheng J., Hou L. Drug development for Alzheimer's disease: microglia induced neuroinflammation as a target? Int. J. Mol. Sci. 2019;20:558. doi: 10.3390/ijms20030558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshitake T., Kiyohara Y., Kato I., Ohmura T., Iwamoto H., Nakayama K., Ohmori S., Nomiyama K., Kawano H., Ueda K. Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population: the Hisayama Study. Neurology. 1995;45:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]
- 17.Skoog I., Nilsson L., Persson G., Lernfelt B., Landahl S., Palmertz B., Andreasson L.A., Odén A., Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 18.Corbett A., Pickett J., Burns A., Corcoran J., Dunnett S.B., Edison P., Hagan J.J., Holmes C., Jones E., Katona C., Kearns I., Kehoe P., Mudher A., Passmore A., Shepherd N., Walsh F., Ballard C. Drug repositioning for Alzheimer's disease. Nat. Rev. Drug Discov. 2012;11:833–846. doi: 10.1038/nrd3869. [DOI] [PubMed] [Google Scholar]
- 19.Monacelli F., Cea M., Borghi R., Odetti P., Nencioni A. Do cancer drugs counteract neurodegeneration? Repurposing for Alzheimer's disease. J. Alzheim. Dis. 2017;55:1295–1306. doi: 10.3233/JAD-160840. [DOI] [PubMed] [Google Scholar]
- 20.Guo J., Cheng J., North B.J., Wei W. Functional analyses of major cancer-related signaling pathways in Alzheimer's disease etiology. Biochim. Biophys. Acta Rev. Canc. 2017;1868:341–358. doi: 10.1016/j.bbcan.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brent T.P., Remack J.S. Formation of covalent complexes between human O 6 alkyltransferase and BCNU-treated defined length synthetic oligodeoxynucleotides. Nucleic Acids Res. 1988;16:6779–6788. doi: 10.1093/nar/16.14.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueda-Kawamitsu H., Lawson T.A., Gwilt P.R. In vitro pharmacokinetics and pharmacodynamics of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) Biochem. Pharmacol. 2002;63:1209–1218. doi: 10.1016/s0006-2952(02)00878-x. [DOI] [PubMed] [Google Scholar]
- 23.Hayes C., Dey D., Palavicini J., Wang H., Patkar K., Minond D., Nefzi A., Lakshmana M. Striking reduction of amyloid plaque burden in an Alzheimer's mouse model after chronic administration of carmustine. BMC Med. 2013;11:81. doi: 10.1186/1741-7015-11-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu L., Tang X. Bexarotene: a promising anticancer agent. Cancer Chemother. Pharmacol. 2010;65:201–205. doi: 10.1007/s00280-009-1140-4. [DOI] [PubMed] [Google Scholar]
- 25.Pathak K., Gaire T. Therapeutic links between anti-cancer drugs (Bexarotene) and ALZHEIMER’S disease. Alzheimer's Dementia. 2016;12:P836. [Google Scholar]
- 26.Tobita T. Treatment with a new synthetic retinoid, Am80, of acute promyelocytic leukemia relapsed from complete remission induced by all-trans retinoic acid. Blood. 1997;90 [PubMed] [Google Scholar]
- 27.Qiao A., Li J., Hu Y., Wang J., Zhao Z. Reduction BACE1 expression via suppressing NF-κB mediated signaling by Tamibarotene in a mouse model of Alzheimer's disease. IBRO Neurosci. Rep. 2021;10:153–160. doi: 10.1016/j.ibneur.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin X., Chen C., Zhang Y., Zhang L., Mei Y., Long X., Tan R., Liang W., Sun L. Acitretin modulates HaCaT cells proliferation through STAT1-and STAT3-dependent signaling. Saudi Pharmaceut. J. 2017;25:620–624. doi: 10.1016/j.jsps.2017.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endres K., Fahrenholz F., Lotz J., Hiemke C., Fellgiebel A. Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology. 2014;83:1930–1935. doi: 10.1212/WNL.0000000000001017. [DOI] [PubMed] [Google Scholar]
- 30.Baccarani M., Saglio G., Goldman J., Hochhaus A., Hehlmann R. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2007;108:1809–1820. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 31.Ben Ami E., Demetri G.D. A safety evaluation of imatinib mesylate in the treatment of gastrointestinal stromal tumor. Expet Opin. Drug Saf. 2016;15:571–578. doi: 10.1517/14740338.2016.1152258. [DOI] [PubMed] [Google Scholar]
- 32.Cancino G.I., Arce K., Castro P.U., Toledo E.M., Alvarez A.R. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging. 2011;32:1249–1261. doi: 10.1016/j.neurobiolaging.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Wang J., Ho L., Chen L., Zhao Z., Wei Z., Qian X., Humala N., Seror I., Bartholomew S., Rosendorff C. Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J. Clin. Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang W.N., Hu X.D., Han H., Shi L.L., Feng G.F., Liu Y., Qian Y.H. The effects of valsartan on cognitive deficits induced by aluminum trichloride and d-galactose in mice. Neurol. Res. 2014;36:651–658. doi: 10.1179/1743132813Y.0000000295. [DOI] [PubMed] [Google Scholar]
- 35.Bachmeier C., Beaulieu-Abdelahad D., Mullan M., Paris D. Selective dihydropyiridine compounds facilitate the clearance of β-amyloid across the blood-brain barrier. Eur. J. Pharmacol. 2011;659:124–129. doi: 10.1016/j.ejphar.2011.03.048. [DOI] [PubMed] [Google Scholar]
- 36.Paris D., Bachmeier C., Patel N., Quadros A., Volmar C.H., Laporte V., Ganey J., Beaulieu-Abdelahad D., Ait-Ghezala G., Crawford F. Selective antihypertensive dihydropyridines lower Aβ accumulation by targeting both the production and the clearance of Aβ across the blood-brain barrier. Mol. Med. 2011;17:149. doi: 10.2119/molmed.2010.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanhanen H., Fa Gard R., Tuomilehto J., Birkenhäger W., Seux M.L., Staessen J.A., Babeanu S., Webster J., Yodfat Y., Forette F. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch. Intern. Med. 2003;12:32–33. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- 38.Batista A.F., Forny-Germano L., Clarke J.R., Silva N., Brito-Moreira J., Boehnke S.E., Winterborn A., Coe B.C., Lablans A., Vital J.F. Wiley-Blackwell Online Open; 2018. The Diabetes Drug Liraglutide Reverses Cognitive Impairment in Mice and Attenuates Insulin Receptor and Synaptic Pathology in a Non-human Primate Model of Alzheimer's Disease; p. 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiciński M., Socha M., Malinowski B., Wódkiewicz E., Walczak M., Górski K., Słupski M., Pawlak-Osińska K. Liraglutide and its Neuroprotective properties—focus on possible biochemical mechanisms in Alzheimer's disease and cerebral ischemic events. Int. J. Mol. Sci. 2019;20:1050. doi: 10.3390/ijms20051050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seabrook T.J., Jiang L., Maier M., Lemere C.A. Minocycline affects microglia activation, Abeta deposition, and behavior in APP-tg mice. Glia. 2006;53:776–782. doi: 10.1002/glia.20338. [DOI] [PubMed] [Google Scholar]
- 41.Biscaro B., Lindvall O., Tesco G., Ekdahl C.T., Nitsch R.M. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer's disease. Neurodegener. Dis. 2012;9 doi: 10.1159/000330363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landmark K., Forsman M., Lindberg K., Ryman T., Martmann-Moe K., Haaverstad S., Wiel S. Nitrendipine and mefruside in elderly hypertensives: effects on blood pressure, cardiac output, cerebral blood flow and metabolic parameters. J. Hum. Hypertens. 1995;9:281–285. [PubMed] [Google Scholar]
- 43.Mucke H. The case of galantamine: repurposing and late blooming of a cholinergic drug. Futur. Sci. OA. 2015;1 doi: 10.4155/fso.15.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irwin R.L., Smith H.J. Cholinesterase inhibition by galanthamine and lycoramine. Biochem. Pharmacol. 1960;3:147–148. doi: 10.1016/0006-2952(60)90030-7. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney J.E., Bachman E.S., Coyle J.T. Effects of different doses of galanthamine, a long-acting acetylcholinesterase inhibitor, on memory in mice. Psychopharmacology. 1990;102:191–200. doi: 10.1007/BF02245921. [DOI] [PubMed] [Google Scholar]
- 46.Khan M.B., Palaka B.K., Sapam T.D., Subbarao N., Ampasala D.R. Screening and analysis of acetyl-cholinesterase (AChE) inhibitors in the context of Alzheimer's disease. Bioinformation. 2018;14:414–428. doi: 10.6026/97320630014414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson C.A. FDA approves galantamine for Alzheimer's disease. Am. J. Health Syst. Pharm. 2001;58 doi: 10.1093/ajhp/58.8.649a. 649-649. [DOI] [PubMed] [Google Scholar]
- 48.Galantamine Additional benefits to patients with Alzheimer's disease. Dement. Geriatr. Cognit. Disord. 2000;11:19–27. doi: 10.1159/000051228. [DOI] [PubMed] [Google Scholar]
- 49.Levin E.D., Simon B.B. Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology. 1998;138:217–230. doi: 10.1007/s002130050667. [DOI] [PubMed] [Google Scholar]
- 50.Wilkinson G. David. Galantamine: a new treatment for Alzheimer's disease. Expert Rev. Neurother. 2001;1:153. doi: 10.1586/14737175.1.2.153. [DOI] [PubMed] [Google Scholar]
- 51.Krzystanek M., Pałasz A. Possibility of a new indication for amantadine in the treatment of bipolar depression-case series study. Pharmaceuticals. 2020;13 doi: 10.3390/ph13100326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sawyer E., Mauro L.S., Ohlinger M.J. Amantadine enhancement of arousal and cognition after traumatic brain injury. Ann. Pharmacother. 2008;42:247–252. doi: 10.1345/aph.1K284. [DOI] [PubMed] [Google Scholar]
- 53.Schwab R.S., England A.C., Poskanzer D.C., Young R.R. Amantadine in the treatment of Parkinson's disease. JAMA. 1969;208:1168–1170. [PubMed] [Google Scholar]
- 54.Stoof J.C., Booij J., Drukarch B. Amantadine as N-methyl-D-aspartic acid receptor antagonist: new possibilities for therapeutic applications? Clin. Neurol. Neurosurg. 1992;94:S4–S6. doi: 10.1016/0303-8467(92)90006-o. [DOI] [PubMed] [Google Scholar]
- 55.Shahnawaz Khan M., Tabrez S., Priyadarshini M., Priyamvada S., M Khan M.J.C., Targets N.D.-D. Vol. 11. 2012. pp. 369–380. (Targeting Parkinson's-Tyrosine Hydroxylase and Oxidative Stress as Points of Interventions). [DOI] [PubMed] [Google Scholar]
- 56.Rascol O., Fabbri M., Poewe W. Vol. 20. 2021. Amantadine in the Treatment of Parkinson's Disease and Other Movement Disorders; pp. 1048–1056. [DOI] [PubMed] [Google Scholar]
- 57.Sharma V.D., Lyons K.E., Pahwa R. Amantadine extended-release capsules for levodopa-induced dyskinesia in patients with Parkinson's disease. Therapeut. Clin. Risk Manag. 2018;14:665–673. doi: 10.2147/TCRM.S144481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nowak-Sliwinska P., Scapozza L., Altaba A. Drug repurposing in oncology: compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Rev. Canc. 2019;2:434–454. doi: 10.1016/j.bbcan.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corsello S.M., Nagari R.T., Spangler R.D., Rossen J., Kocak M., Bryan J.G., Humeidi R., Peck D., Wu X., Tang A.A., Wang V.M., Bender S.A., Lemire E., Narayan R., Montgomery P., Ben-David U., Garvie C.W., Chen Y., Rees M.G., Lyons N.J., McFarland J.M., Wong B.T., Wang L., Dumont N., O'Hearn P.J., Stefan E., Doench J.G., Harrington C.N., Greulich H., Meyerson M., Vazquez F., Subramanian A., Roth J.A., Bittker J.A., Boehm J.S., Mader C.C., Tsherniak A., Golub T.R. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Can. 2020;1:235–248. doi: 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naccarati A., Polakova B., Fau Pardini V., Pardini L., Fau Vodickova B., Vodickova K., Fau Hemminki L., Hemminki R., Fau Kumar K., Kumar P., Fau Vodicka R., Vodicka P. Vol. 2. 2012. pp. 211–218. (Mutations and Polymorphisms in TP53 Gene-Aan Overview on the Role in Colorectal Cancer). [DOI] [PubMed] [Google Scholar]
- 61.Lee W.Y., Lee W.T., Cheng C.H., Chen K.C., Lin C.W. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget. 2015;6:27580–27595. doi: 10.18632/oncotarget.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lan S., Xiang S., Fu Z., Yang G., Li J. The fundamental role of the p53 pathway in tumor metabolism and its implication in tumor therapy. Clin. Can. Res. Official J. Am. Assoc. Can. Res. 2012;18:1561–1567. doi: 10.1158/1078-0432.CCR-11-3040. [DOI] [PubMed] [Google Scholar]
- 63.Lamb J. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 64.Chen M.H., Yang Wl Fau - Lin K.-T., Lin Kt Fau - Liu C.-H., Liu Ch Fau - Liu Y.-W., Liu Yw Fau - Huang K.-W., Huang Kw Fau - Chang P.M.-H., Chang Pm Fau - Lai J.-M., Lai Jm Fau - Hsu C.-N., Hsu Cn Fau - Chao K.-M., Chao Km Fau - Kao C.-Y., Kao Cy Fau - Huang C.-Y.F., Huang C.Y. Vol. 6. 2011. p. 27186. (Gene Expression-Based Chemical Genomics Identifies Potential Therapeutic Drugs in Hepatocellular Carcinoma). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nyce J.W., Magee P.N., Hard G.C., Schwartz A.G. Inhibition of 1,2-dimethylhydrazine-induced colon tumorigenesis in Balb/c mice by dehydroepiandrosterone. Carcinogenesis. 1984:57–62. doi: 10.1093/carcin/5.1.57. [DOI] [PubMed] [Google Scholar]
- 66.Chuang H.Y., Chang Y.F., Hwang J.J. Antitumor effect of orlistat, a fatty acid synthase inhibitor, is via activation of caspase-3 on human colorectal carcinoma-bearing animal. Biomed. Pharmacother. 2011;65:286–292. doi: 10.1016/j.biopha.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 67.Jin B.-R., Kim H.-J., Sim S.-A., Lee M., An H.-J. Anti-obesity drug orlistat alleviates Western-Diet-Driven colitis-associated colon cancer via inhibition of STAT3 and NF-κB-Mediated signaling. Cells. 2021;10:2060. doi: 10.3390/cells10082060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeng X., Liu L., Zheng M., Sun H., Duan Q. Pantoprazole, an FDA-approved proton-pump inhibitor, suppresses colorectal cancer growth by targeting T-cell-originated protein kinase. Oncotarget. 2016;7:22460–22473. doi: 10.18632/oncotarget.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Warburg O. [Origin of cancer cells] Oncologia. 1956;9:75–83. [PubMed] [Google Scholar]
- 70.Ho N., Coomber B.L. Pyruvate dehydrogenase kinase expression and metabolic changes following dichloroacetate exposure in anoxic human colorectal cancer cells. Exp. Cell Res. 2015;331:73–81. doi: 10.1016/j.yexcr.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Janakiram N.B., Steele V.E., Rao C.V. Estrogen receptor-beta as a potential target for colon cancer prevention: chemoprevention of azoxymethane-induced colon carcinogenesis by raloxifene in F344 rats. Cancer Prev. Res. 2009;2:52–59. doi: 10.1158/1940-6207.CAPR-08-0140. [DOI] [PubMed] [Google Scholar]
- 72.Moore N., Houghton J., Lyle S. Slow-cycling therapy-resistant cancer cells. Stem Cell. Dev. 2012;21:1822–1830. doi: 10.1089/scd.2011.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Venkatakrishnan K., Von Moltke L.L., Greenblatt D.J. Effects of the antifungal agents on oxidative drug metabolism. Clin. Pharmacokinet. 2000;38:111–180. doi: 10.2165/00003088-200038020-00002. [DOI] [PubMed] [Google Scholar]
- 74.Buczacki S.J.A., Semiramis P., Emma B., Chrysa K., Jon B., Louis V., Lee H., Hayley F., Garnett M.J., Winton D.J. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J. Exp. Med. 2018 doi: 10.1084/jem.20171385. jem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Popova S.A., Buczacki S.J. Itraconazole perturbs colorectal cancer dormancy through SUFU-mediated WNT inhibition. Molecul. Cell. Oncol. 2018;5 doi: 10.1080/23723556.2018.1494950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y., Zhang L., Chu W., Wang B., Zhang J., Zhao M., Li X., Li B., Lu Y., Yang B. Tanshinone IIA inhibits miR-1 expression through p38 MAPK signal pathway in post-infarction rat cardiomyocytes. Cell. Physiol. Biochem. 2010;26:991–998. doi: 10.1159/000324012. [DOI] [PubMed] [Google Scholar]
- 77.Jieensinue S., Zhu H., Li G., Dong K., Liang M., Li Y. Tanshinone IIA reduces SW837 colorectal cancer cell viability via the promotion of mitochondrial fission by activating JNK-Mff signaling pathways. BMC Cell Biol. 2018;19:21. doi: 10.1186/s12860-018-0174-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qian J., Cao Y., Zhang J., Li L., Wu J., Wei G., Yu J., Huo J. Tanshinone IIA induces autophagy in colon cancer cells through MEK/ERK/mTOR pathway. Transl. Cancer Res. 2020;9:6919–6928. doi: 10.21037/tcr-20-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kussie P.H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A.J. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 80.Patil S.P., Pacitti M.F., Gilroy K.S., Ruggiero J.C., Griffin J.D., Butera J.J., Notarfrancesco J.M., Tran S., Stoddart J.W. Identification of antipsychotic drug fluspirilene as a potential p53-MDM2 inhibitor: a combined computational and experimental study. J. Comput. Aided Mol. Des. 2015;29:155–163. doi: 10.1007/s10822-014-9811-6. [DOI] [PubMed] [Google Scholar]
- 81.Weiderpass E., Labrèche F. Malignant tumors of the female reproductive system. Saf. Health Work. 2012;3:166–180. doi: 10.5491/SHAW.2012.3.3.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tyagi S., Gupta P., Saini A.S., Kaushal C., Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. "J. Adv. Pharm. Technol. Research"" (JAPTR)". 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balakumar P., Rose M., Ganti S.S., Krishan P., Singh M. PPAR dual agonists: are they opening Pandora's Box? Pharmacol. Res. 2007;56:91–98. doi: 10.1016/j.phrs.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 84.Issemann I., Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 85.Nicol C.J., Yoon M., Ward J.M., Yamashita M., Fukamachi K., Peters J.M., Gonzalez F.J. PPARγ influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- 86.Banno K., Iida M., Yanokura M., Irie H., Masuda K., Kobayashi Y., Tominaga E., Aoki D. Drug repositioning for gynecologic tumors: a new therapeutic strategy for cancer. Sci. World J. 2015 doi: 10.1155/2015/341362. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yokoyama Y., Xin B., Shigeto T., Umemoto M., Kasai-Sakamoto A., Futagami M., Tsuchida S., Al-Mulla F., Mizunuma H. Clofibric acid, a peroxisome proliferator–activated receptor α ligand, inhibits growth of human ovarian cancer. Mol. Cancer Therapeut. 2007;6:1379. doi: 10.1158/1535-7163.MCT-06-0722. [DOI] [PubMed] [Google Scholar]
- 88.Yokoyama Y., Shigeto T., Miura R., Kobayashi A., Mizunuma M., Yamauchi A., Futagami M., Mizunuma H. A strategy using photodynamic therapy and clofibric acid to treat peritoneal dissemination of ovarian cancer. Asian Pac. J. Cancer Prev. APJCP. 2016;17:775–779. doi: 10.7314/apjcp.2016.17.2.775. [DOI] [PubMed] [Google Scholar]
- 89.Dewan Z.M. Efficient intervention of growth and infiltration of primary adult T-cell leukemia cells by an HIV protease inhibitor, ritonavir. Blood. 2006;107:716–724. doi: 10.1182/blood-2005-02-0735. [DOI] [PubMed] [Google Scholar]
- 90.Ikezoe T., Daar E.S., Hisatake J., Taguchi H., Koeffler H.P. HIV-1 protease inhibitors decrease proliferation and induce differentiation of human myelocytic leukemia cells. Blood. 2000;96:3553. [PubMed] [Google Scholar]
- 91.Kumar S., Bryant C.S., Chamala S., Qazi A., Seward S., Pal J., Steffes C.P., Weaver D.W., Morris R., Malone J.M. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol. Cancer. 2009;8 doi: 10.1186/1476-4598-8-26. 26-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kobayashi Y., Kashima H., Wu R.-C., Jung J.-G., Kuan J.-C., Gu J., Xuan J., Sokoll L., Visvanathan K., Shih I.-M., Wang T.-L. Mevalonate pathway antagonist suppresses formation of serous tubal intraepithelial carcinoma and ovarian carcinoma in mouse models. Clin. Cancer Res. 2015;21:4652. doi: 10.1158/1078-0432.CCR-14-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hashimoto K., Morishige K.I., Sawada K., Tahara M., Murata Y. Alendronate inhibits intraperitoneal dissemination in in vivo ovarian cancer model. Cancer Res. 2005;65:540–545. [PubMed] [Google Scholar]
- 94.Kobayashi Y., Kashima H., Rahmanto Y.S., Banno K., Yu Y., Matoba Y., Watanabe K., Iijima M., Takeda T., Kunitomi H., Iida M., Adachi M., Nakamura K., Tsuji K., Masuda K., Nomura H., Tominaga E., Aoki D. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget. 2017;8:72147–72156. doi: 10.18632/oncotarget.20046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goldstein J.L., Brown M.S. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 96.Wong W.L., Dimitroulakos J., Minden M., Penn L.Z. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16:508–519. doi: 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- 97.Kornblau S.M., Banker D.E., Stirewalt D., Shen D., Lemker E., Verstovsek S., Estrov Z., Faderl S., Cortes J., Beran M. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood. 2007;109:2999. doi: 10.1182/blood-2006-08-044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sassano A., Platanias L.C. Statins in tumor suppression. Cancer Lett. 2008;260:11–19. doi: 10.1016/j.canlet.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 99.Schmidmaier R., Baumann I., Fau Bumeder P., Bumeder G., Fau Meinhardt I., Meinhardt C., Fau Straka G., Straka B., Fau Emmerich C., Emmerich B. Vol. 79. 2007. pp. 240–243. (First Clinical Experience with Simvastatin to Overcome Drug Resistance in Refractory Multiple Myeloma). [DOI] [PubMed] [Google Scholar]
- 100.Martirosyan A., Clendening J.W., Goard C.A., Penn L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103. doi: 10.1186/1471-2407-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X., Song Y., Ci X., An N., Ju Y., Li H., Wang X., Han C., Cui J., Deng X. Ivermectin inhibits LPS-induced production of inflammatory cytokines and improves LPS-induced survival in mice. Inflamm. Res. 2008;57:524–529. doi: 10.1007/s00011-008-8007-8. [DOI] [PubMed] [Google Scholar]
- 102.Kodama M., Kodama T., Newberg J.Y., Katayama H., Kobayashi M., Hanash S.M., Yoshihara K., Wei Z., Tien J.C., Rangel R., Hashimoto K., Mabuchi S., Sawada K., Kimura T., Copeland N.G., Jenkins N.A. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene. Epithelial Ovarian Can. 2017;114:7301–7310. doi: 10.1073/pnas.1705441114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang M., Hu X., Wang Y., Yao X., Fang Q. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol. Res. 2020;163 doi: 10.1016/j.phrs.2020.105207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bowker S.L., Majumdar S.R., Veugelers P., Johnson J.A. Increased cancer-related mortality for patients with type 2 diabetes Who use sulfonylureas or insulin. Diabetes Care. 2006;29:254. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 105.Landman G.W.D., Kleefstra N., van Hateren K.J.J., Groenier K.H., Gans R.O.B., Bilo H.J.G. Metformin associated with lower cancer mortality in type 2 diabetes. Diabetes Care. 2010;33:322. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Libby G., Donnelly L., Donnan P., Alessi D., Morris A., Evans J. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leonardi G.C., Falzone L., Salemi R., Zanghì A., Spandidos D.A., McCubrey J.A., Candido S., Libra M. Vol. 52. 2018. Cutaneous Melanoma: From Pathogenesis to Therapy (Review) pp. 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Niu G., Bowman M., Fau Huang T., Huang S., Fau Shivers M., Shivers D., Fau Reintgen S., Reintgen A., Fau Daud D., Daud A., Fau Chang A., Chang A., Fau Kraker A., Kraker R., Fau Jove A., Jove H., Fau Yu R., Yu H. Vol. 21. 2002. pp. 7001–7010. (Roles of Activated Src and Stat3 Signaling in Melanoma Tumor Cell Growth). [DOI] [PubMed] [Google Scholar]
- 109.McCusker Jp D.M., Yan R., He S., Dordick J.S., McGuinness D.L. Finding melanoma drugs through a probabilistic knowledge graph. Peer J. Comp. Sci. 2017;3:e106. doi: 10.7717/peerj-cs.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Riedel T., Demaria O., Zava O., Joncic A., Gilliet M., Dyson P. Drug repurposing approach identifies a synergistic drug combination of an antifungal agent and an experimental organometallic drug for melanoma treatment. Mol. Pharm. 2017;15 doi: 10.1021/acs.molpharmaceut.7b00764. [DOI] [PubMed] [Google Scholar]
- 111.Giovanelli A., Silva Cl Fau Medeiros L., Medeiros M.C.d., Fau Vasconcellos L., Vasconcellos M.C. The molluscicidal activity of niclosamide (Bayluscide WP70(R)) on Melanoides tuberculata (Thiaridae), a snail associated with habitats of Biomphalaria glabrata (Planorbidae) Mem. Inst. Oswaldo Cruz. 2002;97:743–745. doi: 10.1590/s0074-02762002000500027. [DOI] [PubMed] [Google Scholar]
- 112.Enna S.J., Bylund D.B. In: xPharm: the Comprehensive Pharmacology Reference. Enna S.J., Bylund D.B., editors. Elsevier; New York: 2007. Niclosamide; pp. 1–2. [Google Scholar]
- 113.Zhu Y., Zuo W., Chen L., Bian S., Jing J., Gan C., Wu X., Liu H., Su X., Hu W., Guo Y., Wang Y., Ye T. Repurposing of the anti-helminthic drug niclosamide to treat melanoma and pulmonary metastasis via the STAT3 signaling pathway. Biochem. Pharmacol. 2019;169 doi: 10.1016/j.bcp.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 114.Ren X., Duan L., He Q., Zhang Z., Zhou Y., Wu D., Pan J., Pei D., Ding K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med. Chem. Lett. 2010;1:454–459. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pampori N.A., Govind S., Srivastava V. Cotugnia digonopora: carbohydrate metabolism and effect of anthelmintics on immature worms. J. Helminthol. 1984;58:39–47. doi: 10.1017/s0022149x00028042. [DOI] [PubMed] [Google Scholar]
- 116.Chen H., Yang Z., Ding C., Chu L., Zhang Y., Terry K., Liu H., Qiang S., Jia Z. Discovery of O-Alkylamino-Tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med. Chem. Lett. 2013;4 doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goenka S., Simon S.R. Organogold drug Auranofin exhibits anti-melanogenic activity in B16F10 and MNT-1 melanoma cells. Arch. Dermatol. Res. 2019:312. doi: 10.1007/s00403-019-01974-1. [DOI] [PubMed] [Google Scholar]
- 118.None Spontaneous regression of breast cancer. Arch. Intern. Med. 2009;169:47. doi: 10.1001/archinternmed.2009.89. [DOI] [PubMed] [Google Scholar]
- 119.Morrison B.W., Doudican N.A., Patel K.R., Orlow S.J. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010;20:11. doi: 10.1097/CMR.0b013e328334131d. [DOI] [PubMed] [Google Scholar]
- 120.Cen D., Gonzalez R.I., Buckmeier J.A., Kahlon R.S., Tohidian N.B., Meyskens F.L. Disulfiram induces apoptosis in human melanoma cells: a redox-related process. Mol. Cancer Therapeut. 2002;1:197–204. [PubMed] [Google Scholar]
- 121.Rg M., Jd M., Lyon M., Dh M. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND), Amyotrophic Lateral Sclerosis & Other Motor. Neuron Disorders. 2000;4:191–206. [PubMed] [Google Scholar]
- 122.Namkoong Shin S.S., Lee H.J., Marin Y.E., Wall B.A. Goydos, Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67(5):2298–2305. doi: 10.1158/0008-5472.CAN-06-3665. 2007. [DOI] [PubMed] [Google Scholar]
- 123.Mehnert J.M., Wen Y., Lee J.H., Dudek L., Goydos J.S. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011;29:8557. doi: 10.1111/pcmr.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kaplan M.J. Leflunomide Aventis pharma. Curr. Opin. Invest. Drugs. 2001;2:222–230. [PubMed] [Google Scholar]
- 125.Mclean J.E., Neidhardt E., Grossman T.H., Hedstrom L. Multiple inhibitor analysis of the brequinar and leflunomide binding sites on human dihydroorotate dehydrogenase. Biochemistry. 2001;40:2194–2200. doi: 10.1021/bi001810q. [DOI] [PubMed] [Google Scholar]
- 126.Schiff M.H., Strand V., Oed C., Loew-Friedrich I. Leflunomide: efficacy and safety in clinical trials for the treatment of rheumatoid arthritis. Drugs Today. 2000;36:383–394. doi: 10.1358/dot.2000.36.6.584259. [DOI] [PubMed] [Google Scholar]
- 127.White R.M., Cech J., Ratanasirintrawoot S., Lin C.Y., Rahl P.B., Burke C.J., Langdon E., Tomlinson M.L., Mosher J., Kaufman C. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2015;471:518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Riedel T., Demaria O., Zava O., Joncic A., Gilliet M., Dyson P.J. Drug repurposing approach identifies a synergistic drug combination of an antifungal agent and an experimental organometallic drug for melanoma treatment. Mol. Pharm. 2017:116–126. doi: 10.1021/acs.molpharmaceut.7b00764. [DOI] [PubMed] [Google Scholar]
- 129.Hideshima T., Anderson K.C. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat. Rev. Cancer. 2002;2:927–937. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- 130.Singhal S., Mehta R., Fau Desikan J., Desikan D., Fau Ayers R., Ayers P., Fau Roberson D., Roberson P., Fau Eddlemon P., Eddlemon N., Fau Munshi P., Munshi E., Fau Anaissie N., Anaissie C., Fau Wilson E., Wilson M., Fau Dhodapkar C., Dhodapkar J., Fau Zeddis M., Zeddis B., Fau Barlogie J., Barlogie B. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999;341:1565–1571. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 131.Gupta D., Treon S., Shima Y., Hideshima T., Podar K., Tai Y., Lin B., Lentzsch S., Davies F., Chauhan D. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15:1950–1961. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- 132.Miao X., Yu H., Lei S., Peng J. Therapeutic effects of thalidomide in hematologic disorders: a review. Front. Med. 2013;7 doi: 10.1007/s11684-013-0277-z. [DOI] [PubMed] [Google Scholar]
- 133.Ray A., Rice L.B. Wildcatters welcome: the need for new antimicrobial agents. Therapy. 2004;1:1–5. [Google Scholar]
- 134.Jackson N., Czaplewski L., Piddock L.J.V. Discovery and development of new antibacterial drugs: learning from experience? J. Antimicrob. Chemother. 2018;73:1452–1459. doi: 10.1093/jac/dky019. [DOI] [PubMed] [Google Scholar]
- 135.Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2006;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 136.Tande A.J., Osmon D.R., Greenwood-Quaintance K.E., Mabry T.M., Hanssen A.D., Patel R. Clinical characteristics and outcomes of prosthetic joint infection caused by small colony variant staphylococci. mBio. 2014;5 doi: 10.1128/mBio.01910-14. e01910-01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Trombetta R.P., Dunman P.M., Schwarz E.M., Kates S.L., Awad H.A.J.M. A high-throughput screening approach to repurpose FDA-approved drugs for bactericidal applications against. Staphylococcus Aureus Small-colony Variants. 2018;3:422–450. doi: 10.1128/mSphere.00422-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Antoniani D., Rossi E., Rinaldo S. The immunosuppressive drug azathioprine inhibits biosynthesis of the bacterial signal molecule cyclic-di-GMP by interfering with intracellular nucleotide pool availability. Appl. Microbiol. Biotechnol. 2013;97:7325–7336. doi: 10.1007/s00253-013-4875-0. [DOI] [PubMed] [Google Scholar]
- 139.Gooyit M., Janda K.D. Reprofiled anthelmintics abate hypervirulent stationary-phase Clostridium difficile. Sci. Rep. 2016;6:33642. doi: 10.1038/srep33642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ayerbe-Algaba R., Gil-Marqués M.L., Jiménez-Mejías M.E., Sánchez-Encinales V., Parra-Millán R., Pachón-Ibáñez M.E., Pachón J., Smani Y. Synergistic activity of niclosamide in combination with colistin against colistin-susceptible and colistin-resistant Acinetobacter baumannii and Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018;8 doi: 10.3389/fcimb.2018.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mazumdar K., Kumar K.A., Dutta N.K. Potential role of the cardiovascular non-antibiotic (helper compound) amlodipine in the treatment of microbial infections: scope and hope for the future. Int. J. Antimicrob. Agents. 2010;36:295–302. doi: 10.1016/j.ijantimicag.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 142.Kumar K.A., Ganguly K., Mazumdar K., Dutta N.K., Dastidar S.G., Chakrabarty A.N. Amlodipine: a cardiovascular drug with powerful antimicrobial property. Acta Microbiol. Pol. 2003;52:285–292. [PubMed] [Google Scholar]
- 143.Foye W.O. Lippincott williams & wilkins; 2008. Foye's Principles of Medicinal Chemistry. [Google Scholar]
- 144.Farha M.A., Brown E.D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 2019;4:565–577. doi: 10.1038/s41564-019-0357-1. [DOI] [PubMed] [Google Scholar]
- 145.Zhai B., Wu C., Wang L., Sachs M.S., Lin X. The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrob. Agents Chemother. 2012;56:3758–3766. doi: 10.1128/AAC.00212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Silva R.C., Silva C., Batista D., Silva A., Campos R.S., Sampaio L.S., Nascimento F.D., Gaspar B.S., Fonseca S.C., Josino M. In vitro anti-Candida activity of selective serotonin reuptake inhibitors against fluconazole-resistant strains and their activity against biofilm-forming isolates. Microb. Pathog. 2017;107:341. doi: 10.1016/j.micpath.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 147.Sedlacek M., Gemery J.M., Cheung A.L., Bayer A.S., Remillard B.D. Aspirin treatment is associated with a significantly decreased risk of Staphylococcus aureus bacteremia in hemodialysis patients with tunneled catheters. Am. J. Kidney Diseases Off. J. Nat. Kidney Found. 2007;49:401–408. doi: 10.1053/j.ajkd.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 148.Alem M., Douglas L.J. Effects of aspirin and other nonsteroidal anti-inflammatory drugs on biofilms and planktonic cells of Candida albicans. Antimicrob. Agents Chemother. 2004;48:41–47. doi: 10.1128/AAC.48.1.41-47.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ehmann T.S., Delva N.J., Beninger R.J. Flupenthixol in chronic schizophrenic inpatients: a controlled comparison with haloperidol. J. Clin. Psychopharmacol. 1987;7:173–175. [PubMed] [Google Scholar]
- 150.Reilly J.G., Ayis S.A., Ferrier I.N., Jones S.J., Thomas S. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048–1052. doi: 10.1016/s0140-6736(00)02035-3. [DOI] [PubMed] [Google Scholar]
- 151.Thanacoody H.K.R. Thioridazine: resurrection as an antimicrobial agent? Br. J. Clin. Pharmacol. 2010;64:566–574. doi: 10.1111/j.1365-2125.2007.03021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen R.E., Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2007;1773:1311. doi: 10.1016/j.bbamcr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dannaoui E., Schwarz P., Lortholary O. In vitro interactions between antifungals and immunosuppressive drugs against zygomycetes. Antimicrob. Agents Chemother. 2009;53:3549–3551. doi: 10.1128/AAC.00184-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Basu L.R., Mazumdar K., Dutta N.K., Karak P., Dastidar S.G. Antibacterial property of the antipsychotic agent prochlorperazine, and its synergism with methdilazine. Microbiol. Res. 2005;160:95–100. doi: 10.1016/j.micres.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 155.Kaatz G.W., Moudgal V.V., Seo S.M., Kristiansen J.E. Phenothiazines and Thioxanthenes inhibit multidrug Efflux pump activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 2003;47:719. doi: 10.1128/AAC.47.2.719-726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cristofori P., Lanzoni A., Quartaroli M., Pastorino A.M., Zan Ca Naro C., Cominacini L., Gaviraghi G., Turton J. The calcium-channel blocker lacidipine reduces the development of atherosclerotic lesions in the apoE-deficient mouse. J. Hypertens. 2000;18:1429–1436. doi: 10.1097/00004872-200018100-00010. [DOI] [PubMed] [Google Scholar]
- 157.Dasgupta A., Jeyaseeli L., Dutta N.K., Mazumdar K., Shirataki Y. Studies on the antimicrobial potential of the cardiovascular drug lacidipine. vivo (Athens, Greece) 2007;21:847–850. [PubMed] [Google Scholar]
- 158.Truong M., Monahan L.G., Carter D., Charles I.G. Repurposing drugs to fast-track therapeutic agents for the treatment of cryptococcosis. PeerJ. 2018;6 doi: 10.7717/peerj.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sweta Samantaray, Joao N., Correia Mariam, Garelnabi Kerstin, Voelz Robin. Novel cell-based in vitro screen to identify small-molecule inhibitors against intracellular replication of Cryptococcus neoformans in macrophages. Int. J. Antimicrob. Agents. 2016;48:69–77. doi: 10.1016/j.ijantimicag.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Horwitz J.P., Chua J., Noel M. Nucleosides. V. The Monomesylates of 1-(2'-Deoxy-β-D-lyxofuranosyl)thymine1,2. J. Org. Chem. 1964;29:806–810. [Google Scholar]
- 161.Lin Y.-W., Abdul Rahim N., Zhao J., Han M.-L., Yu H.H., Wickremasinghe H., Chen K., Wang J., Paterson D.L., Zhu Y.J.A.a. NDM-producing multidrug-resistant Klebsiella pneumoniae. Vol. 63. 2019. chemotherapy, Novel polymyxin combination with the antiretroviral zidovudine exerts synergistic killing against; pp. 2176–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thomson J.M., Lamont I.L. Nucleoside analogues as antibacterial agents. Front. Microbiol. 2019;10:952. doi: 10.3389/fmicb.2019.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Younis W., Thangamani S., Seleem M.N. Repurposing non-antimicrobial drugs and clinical molecules to treat bacterial infections. Curr. Pharmaceut. Des. 2015;21:4106–4111. doi: 10.2174/1381612821666150506154434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Breger J., Fuchs B.B., Aperis G., Moy T.I., Ausubel F.M., Mylonakis E. Antifungal chemical compounds identified using a C. elegans pathogenicity assay. PLoS Pathog. 2007;3:e18. doi: 10.1371/journal.ppat.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Butts A., Krysan D.J., Goldman W.E. Antifungal drug discovery: something old and something new. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ejim L., Farha M.A., Falconer S.B., Wildenhain J., Coombes B.K., Tyers M., Brown E.D., Wright G.D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011;7:348–350. doi: 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- 167.You Z., Ran X., Dai Y., Ran Y. Vol. 28. 2018. pp. 492–501. (Clioquinol, an Alternative Antimicrobial Agent against Common Pathogenic Microbe). [DOI] [PubMed] [Google Scholar]
- 168.Bareggi S.R., Cornelli U. Clioquinol: review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 2012;18:41–46. doi: 10.1111/j.1755-5949.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Alsterholm M., Karami J., Fau Faergemann N., Faergemann J. Vol. 90. 2010. pp. 239–245. (Antimicrobial Activity of Topical Skin Pharmaceuticals - an in Vitro Study). [DOI] [PubMed] [Google Scholar]
- 170.Pippi B., Lopes W., Reginatto P., Silva F.É.K., Joaquim A.R., Alves R.J., Silveira G.P., Vainstein M.H., Andrade S.F., Fuentefria A.M. Vol. 27. 2019. pp. 41–48. (New Insights into the Mechanism of Antifungal Action of 8-hydroxyquinolines). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nosengo N. Can you teach old drugs new tricks? Nature. 2016;534:314–316. doi: 10.1038/534314a. [DOI] [PubMed] [Google Scholar]
- 172.Barrows N.J., Campos R.K., Powell S.T., Prasanth K.R., Schott-Lerner G., Soto-Acosta R., Galarza-Muñoz G., McGrath E.L., Urrabaz-Garza R., Gao J., Wu P., Menon R., Saade G., Fernandez-Salas I., Rossi S.L., Vasilakis N., Routh A., Bradrick S.S., Garcia-Blanco M.A. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe. 2016;20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xu M., Lee E.M., Wen Z., Cheng Y., Huang W.-K., Qian X., Tcw J., Kouznetsova J., Ogden S.C., Hammack C., Jacob F., Nguyen H.N., Itkin M., Hanna C., Shinn P., Allen C., Michael S.G., Simeonov A., Huang W., Christian K.M., Goate A., Brennand K.J., Huang R., Xia M., Ming G.-L., Zheng W., Song H., Tang H. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tang H., Hammack C., Ogden Sarah C., Wen Z., Qian X., Li Y., Yao B., Shin J., Zhang F., Lee Emily M., Christian Kimberly M., Didier Ruth A., Jin P., Song H., Ming G.-l. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Diamond M.S., Zachariah M., Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- 176.Allison A. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14:2–8. doi: 10.1191/0961203305lu2109oa. [DOI] [PubMed] [Google Scholar]
- 177.Barrows N., Campos R., Powell S.T., Prasanth K., Schott-Lerner G., Soto-Acosta R., Galarza-Muñoz G., Mcgrath E., Urrabaz-Garza R., Gao J. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe. 2016:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Balasubramanian A., Teramoto T., Kulkarni A.A., Bhattacharjee A.K., Padmanabhan R. Antiviral activities of selected antimalarials against dengue virus type 2 and Zika virus. Antivir. Res. 2017;137 doi: 10.1016/j.antiviral.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 179.Albulescu I.C., Hoolwerff M.V., Wolters L.A., Bottaro E., Hemert M. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antivir. Res. 2015;121:39–46. doi: 10.1016/j.antiviral.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 180.Bhatia H.K., Singh H., Grewal N., Natt N.K., Sofosbuvir A novel treatment option for chronic hepatitis C infection. J. Pharmacol. Pharmacother. 2014;5:278–284. doi: 10.4103/0976-500X.142464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sacramento C.Q., de Melo G.R., de Freitas C.S., Rocha N., Hoelz L.V.B., Miranda M., Fintelman-Rodrigues N., Marttorelli A., Ferreira A.C., Barbosa-Lima G., Abrantes J.L., Vieira Y.R., Bastos M.M., de Mello Volotão E., Nunes E.P., Tschoeke D.A., Leomil L., Loiola E.C., Trindade P., Rehen S.K., Bozza F.A., Bozza P.T., Boechat N., Thompson F.L., de Filippis A.M.B., Brüning K., Souza T.M.L. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017;7:40920. doi: 10.1038/srep40920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Furuta Y., Komeno T., Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Japan Academy, Series B. 2017;93:449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Guedj J., Piorkowski G., Jacquot F., Madelain V., Lamballerie X.D. Antiviral efficacy of favipiravir against Ebola virus: a translational study in cynomolgus macaques. PLoS Med. 2018;15 doi: 10.1371/journal.pmed.1002535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sun W., He S., Martínez-Romero C., Kouznetsova J., Tawa G., Xu M., Shinn P., Fisher E., Long Y., Motabar O., Yang S., Sanderson P.E., Williamson P.R., García-Sastre A., Qiu X., Zheng W. Synergistic drug combination effectively blocks Ebola virus infection. Antivir. Res. 2017;137:165–172. doi: 10.1016/j.antiviral.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roberts D.M., Gallapatthy G., Dunuwille A., Chan B.S. Pharmacological treatment of cardiac glycoside poisoning. Br. J. Clin. Pharmacol. 2016;81:488–495. doi: 10.1111/bcp.12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Reddy D., Kumavath R., Barh D., Azevedo V., Ghosh P. Anticancer and antiviral properties of cardiac glycosides: a review to explore the mechanism of actions. Molecules. 2020;25:3596. doi: 10.3390/molecules25163596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kapoor A., Cai H., Forman M., He R., Shamay M., Arav-Boger R. Human cytomegalovirus inhibition by cardiac glycosides: evidence for involvement of the hERG gene. Antimicrob. Agents Chemother. 2012;56:4891–4899. doi: 10.1128/AAC.00898-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mukhopadhyay R., Roy S., Venkatadri R., Su Y.P., Arav-Boger R. Efficacy and mechanism of action of low dose emetine against human cytomegalovirus. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xu J., Shi P.-Y., Li H., Zhou J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect. Dis. 2020;6:909–915. doi: 10.1021/acsinfecdis.0c00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rossignol J.F. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014;110:94–103. doi: 10.1016/j.antiviral.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Britt W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008;325:417–470. doi: 10.1007/978-3-540-77349-8_23. [DOI] [PubMed] [Google Scholar]
- 192.Wiebusch L., Hagemeier C. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1) J. Virol. 1999;73:9274–9283. doi: 10.1128/jvi.73.11.9274-9283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mercorelli B., Luganini A., Celegato M., Palù G., Gribaudo G., Loregian A. Repurposing the clinically approved calcium antagonist manidipine dihydrochloride as a new early inhibitor of human cytomegalovirus targeting the Immediate-Early 2 (IE2) protein. Antivir. Res. 2017;150:130–136. doi: 10.1016/j.antiviral.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 194.Alexander Ploss, Jean Dubuisson. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut. 2012;61:i25–i35. doi: 10.1136/gutjnl-2012-302048. [DOI] [PubMed] [Google Scholar]
- 195.Sato A., Saito Y., Sugiyama K., Sakasegawa N., Muramatsu T., Fukuda S., Yoneya M., Kimura M., Ebinuma H., Hibi T., Ikeda M., Kato N., Saito H. Suppressive effect of the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) on hepatitis C virus replication. J. Cell. Biochem. 2013;114:1987–1996. doi: 10.1002/jcb.24541. [DOI] [PubMed] [Google Scholar]
- 196.Wang Y.C., Yang X., Xing L.H., Kong W.Z. Effects of SAHA on proliferation and apoptosis of hepatocellular carcinoma cells and hepatitis B virus replication. World J. Gastroenterol. 2013;19:5159–5164. doi: 10.3748/wjg.v19.i31.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Valencia D.N. Brief review on COVID-19: the 2020 pandemic caused by SARS-CoV-2. Cureus. 2020:12. doi: 10.7759/cureus.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Suthar T.R., Gaikwad S.T., Suthar A.D. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): a review. Int. J. Curr. Microbiol. Appl. Sci. 2020;9:3201–3208. [Google Scholar]
- 199.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y., Zhao Y., Li Y., Wang X., Peng Z. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Indu P., Ragavan R.M., Narasingam A., Al-Dhabi N.A., Ignacimuthu S. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: a molecular docking and drug repurposing approach. J. Infection Public Health. 2020;13 doi: 10.1016/j.jiph.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen Y.W., Yiu C., Wong K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000 Research. 2020;9:129. doi: 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lo M.K., Jordan R., Arvey A., Sudhamsu J., Shrivastava-Ranjan P., Hotard A.L., Flint M., Mcmullan L.K., Siegel D., Clarke M.O. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. For. Rep. 2017;7:43395. doi: 10.1038/srep43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sheahan T.P., Sims A.C., Graham R.L., Menachery V.D., Gralinski L.E., Case J.B., Leist S.R., Pyrc K., Feng J.Y., Trantcheva I. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., Smith E.C., Case J.B., Feng J.Y., Jordan R., Ray A.S., Cihlar T., Siegel D., Mackman R.L., Clarke M.O., Baric R.S., Denison M.R. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading Exoribonuclease. mBio. 2018;9 doi: 10.1128/mBio.00221-18. e00221-00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sheahan T., Sims A.C., Leist S.R., Schäfer A., Baric R.S. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020;11:222. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mothay D., Ramesh K.V. Binding site analysis of potential protease inhibitors of COVID-19 using AutoDock. VirusDisease. 2020;31:194–199. doi: 10.1007/s13337-020-00585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ge Y., Tian T., Huang S., Wan F., Li J., Li S., Yang H., Hong L., Wu N., Yuan E., Cheng L., Lei Y., Shu H., Feng X., Jiang Z., Chi Y., Guo X., Cui L., Xiao L., Li Z., Yang C., Miao Z., Tang H., Chen L., Zeng H., Zhao D., Zhu F., Shen X., Zeng J. A Data-Driven Drug Repositioning Framework Discovered a Potential Therapeutic Agent Targeting COVID-19. bioRxiv; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Berger N.A., Besson V.C., Boulares A.H., Bürkle A., Chiarugi A., Clark R.S., Curtin N.J., Cuzzocrea S., Dawson T.M., Dawson V.L., Haskó G., Liaudet L., Moroni F., Pacher P., Radermacher P., Salzman A.L., Snyder S.H., Soriano F.G., Strosznajder R.P., Sümegi B., Swanson R.A., Szabo C. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018;175:192–222. doi: 10.1111/bph.13748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jagtap P., Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 212.Lin S.Y., Liu C.L., Chang Y.M., Zhao J., Perlman S., Hou M.H. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J. Med. Chem. 2014;57:2247. doi: 10.1021/jm500089r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Garimella T., Tao X., Sims K., Chang Y.-T., Rana J., Myers E., Wind-Rotolo M., Bhatnagar R., Eley T., LaCreta F., AbuTarif M. Effects of a fixed-dose Co-formulation of Daclatasvir, Asunaprevir, and beclabuvir on the pharmacokinetics of a cocktail of cytochrome P450 and drug transporter substrates in healthy subjects. Drugs R. 2018;18:55–65. doi: 10.1007/s40268-017-0222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sekhar T. Molecular docking and virtual screening based prediction of drugs for COVID-19. Comb. Chem. High Throughput Screen. 2021;24:716–728. doi: 10.2174/1386207323666200814132149. [DOI] [PubMed] [Google Scholar]
- 215.Hall D.C., Jr., Ji H.-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Trav. Med. Infect. Dis. 2020;35 doi: 10.1016/j.tmaid.2020.101646. 101646-101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods - ScienceDirect. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mohamed K., Yazdanpanah N., Saghazadeh A., Rezaei N. Computational drug discovery and repurposing for the treatment of COVID-19: a systematic review. Bioorg. Chem. 2021;106 doi: 10.1016/j.bioorg.2020.104490. 104490-104490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Smith M., Smith J. Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. ChemRxiv; 2020. [Google Scholar]
- 219.Fujimiya H., Nakashimd S., Miyata H., Nozawa Y. Effect of a novel antiallergic drug, pemirolast, on activation of rat peritoneal mast cells: inhibition of exocytotic response and membrane phospholipid turnover. Int. Arch. Allergy Immunol. 2009;96:62–67. doi: 10.1159/000235536. [DOI] [PubMed] [Google Scholar]
- 220.Timmins G.S., Deretic V. Mechanisms of action of isoniazid. Mol. Microbiol. 2010;62:1220–1227. doi: 10.1111/j.1365-2958.2006.05467.x. [DOI] [PubMed] [Google Scholar]
- 221.Ley J.P., Krammer G., Reinders G., Gatfield I.L., Bertram H.J. Evaluation of bitter masking flavanones from herba santa (Eriodictyon californicum (H. And A.) Torr., hydrophyllaceae) J. Agric. Food Chem. 2005;53:6061. doi: 10.1021/jf0505170. [DOI] [PubMed] [Google Scholar]
- 222.Khalili J.S., Zhu H., Mak N.S.A., Yan Y., Zhu Y. Novel coronavirus treatment with ribavirin: groundwork for an evaluation concerning COVID-19. J. Med. Virol. 2020;92 doi: 10.1002/jmv.25798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (London, England) 2020;395:1695–1704. doi: 10.1016/S0140-6736(20)31042-4. 10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zeng D., Debabov D., Hartsell T.L., Cano R.J., Adams S., Schuyler J.A., McMillan R., Pace J.L. Approved glycopeptide antibacterial drugs: mechanism of action and resistance. Cold Spring Harb. Perspect Med. 2016;6:a026989. doi: 10.1101/cshperspect.a026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nan Z., Pan T., Zhang J., Li Q., Xue Z., Bai C., Feng H., Tao P., Zhang J., Chao L. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of Ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biol. Chem. 2016;291:9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Colson P., Raoult D. Fighting viruses with antibiotics: an overlooked path. Int. J. Antimicrob. Agents. 2016:349–352. doi: 10.1016/j.ijantimicag.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang J., Ma X., Yu F., Liu J., Zhang H. 2020. Teicoplanin Potently Blocks the Cell Entry of 2019-nCoV. [Google Scholar]
- 228.Ceccarelli G., Alessandri F., Oliva A., Borrazzo C., Dell'Isola S., Ialungo A.M., Rastrelli E., Pelli M., Raponi G., Turriziani O., Ruberto F., Rocco M., Pugliese F., Russo A., d'Ettorre G., Venditti M. The role of teicoplanin in the treatment of SARS-CoV-2 infection: a retrospective study in critically ill COVID-19 patients (Tei-COVID study) J. Med. Virol. 2021;93:4319–4325. doi: 10.1002/jmv.26925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yamamoto N., Matsuyama S., Hoshino T., Yamamoto N. 2020. Nelfinavir Inhibits Replication of Severe Acute Respiratory Syndrome Coronavirus 2 in Vitro. [Google Scholar]
- 230.Xu Z., Yao H., Shen J., Wu N., Xu Y., Lu X., zhu w., Li L.-J. Nelfinavir Is Active against SARS-CoV-2 in Vero E6 Cells. ChemRxiv; 2020. [Google Scholar]
- 231.Multidrug Treatment with Nelfinavir and Cepharanthine against COVID-19. 2020. [Google Scholar]
- 232.Li C., Zhu X., Quanquin N., Deng Y.Q., Ji X., Tian M., Aliyari R., Zuo X., Yuan L., Afridi S.K. EBioMedicine; 2017. Chloroquine, a FDA-Approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice; pp. 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wolfe M.S., Cordero J.F. Safety of chloroquine in chemosuppression of malaria during pregnancy. Br. Med. J. 1985;290:1466–1467. doi: 10.1136/bmj.290.6480.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hsieh S.C., Wu Y.C., Zou G., Nerurkar V.R., Shi P.Y., Wang W.K. Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J. Biol. Chem. 2014;289:33149. doi: 10.1074/jbc.M114.610428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kashour Z., Riaz M., Garbati M.A., AlDosary O., Tlayjeh H., Gerberi D., Murad M.H., Sohail M.R., Kashour T., Tleyjeh I.M. Efficacy of chloroquine or hydroxychloroquine in COVID-19 patients: a systematic review and meta-analysis. J. Antimicrob. Chemother. 2020;76:30–42. doi: 10.1093/jac/dkaa403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tortorici M.A., Walls A.C., Lang Y., Wang C., Li Z., Koerhuis D., Boons G.-J., Bosch B.-J., Rey F.A., de Groot R.J., Veesler D. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019;26:481–489. doi: 10.1038/s41594-019-0233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A.J.c. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven. Protease Inhibitor. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Capoluongo E.D., Amato F., Castaldo G. The friendly use of chloroquine in the COVID-19 disease: a warning for the G6PD-deficient males and for the unaware carriers of pathogenic alterations of the G6PD gene. Clin. Chem. Lab. Med. 2020;58:1162–1164. doi: 10.1515/cclm-2020-0442. [DOI] [PubMed] [Google Scholar]