

Abstract
P53 has been implicated in the pathogenesis of obesity and diabetes; however, the mechanisms and tissue sites of action are incompletely defined. Therefore, we investigated the role of hepatocyte p53 in metabolic homeostasis using a hepatocyte-specific p53 knockout mouse model. To gain further mechanistic insight we studied mice under two complementary conditions of restricted weight gain: vertical sleeve gastrectomy (VSG) or food restriction. VSG or sham surgery was performed in high fat diet-fed male hepatocyte-specific p53 wild-type and knockout littermates. Sham-operated mice were fed ad libitum or food restricted to match their body weight to VSG-operated mice. Hepatocyte-specific p53 ablation in sham-operated ad libitum-fed mice impaired glucose homeostasis, increased body weight and decreased energy expenditure without changing food intake. The metabolic deficits induced by hepatocyte-specific p53 ablation were corrected, in part by food restriction, and completely by VSG. Unlike food restriction, VSG corrected the effect of hepatocyte p53 ablation to lower energy expenditure, resulting in a greater improvement in glucose homeostasis compared with food restricted mice. These data reveal an important new role for hepatocyte p53 in the regulation of energy expenditure and body weight and suggest that VSG can improve alterations in energetics associated with p53 dysregulation.
Keywords: Body weight, energy expenditure, bariatric surgery, p53
INTRODUCTION
The transcription factor, p53, is a tumor suppressor with a well-defined role in the maintenance of normal cell growth and genomic stability through the regulation of proliferation, apoptosis, and senescence (1). More recently, it has been established that p53 is also a key regulator of cellular metabolism (1). In particular, p53 suppresses aerobic glycolysis, upregulates mitochondrial oxidative phosphorylation, and promotes β-oxidation of fatty acids (2). These p53 activities counteract the Warburg effect in which cancer cells favor aerobic glycolysis over mitochondrial oxidative phosphorylation to gain a survival advantage (3). Thus, disruption of p53 signaling is a key pathogenic mediator of cancer, with over 50% of all cancer types being associated with somatic point mutations in p53 (4). However, the metabolic actions of p53 have been shown to be highly relevant in the maintenance of metabolic homeostasis in non-cancerous states as well. Numerous preclinical studies in p53 knockout mouse models reveal that p53 ablation promotes metabolic impairment (5–8). Furthermore, impairments in p53 signaling in humans are associated with metabolic disease. For example, certain p53 single nucleotide polymorphisms are associated with diabetes in humans and promote obesity and glucose dysregulation when introduced into mice (9). However, the mechanisms and key tissue sites of action by which p53 regulates whole body metabolic homeostasis remain incompletely defined.
Our understanding of the metabolic role of p53 is limited by a lack of studies conducted in tissue-specific knockout mouse models, a lack of studies performed in nontransformed cells and tissues, and a lack of studies assessing p53 function in complementary conditions of metabolic stress and intervention. Several preclinical studies report that dysregulation of p53 signaling results in disruptions in hepatic metabolism (10, 11). Furthermore, metabolic stressors, such as high fat diet (HFD) feeding, increase hepatic p53 expression (9, 12), suggesting that p53 may function as a compensatory mechanism against obesity-induced hepatic metabolic dysregulation. Therefore, we sought to define the role of hepatocyte p53 in metabolic homeostasis using a HFD-fed hepatocyte-specific p53 knockout mouse model. In parallel, we studied HFD-fed wild-type and knockout mice under two complementary conditions of restricted weight gain to define the key mechanisms by which hepatocyte p53 regulates whole body metabolism. Specifically, we studied mice after vertical sleeve gastrectomy (VSG) or food restriction that was tailored to match body weight to VSG-operated mice.
Bariatric surgery, such as VSG, is defined as a surgical manipulation of the gut that is performed for the purpose of body weight loss. It is currently the most effective long-term treatment for obesity and often results in remission of type 2 diabetes prior to weight loss (13, 14). While these metabolic improvements are partially related to body weight loss, these effects are also mediated by factors independent of weight loss and restricted caloric intake (15). Unlike food restriction, bariatric surgery improves energy homeostasis at the cellular level, with patients demonstrating increased cellular energetic efficiency and increased energy expenditure (16–19). Thus, we hypothesized that hepatocyte-specific ablation of p53 would result in metabolic impairments that would be corrected, in part, by food restriction and to a greater extent by VSG. Herein, we report that genetic ablation of hepatocyte p53 results in remarkable increases in body weight with an associated decrease in energy expenditure. We found that the metabolic deficits induced by hepatocyte-specific p53 ablation were corrected, in part by food restriction, and completely by VSG and the associated restoration of energy expenditure.
MATERIALS AND METHODS
Animals and Diets
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Cornell University. Hepatocyte-specific p53 knockout mice on a C57BL/6J background were generated by crossing an albumin Cre mouse line (B6N.Cg-Tg(Alb-cre)21Mgn/J) with a floxed p53 mouse line (B6.129P2-Trp53tm1Brn/J) (Jackson Laboratories; Bar Harbor, ME). Floxed p53 mice were generated as previously described and have been previously demonstrated to exhibit deletion of the p53 gene from exon 2 to exon 10 when crossed with the appropriate cre driver (20). qPCR analysis of p53 gene expression in mRNA extracted from liver indicated 50% knockdown efficiency in this mouse model (Supplemental Figure 1). Study mice were individually housed and maintained in a temperature and humidity-controlled room, with a 14:10-hour light-dark cycle. Starting at one month of age male p53Hep+/+ (WT) and p53Hep−/− (KO) littermates were fed a HFD consisting of ground chow (5012 LabDiets; St. Louis, MO) supplemented with 3.4% butter fat, 8.5% tallow, 13.1% soybean oil, 3.5% mineral mix, and 1% vitamin mix (Dyets; Bethlehem, PA) by weight for two months to produce an obese insulin resistant phenotype. At three months of age, mice underwent sham or VSG surgery, as previously described (21, 22). Sham-operated animals were either fed ad libitum (S-AL) or weight matched (S-WM) to VSG-operated mice. Weight matching was performed as previously described (23). Mice were maintained on HFD throughout the study. The following groups were studied: S-AL-WT (n=10), S-WM-WT (n=9), VSG-WT (n=11), S-AL-KO (n=9), S-WM-KO (n=10), VSG-KO (n=12). Food intake and body weight were measured twice per week. A fasting blood sample was collected at baseline (1 week before surgery), one month and three months after surgery for assessment of glucose concentrations, serum triglycerides and serum cholesterol. An oral glucose tolerance test was performed at 2 months after surgery, after a 6 hour fast (1 g/kg body weight gavage with dextrose), as previously described (21). Oral glucose tolerance test glucose measurements were made using a glucometer (One-Touch Ultra, Lifescan; Milpitas, CA). Serum insulin concentrations were measured in a sub-set of mice (n=7–9 per group) by ELISA (Millipore; Burlington, MA). Circulating triglyceride and cholesterol concentrations were measured using enzymatic colorimetric assays (ThermoFischer Scientific; Waltham, MA). Indirect calorimetry was performed at 2.5 months after surgery using a Promethion Indirect Calorimeter (Sable Systems, Las Vegas, NV). Six-hour fasted mice were euthanized 3 months after surgery by an overdose of pentobarbital (200 mg/kg IP) for tissue collection. At the time of sacrifice, three mice in the VSG-WT group and five mice in the VSG-KO group had a mild contained granuloma and/or adherent tissue attached to the stomach. However, study outcomes do not differ whether these mice are included or excluded from data analysis, so these mice have been included in the data presented.
Cell Culture
Primary adipocytes were cultured by isolating and culturing the stromal vascular fraction from inguinal fat from 5–6-week old, male, C57BL/6J mice, as previously described (24). Once the cells were differentiated, cells were treated overnight with media that had been conditioned with the plasma obtained from all experimental groups. mRNA was extracted using Trizol (Invitrogen; Carlsbad, CA) and RNeasy kits (Qiagen; Hilden, Germany).
Histopathology
Liver samples were fixed in 4% PFA at the time of euthanasia and paraffin embedded. Representative sections were obtained and stained with hematoxylin and eosin for scoring of histologic changes. Sections were scored in a blinded fashion by a board-certified anatomic pathologist for cell size variation, karyomegaly, lipidosis, lipid distribution, degeneration and necrosis, cell alteration and hyperplasia, and oval cell hyperplasia.
Real-time quantitative PCR (qPCR)
Differential gene expression was measured by qPCR. Total RNA was extracted from liver tissue using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX), then reverse-transcribed for qPCR as previously described (25). qPCR of p53, Ppary, Prdm16, Upc1, Puma, Pparα, p21, Gck, and Sco2 was performed using SYBR Green detection and threshold values were normalized to the expression of the housekeeping gene. Tbp was used as the housekeeping gene for hepatic tissues and mouse primary white adipocytes, and to Ppia for WAT. Primer sequences used are presented in Table 1.
Table 1.
Primers used for Quantitative real-time PCR.
| Gene Name | Primer Sequence (5’ → 3’) |
|---|---|
| Tbp | F: ctg gaa ttg tac cgc agc tt R: gca aat cgc ttg gga tta tat tca g |
| Ppia | F: cgc gtc tcc ttc gag ctg tttg R: tgt aaa gtc acc acc ctg gca cat |
| Ppary | F: gtg cca gtt tcg atc cgt aga R: ggc cag cat cgt gta gat ga |
| Prdm16 | F: gac ttg gac act acc acg gg R: aga tgc acc ccc aaa ctc ag |
| Ucp1 | F: act gcc aca cct cca gtc att R: ctt tgc ctc act cag gat tgg |
| Puma | F: cct agt tgg gct cca ttt ctg R: acct ca acg cgc agt ac |
| Pparα | F: ctg aac atc gag tgt cga ata R: ccg aaa gaa gcc ctt aca |
| p21 | F: ctt gtc gct gtc ttg cac tc R: tag aaa tct gtc agg ctg gtc |
| Gck | F: tca gga ggc cag tgt aaa R: ccc agg tct aag gag aga aa |
| Sco2 | F: agg gct gag aag gaa cag tg R: ctc atc ggg gca aat atc ag |
| p53 | F: ctc gaa gac tgg atg act c R: aca gat cgt cca tgc act gac |
F, forward; R, reverse.
Statistics and data analysis
Data are presented as mean ± SEM. All statistical analyses were performed using GraphPad Prism 8.00 for Mac (GraphPad Software). Data were analyzed by two-factor or three-factor ANOVA with Bonferroni’s post-test, ANCOVA or Student’s t-test, as indicated. Differences were considered significant at p≤0.05.
RESULTS
Hepatocyte-specific p53 ablation increases body weight and adiposity which is normalized by VSG and food restriction
Cumulative food intake did not differ between genotypes (Figure 1A–B). However, S-AL-KO mice exhibited dramatically elevated body weight and total white adipose tissue (WAT) weight compared with S-AL-WT mice (Figure 1C–D, p<0.05). VSG reduced cumulative food intake during the first 27 days after surgery compared to S-AL mice, independently of genotype (Figure 1A, p<0.01); however, VSG-induced reductions in total cumulative food intake only reached significance in VSG-WT mice (Figure 1B, p<0.01). S-WM mice had to be food restricted to a greater degree than VSG-operated mice to match body weight to the VSG-operated mice (Figure 1A–C, p<0.05). VSG and S-WM lowered body weight independently of genotype (Figure 1C, p<0.05). WAT weight was elevated in S-AL-KO compared with S-AL-WT (Figure 1D, p<0.001). VSG reduced WAT weight compared to S-AL controls, independently of genotype (Figure 1D–E, p<0.05). Adipose depot weights did not differ between the S-WM and VSG-operated groups in either genotype (Figure 1D–E). These data demonstrate that hepatocyte-specific p53 ablation increases body weight and adiposity independently of food intake and that this effect is corrected by VSG.
Figure 1. Hepatocyte-specific p53 ablation increases body weight and adiposity which is normalized by VSG and food restriction.
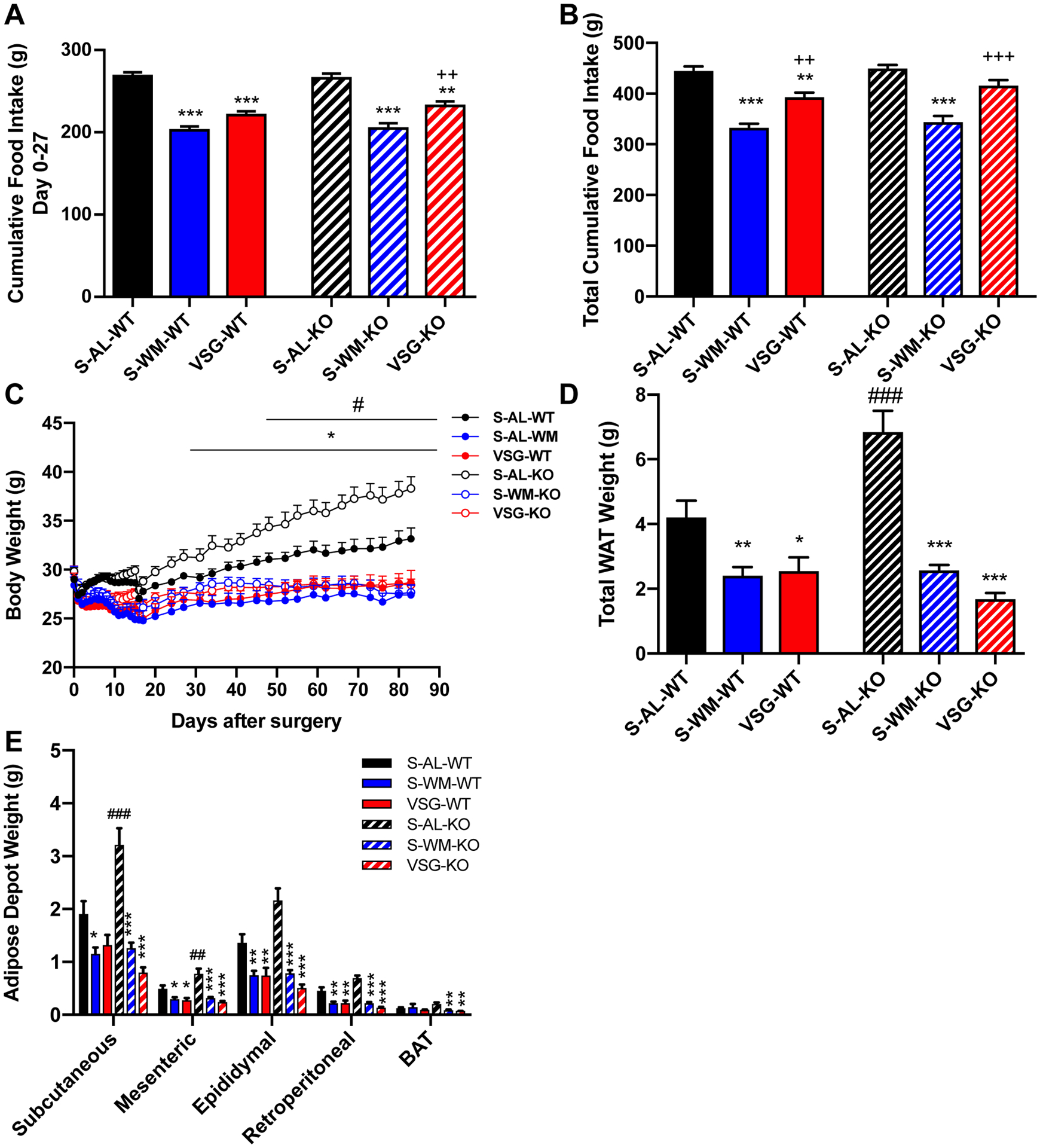
A) Cumulative food intake during days 0–27 after surgery and B) throughout study. C) Body weight, D) total white adipose tissue (WAT) weight and E) WAT depot and brown adipose tissue (BAT) weights. *p<0.05, **p<0.01, ***p<0.001 compared with S-AL; ++p<0.01, +++p<0.001 compared with S-WM; ##p<0.01, ###p<0.001 compared with WT by two-factor or three-factor ANOVA.
Hepatocyte-specific p53 ablation decreases energy expenditure which is normalized by VSG
Given that hepatocyte-specific p53 ablation increased body weight and adiposity independently of food intake, we assessed energy expenditure. Consistent with the difference in body weight, 24-hour energy expenditure was decreased in S-AL-KO mice compared with S-AL-WT mice (Figure 2A–B, p<0.05), suggesting that reduced energy expenditure contributes to the effect of hepatocyte-specific p53 ablation to increase body weight. VSG increased 24-hour energy expenditure by 17% compared to S-WM in p53Hep+/+ mice (Figure 2A–B, p<0.05). VSG corrected hepatocyte-specific p53 ablation-induced impairments in energy expenditure, as VSG increased total 24-hour energy expenditure by 35% in p53Hep−/− mice compared to S-AL controls (Figure 2A–B, p<0.01). Energy expenditure, during any period, did not differ between S-WM and VSG by genotype. S-WM exhibited a decrease in VO2 and VCO2 compared to S-AL groups in both genotypes (Supplemental Figure 2A–D, p<0.001). However, this effect was normalized by VSG in both genotypes (Supplemental Figure 2A–D, p<0.001). Despite these differences in VO2 and VCO2, there was no difference in respiratory quotient between groups (Supplemental Figure 2E–F).
Figure 2. Hepatocyte-specific p53 ablation decreases energy expenditure which is normalized by VSG.
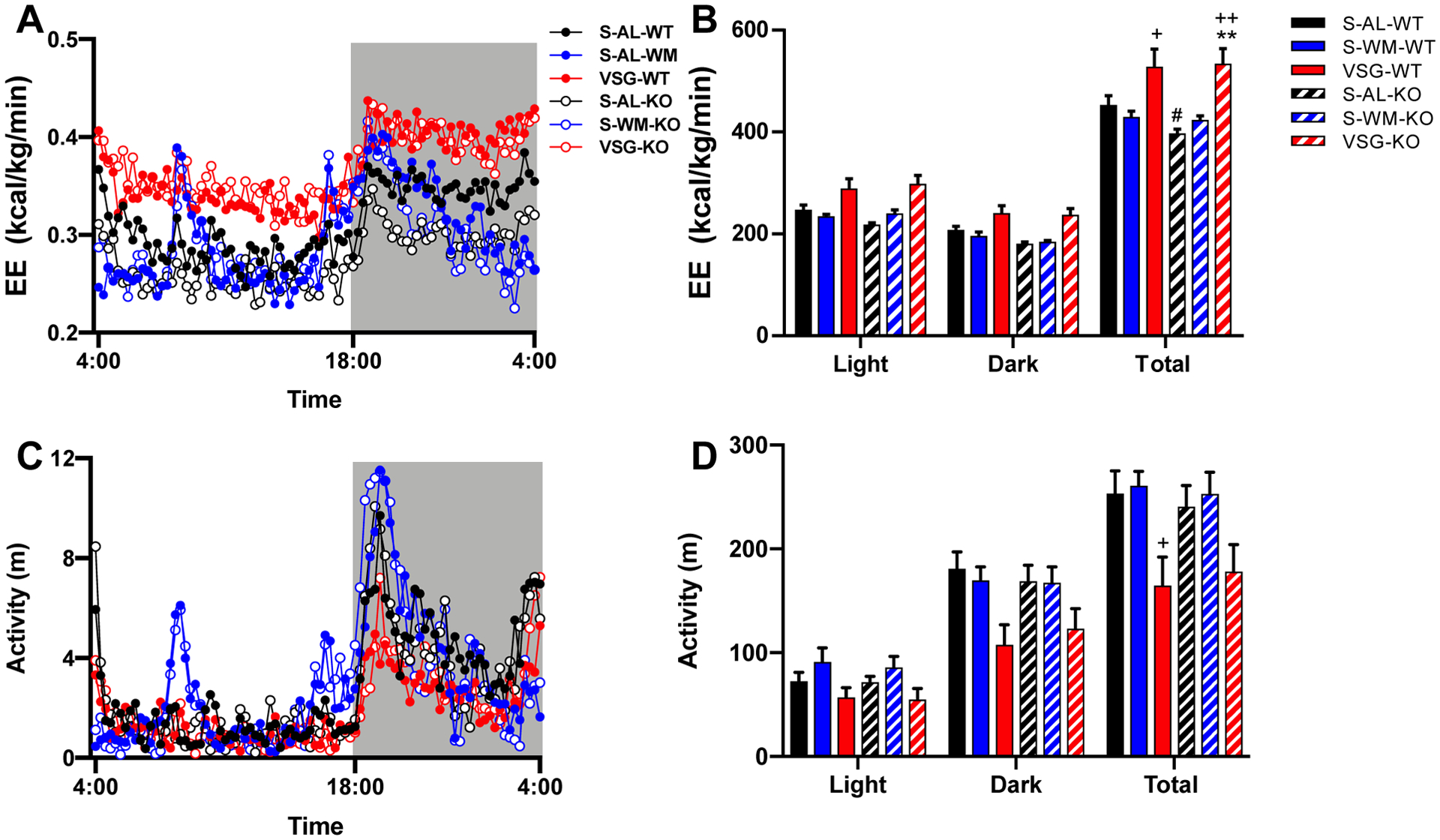
A) Energy expenditure (EE) over time, B) total EE during the light cycle, dark cycle, and full 24 hour cycle, C) activity over time, and D) total activity during the light cycle, dark cycle, and full 24 hour cycle. **p<0.01 compared with S-AL; +p<0.05, ++p<0.01 compared with S-WM; #p<0.05 compared with S-AL-WT by three-factor ANOVA and ANCOVA (using total body mass as the co-variate).
Activity did not differ between genotypes within the three conditions (Figure 2C–D). Activity in VSG-operated p53Hep+/+ mice was decreased compared to S-WM (Figure 2D, p<0.05). Although there was a trend for a decrease in activity in VSG-operated p53Hep−/− mice compared to S-AL and S-WM groups, this did not reach significance (Figure 2D). Together, these data demonstrate that hepatocyte p53 regulates body weight through determination of energy expenditure. Furthermore, these data show that the effect of VSG to increase energy expenditure occurs via pathways other than hepatocyte p53.
Hepatocyte-specific p53 ablation impairs glucose tolerance which is improved by VSG and food restriction
To assess the role of hepatocyte p53 in glucose regulation, we measured fasting blood glucose concentrations throughout the study and performed an oral glucose tolerance test (OGTT). Fasting blood glucose concentrations did not differ at any time point between genotype within the three conditions (Figure 3A). VSG and S-WM exhibited reduced fasting blood glucose concentrations compared with S-AL in both genotypes at 1 and 3 months after surgery (Figure 3A, p<0.05). S-AL-KO mice exhibited significantly elevated fasting insulin concentrations compared with S-AL-WT mice (Figure 3B, p<0.001). VSG and S-WM exhibited reduced fasting insulin concentrations compared with S-AL in p53Hep−/− mice (p<0.001); however, this effect did not reach significance in p53Hep+/+ mice (Figure 3B).
Figure 3. Hepatocyte-specific p53 ablation impairs glucose homeostasis which is normalized by VSG and food restriction.
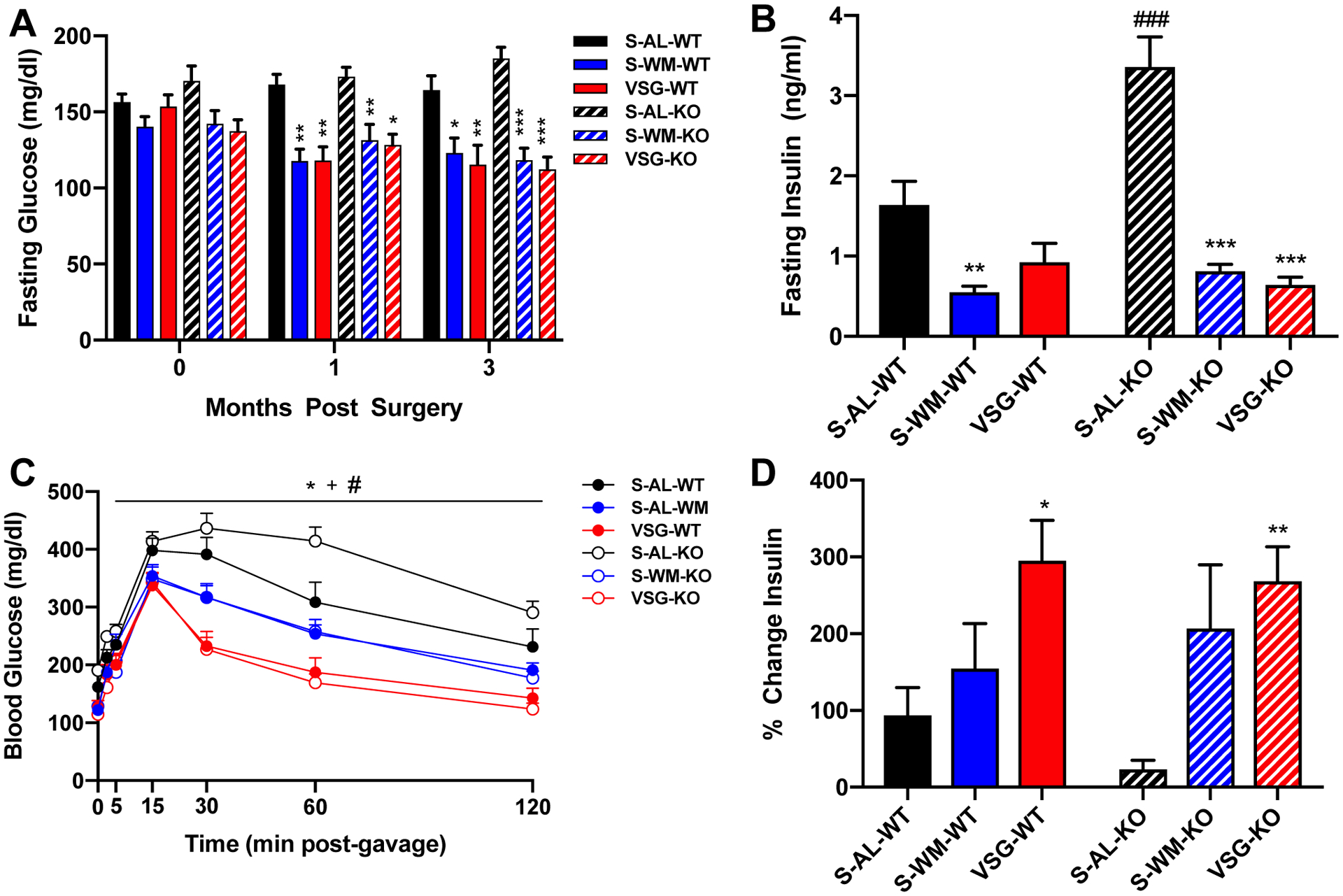
A) Fasting blood glucose concentrations at baseline, one month and three months after surgery. B) Fasting serum insulin concentrations at the time of OGTT. C) Blood glucose concentrations D) and percent change in insulin from baseline to 15 minutes after the glucose gavage during an OGTT at 2 months after surgery. *p<0.05, **p<0.01, ***p<0.001 VSG compared with S-AL; +p<0.05 VSG compared with S-WM; #p<0.05, ###p<0.001 S-AL-KO compared with S-AL-WT by two-factor or three-factor ANOVA.
S-AL-KO mice exhibited an increase in glucose excursions during the OGTT compared with S-AL-WT mice (Figure 3C, p<0.05). S-WM mice exhibited reduced blood glucose excursions compared with S-AL in both genotypes (Figure 3C, p<0.05). VSG reduced blood glucose excursions and the blood glucose area under the curve during the OGTT compared with S-AL and S-WM in both genotypes (Figure 3C, Supplemental Figure 3, p<0.05). Furthermore, VSG increased GSIS compared with S-AL in both genotypes, demonstrating an improvement in islet function (Figure 3D, p<0.05). Together, these data show that hepatocyte-specific p53 ablation impairs glucose tolerance, which can be improved by food restriction alone. However, the magnitude of improvement in glucose tolerance was greater in the VSG-KO compared to S-WM-KO (Supplemental Figure 3, p<0.05), demonstrating that VSG improves the metabolic impairment resulting from hepatocyte-specific p53 ablation, at least in part, independently of body weight.
Hepatocyte-specific p53 ablation enhances cell size variation, karyomegaly and hepatic expression of downstream p53 mediators
In order to determine the impact of hepatocyte-specific p53 ablation and VSG on hepatic histomorphology, liver sections were stained with H&E and scored by a board-certified pathologist, in a blinded fashion. As p53 mutations are associated with the development of hepatic neoplasia (26), we assessed all mice for the presence of gross and histopathologic signs of neoplasia or precancerous lesions. No gross or histopathologic signs of neoplasia or precancerous lesions were observed. However, hepatocyte-specific p53 ablation significantly increased cell size variation and karyomegaly scores compared to wild-type groups (Figure 4A–C, p<0.05). Cell size variation and karyomegaly scores did not differ between the three treatment groups for either genotype. Furthermore, inflammation, degeneration and necrosis, hyperplasia, and oval cell hyperplasia scores did not differ between genotype for the three conditions (Figure 4D–G).
Figure 4. Hepatocyte-specific p53 ablation enhances cell size variation, karyomegaly and hepatic expression of downstream p53 mediators.
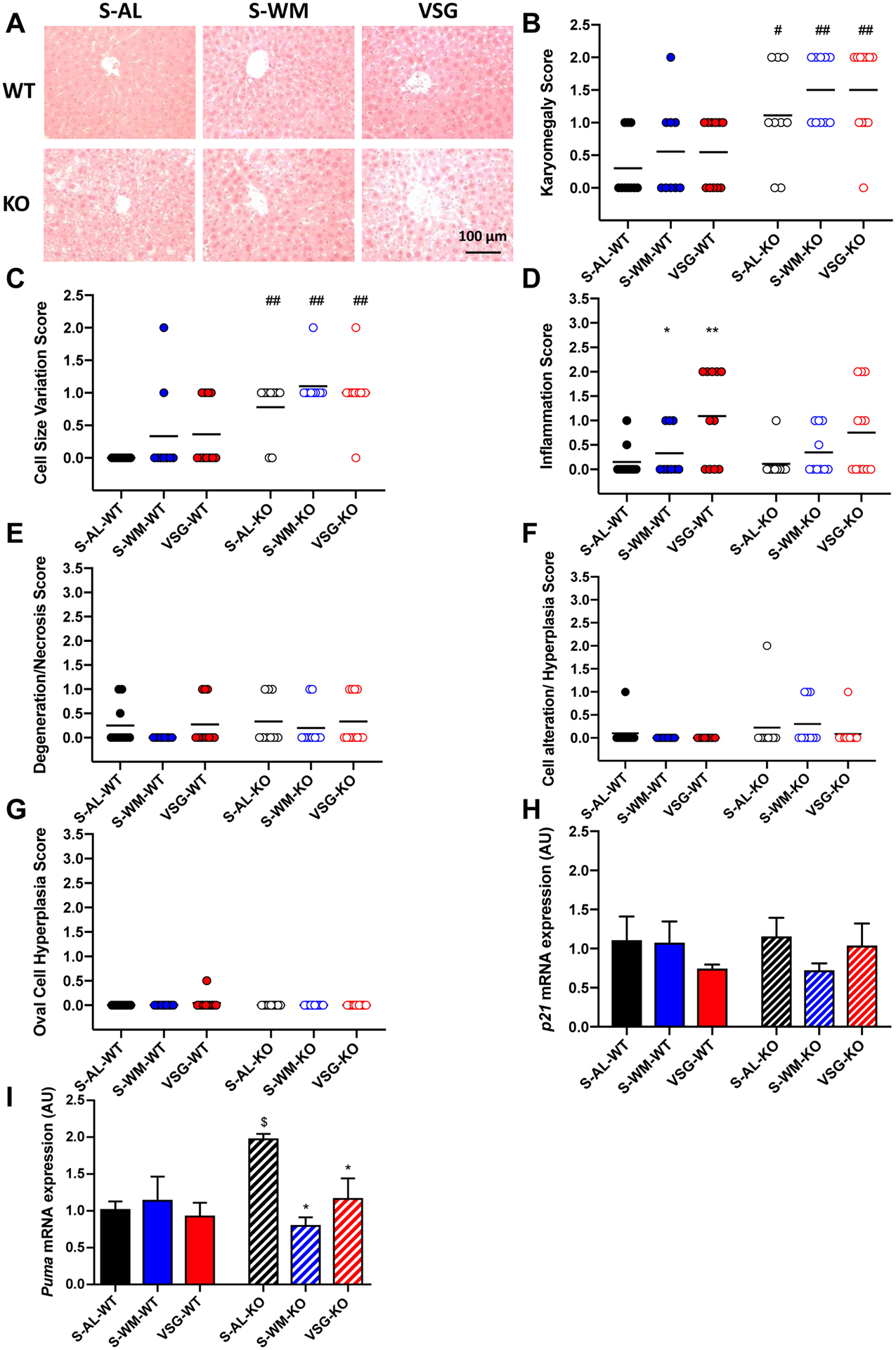
A) Representative images of H&E stained liver sections, B) karyomegaly score, C) cell size variation score, D) inflammation score, E) liver degeneration score, F) cellular alteration and hyperplasia score, G) oval cell hyperplasia score, H) hepatic p21 and I) PUMA mRNA expression. *p<0.05 compared with S-AL; #p<0.05, ##p<0.01 compared with WT by two-factor ANOVA; $p<0.05 compared with S-AL-WT by unpaired two-tailed Student’s t-test.
In order to assess the effects hepatocyte-specific p53 ablation on downstream mediators of p53 signaling, we measured mRNA expression of p21 and Puma. p53 is responsible for the induction of p21, which functions as an inhibitor of cyclin-dependent kinases required for cell cycle progression (27). However, relative p21 mRNA expression did not differ between genotypes within the three conditions (Figure 4H). PUMA (p53 upregulated modulator of apoptosis) is a Bcl-2 homology 3 protein that is activated by p53-dependent and independent pathways, and functions to transduce proapoptotic signals to the mitochondria, where it induces mitochondrial dysfunction and caspase activation (28). Direct comparison of S-AL-KO with S-AL-WT demonstrated that S-AL-KO mice exhibited significantly increased Puma mRNA expression compared with S-AL-WT mice (Figure 4I, p<0.05), suggesting that in the absence of hepatocyte p53, Puma is upregulated to compensate for p53 loss. However, VSG-KO and S-WM-KO mice exhibited decreased Puma mRNA expression compared with S-AL-KO mice (Figure 4I, p<0.05), suggesting that the need for compensation in response to hepatocyte-specific p53 ablation is lost in the presence of food restriction and VSG.
Hepatocyte-specific p53 ablation promotes liver lipid deposition which is normalized by VSG and food restriction
p53 promotes β-oxidation of fatty acids and thus is a critical regulator of lipid metabolism (5, 29). Therefore, we assessed circulating and hepatic lipid concentrations. Liver weight was elevated in S-AL-KO mice compared with S-AL-WT mice (Figure 5A, p<0.05). Liver lipid distribution and lipidosis scores, as well as liver triglyceride content, were higher in S-AL-KO mice compared with S-AL-WT mice (Figure 5B–D, p<0.05). VSG-KO and S-WM-KO mice exhibited decreased liver weight, lipid distribution and lipidosis scores and triglyceride content to normalize liver triglyceride content to that of p53Hep+/+ mice (Figure 5A–D, p<0.05). The effect of S-WM to lower liver lipid deposition was similar to VSG. Circulating triglyceride and cholesterol concentrations did not differ at any time point between genotypes in all three treatment groups (Figure 5E–F). VSG and S-WM exhibited lower circulating cholesterol concentrations compared with S-AL in both genotypes at 3 months post-surgery (Figure 5E, p<0.05). These data demonstrate that hepatocyte-specific p53 ablation increases hepatic lipid deposition primarily through its effects to increase body weight. This effect is reversible by VSG, primarily through its effect to reduce body weight gain.
Figure 5. Hepatocyte-specific p53 ablation promotes liver lipid deposition which is normalized by VSG and food restriction.
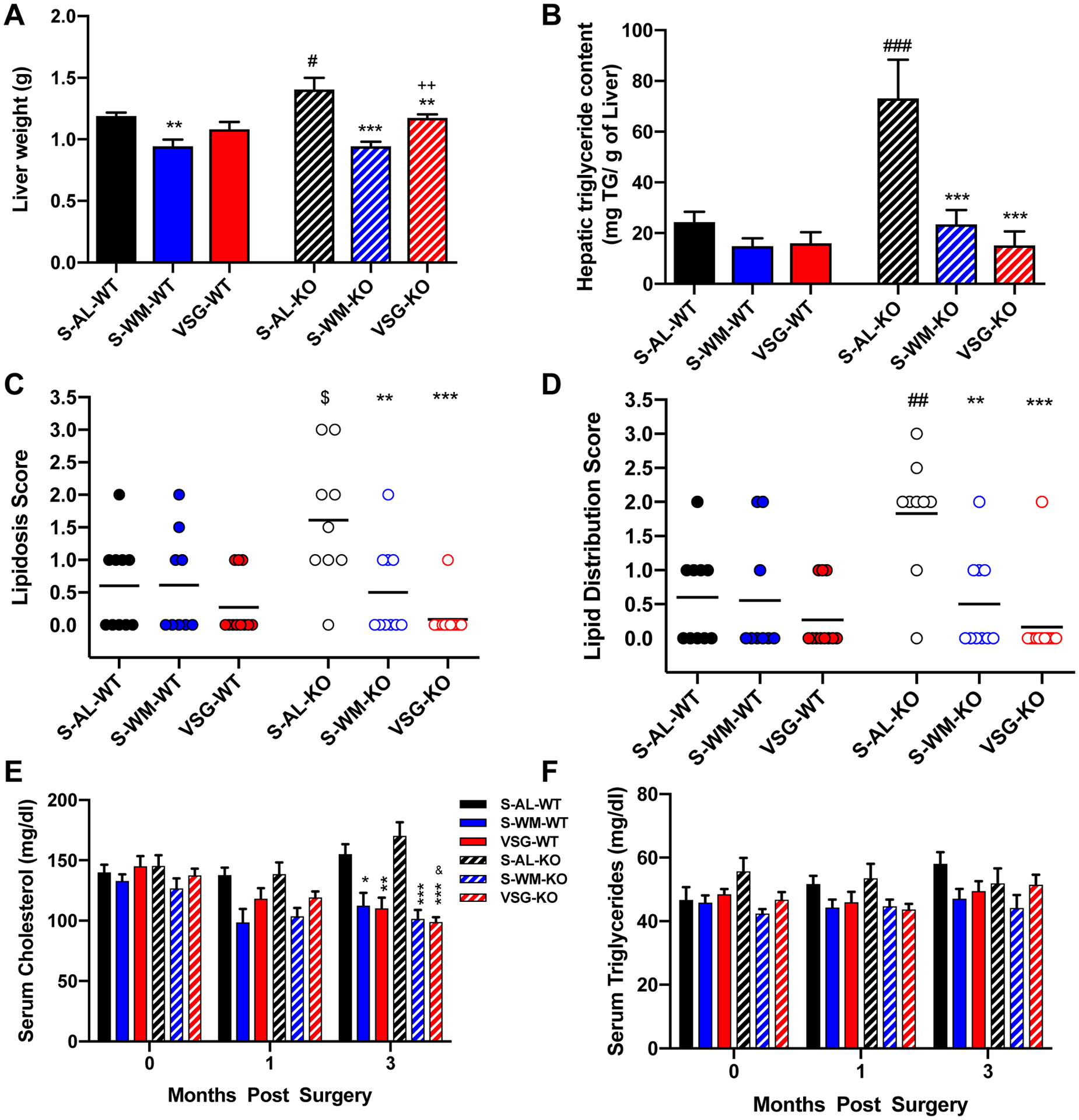
A) Liver weight, B) hepatic triglyceride content, C) liver lipidosis score, D) liver lipid distribution score, E) fasting serum cholesterol concentrations and F) fasting serum triglyceride concentration. &p<0.05 vs VSG-baseline; *p<0.05, **p<0.01, ***p<0.001 compared with S-AL; ++p<0.01 compared with S-WM; #p<0.05, ##p<0.01, ###p<0.001 compared with S-AL-WT by two-factor or three-factor ANOVA; $p<0.05 compared with S-AL-WT by unpaired two-tailed Student’s t-test.
Hepatocyte-specific p53 ablation promotes compensatory alterations in markers of cellular metabolism that are normalized by VSG and food restriction
A major mechanism of energy expenditure regulation is thermogenesis in brown adipose tissue (30). Prompted by recent evidence suggesting a role of p53 in brown fat differentiation (8), we assessed whether hepatocyte p53 may regulate energy expenditure through a factor secreted into the circulation. We measured the expression of markers of beige fat following the treatment of primary mouse white adipocytes with media conditioned with plasma from study mice. Ucp1, Pparγ and Prdm16 mRNA expression in white adipocytes did not differ between groups (Figure 6A–C). These data suggest that VSG and hepatocyte p53 do not impact energy expenditure through a circulating factor; however, it is possible that an endocrine effect was not detected due to limitations of conditioned media experiments, such as factor stability, physiological concentrations, and temporal signaling that are present in vivo. Furthermore, p53 mRNA expression in visceral WAT did not differ between groups or between genotype, demonstrating no compensatory induction of p53 in adipose tissue following hepatocyte-specific p53 ablation (Supplemental Figure 4).
Figure 6. Hepatocyte-specific p53 ablation promotes compensatory alterations in markers of cellular metabolism that are normalized by VSG and food restriction.
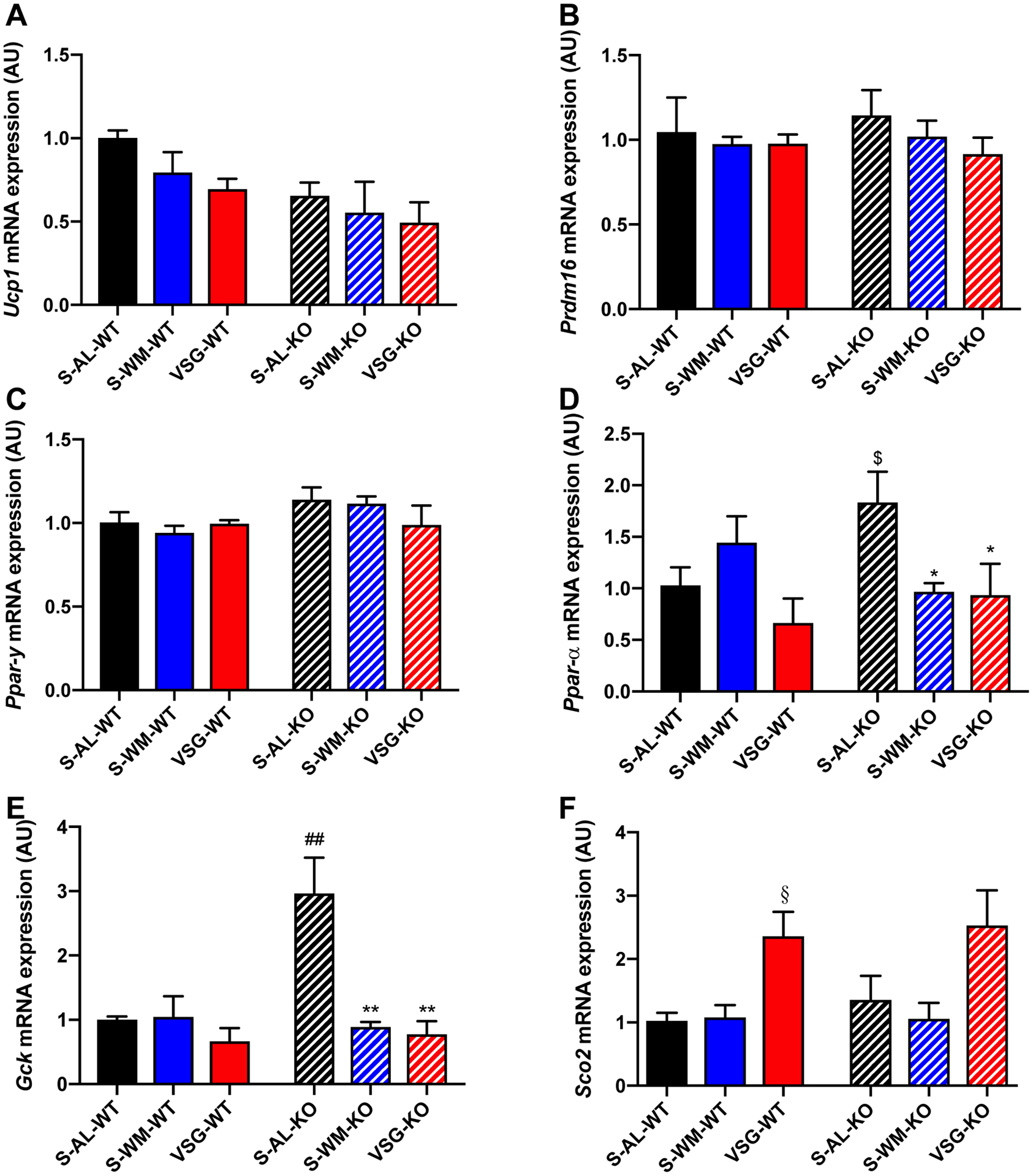
A) Relative Upc1, B) Pdrm16, and C) Pparγ mRNA expression in cultured white adipocytes treated with media conditioned with fasting serum from the indicated groups. D) Hepatic Pparα, E) Gck and F) Sco2 mRNA expression. *p≤0.05, **p<0.01 compared with S-AL; ##p<0.01 compared with S-AL-WT by two-factor ANOVA. $p<0.05 compared with S-AL-WT by unpaired one-tailed Student’s t-test; §p<0.05 compared with S-AL-WT and S-WM-WT by unpaired two-tailed Student’s t-test.
Based on our results demonstrating that ablation of hepatocyte p53 decreases energy expenditure and increases hepatic lipid deposition, we measured the hepatic expression of Pparα (peroxisome proliferator-activated receptor-α), a p53-regluated mitochondrial gene. p53 regulates fatty acid oxidation by facilitating transport of fatty acids into the mitochondria and by enhancing β-oxidation of fatty acids in response to nutrient stress (31). PPARα stimulation increases mitochondrial and peroxisomal fatty acid β-oxidation (32). Pparα mRNA expression was increased in S-AL-KO mice when directly compared with S-AL-WT (Figure 6D, p<0.05), suggesting that in the absence of hepatocyte p53 there is a compensatory response in which Pparα expression is uncoupled from p53 regulation to increase mitochondrial fatty acid β-oxidation to reduce hepatic lipid levels. Pparα mRNA expression did not differ between groups in p53Hep+/+ mice (Figure 6D). However, Pparα mRNA expression was decreased in S-WM-KO and VSG-KO mice compared with S-AL-KO (Figure 6D, p<0.05), suggesting that weight loss corrects the need for compensatory increases in hepatic Pparα mRNA expression.
p53 downregulates a number of critical components of the glycolytic pathway to promote mitochondrial oxidative phosphorylation over aerobic glycolysis (3, 33). GCK (glucokinase) is required for the first step in glycolysis (34). Gck mRNA expression was elevated in S-AL-KO mice compared with S-AL-WT mice, suggesting that Gck is a p53 target (Figure 6E, p<0.01). Similar to Pparα, VSG-KO and S-WM-KO exhibited reduced Gck mRNA expression compared with S-AL-KO to a similar extent (Figure 6E, p<0.01), suggesting that the effect of VSG to alter hepatic Gck expression in p53Hep−/− mice is due to decreased body weight gain. SCO2 (Synthesis of Cytochrome c Oxidase 2), a direct transcriptional target of p53, modulates oxidative phosphorylation through the regulation of the cytochrome c oxidase complex, which is the major site of oxygen utilization (33). Sco2 mRNA expression was increased in VSG-WT mice when directly compared with S-AL-WT and S-WM-WT (Figure 6F, p<0.05). A similar trend was seen in the p53Hep−/− mice, however, this did not reach significance, suggesting that VSG may upregulate oxidative phosphorylation through p53-dependent and independent mechanisms. Finally, as Puma is known to induce mitochondrial dysfunction, increased Puma expression in S-AL-KO mice (Figure 4I, p<0.05) may also contribute to impaired mitochondrial function and energy expenditure. Together, these data demonstrate that hepatocyte-specific p53 ablation promotes transcriptional changes that favor alterations in mitochondrial function, which are corrected by VSG and food restriction.
DISCUSSION
Herein, we provide compelling data that hepatocyte p53 is a key regulator of body weight and glucose homeostasis, largely through regulation of energy expenditure. Food restriction alone was able to combat all of the negative metabolic effects of hepatocyte-specific ablation of p53, except for the reduction in energy expenditure, demonstrating that hepatocyte p53 regulates whole-body metabolic function largely through determination of body weight. However, unlike food restriction, VSG was able to correct the effect of hepatocyte-specific p53 ablation to reduce energy expenditure which was associated with a greater improvement in glucose tolerance than food restriction alone, despite similar body weights between VSG and S-WM groups.
While our data are consistent with previous work demonstrating that p53 contributes to the pathogenesis of metabolic disease in mouse models, our data are the first to demonstrate that p53 action specifically within the hepatocyte plays a profound role in whole body energy balance (7, 8, 35). Considering that the increase in body weight following hepatocyte-specific p53 ablation was independent of food intake and associated with a decrease in energy expenditure, the mechanism of action underlying this impairment likely relates to p53’s role in mitochondrial energetics. In the absence of functional p53, cells exhibit decreased mitochondrial respiration, and consequently, decreased oxygen consumption, and a shift to anaerobic glycolysis for energy production (3, 33, 36). Consistent with our findings that hepatocyte p53 knockout decreases whole body energy expenditure, previous work demonstrates that p53 knockout in cultured primary mouse hepatocytes decreases mitochondrial respiration, whereas overexpression of p53 in skeletal myocytes enhances mitochondrial DNA content (10, 37). Furthermore, we found that hepatocyte-specific p53 ablation increased expression of Gck, suggesting an increased dependence on glycolysis, as compared to mitochondrial respiration for energy production.
Of note, previous work in whole-body knockout mouse models suggest that the role of p53 in metabolic regulation is tissue-specific. Studies in whole body p53 knockout mouse models report conflicting findings on the effect of p53 knockout on body weight and metabolic homeostasis, with some studies finding that p53 knockout decreases body weight and improves glucose regulation in mice on HFD (38) and others finding the opposite (39, 40). However, when p53 knockout is studied using tissue-specific approaches, p53 knockout is found to promote weight gain and metabolic impairment (7, 8, 10). The discrepancy in findings in whole body p53 knockout models, maybe due to, in part, the development of compensatory pathways. These conflicting findings point to the need to generate a more refined understanding of p53 in metabolic regulation through the study of tissue-specific manipulations.
VSG overcame the energetic impairments induced by hepatocyte-specific p53 ablation. The mechanisms by which VSG increases energy expenditure are incompletely defined. Studies in mouse bariatric models and human patients after bariatric surgery reveal a higher total energy expenditure (21, 41–43). Furthermore, in comparing the energetic differences between caloric restriction and bariatric surgery, caloric restriction results in compensatory reductions in energy expenditure (44). Bariatric surgery, on the other hand, overcomes this metabolic adaptation leading to sustained weight loss (23, 41, 44, 45). While our data demonstrate that food restriction corrected much of the metabolic impairment induced by genetic ablation of hepatocyte p53, food restriction did not correct the impairment in energy expenditure. In contrast, VSG increased energy expenditure in hepatocyte-specific p53 knockout mice, which led to greater improvements in glucose homeostasis. This demonstrates that VSG can increase energy expenditure through pathways other than hepatocyte p53. This is in agreement with a recent study in humans that found whole-body p53 expression is downregulated after bariatric surgery (46), such that surgically induced weight loss and remission of type 2 diabetes may suppress the metabolic stress needed for p53 induction. Of note, body weight independent improvements in glucose homeostasis after VSG are also likely the result of post-operative endocrine changes, such as potentiation of glucagon-like peptide-1 and bile acid receptor signaling (21, 22). Overall, weight loss alone is able to correct much of the metabolic impairment induced by hepatocyte-specific p53 ablation; however, VSG produces further body weight-independent improvements in glucose regulation and energy expenditure.
Our findings provide additional insight into the underlying mechanisms by which metabolic dysfunction develops in patients with p53 mutations and p53 dysregulation, such as Li-Fraumeni Syndrome or Ataxia telangiectasia. Furthermore, these data highlight the important interplay between metabolic disease and cancer, suggesting the aberrations in hepatic p53 signaling first cause metabolic impairment that may prime the body for cancer development. Finally, our data demonstrate that VSG overcomes the metabolic impairment induced by hepatocyte-specific p53 ablation. These data suggest that VSG may be an effective method of reducing the risk of obesity-related cancers and has the potential to improve the alterations in mitochondrial energetics associated with p53 dysregulation.
Supplementary Material
Acknowledgements:
We thank the Animal Health and Diagnostic Center Histopathology Core for preparation of samples for histological analysis. We would like to thank Jackie Belliveau for assistance with animal care.
Grant Support:
This research was supported by the Cornell Comparative Cancer Biology Training Program and NIH/NCI R21CA195002.
ABBREVIATIONS:
- AL
Ad libitum
- AUC
area under the curve
- BAT
brown adipose tissue
- GSIS
glucose stimulated insulin secretion
- HFD
high fat diet
- KO
knockout
- OGTT
oral glucose tolerance test
- qPCR
real-time quantitative PCR
- S
sham surgery
- VSG
vertical sleeve gastrectomy
- WM
weight-matched
- WAT
white adipose tissue
- WT
wild-type
Footnotes
Conflicts of Interests: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Berkers CR, Maddocks OD, Cheung EC, Mor I, and Vousden KH (2013) Metabolic regulation by p53 family members. Cell metabolism 18, 617–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, and Yang X (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature cell biology 13, 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O (1956) On the origin of cancer cells. Science (New York, N.Y.) 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ, and Oren M (2009) The first 30 years of p53: growing ever more complex. Nature reviews. Cancer 9, 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Zhao X, Gao X, Mei Y, and Wu M (2013) A new role of p53 in regulating lipid metabolism. Journal of molecular cell biology 5, 147–150 [DOI] [PubMed] [Google Scholar]
- 6.Armata HL, Golebiowski D, Jung DY, Ko HJ, Kim JK, and Sluss HK (2010) Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Molecular and cellular biology 30, 5787–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quiñones M, Al-Massadi O, Folgueira C, Bremser S, Gallego R, Torres-Leal L, Haddad-Tóvolli R, García-Caceres C, Hernandez-Bautista R, Lam BYH, Beiroa D, Sanchez-Rebordelo E, Senra A, Malagon JA, Valerio P, Fondevila MF, Fernø J, Malagon MM, Contreras R, Pfluger P, Brüning JC, Yeo G, Tschöp M, Diéguez C, López M, Claret M, Kloppenburg P, Sabio G, and Nogueiras R (2018) p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nature Communications 9, 3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molchadsky A, Ezra O, Amendola PG, Krantz D, Kogan-Sakin I, Buganim Y, Rivlin N, Goldfinger N, Folgiero V, Falcioni R, Sarig R, and Rotter V (2013) p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell death and differentiation 20, 774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS, and Murphy ME (2016) The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell reports 14, 2413–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prokesch A, Graef FA, Madl T, Kahlhofer J, Heidenreich S, Schumann A, Moyschewitz E, Pristoynik P, Blaschitz A, Knauer M, Muenzner M, Bogner-Strauss JG, Dohr G, Schulz TJ, and Schupp M (2017) Liver p53 is stabilized upon starvation and required for amino acid catabolism and gluconeogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 31, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porteiro B, Fondevila MF, Buque X, Gonzalez-Rellan MJ, Fernandez U, Mora A, Beiroa D, Senra A, Gallego R, Ferno J, Lopez M, Sabio G, Dieguez C, Aspichueta P, and Nogueiras R (2018) Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Molecular metabolism 8, 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Homayounfar R, Jeddi-Tehrani M, Cheraghpour M, Ghorbani A, and Zand H (2015) Relationship of p53 accumulation in peripheral tissues of high-fat diet-induced obese rats with decrease in metabolic and oncogenic signaling of insulin. General and comparative endocrinology 214, 134–139 [DOI] [PubMed] [Google Scholar]
- 13.Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, and Schoelles K (2004) Bariatric surgery: a systematic review and meta-analysis. Jama 292, 1724–1737 [DOI] [PubMed] [Google Scholar]
- 14.Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, Barakat HA, deRamon RA, Israel G, and Dolezal JM (1995) Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Annals of surgery 222, 339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubino F, R’Bibo SL, del Genio F, Mazumdar M, and McGraw TE (2010) Metabolic surgery: the role of the gastrointestinal tract in diabetes mellitus. Nat Rev Endocrinol 6, 102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nijhawan S, Richards W, O’Hea MF, Audia JP, and Alvarez DF (2013) Bariatric surgery rapidly improves mitochondrial respiration in morbidly obese patients. Surgical endoscopy 27, 4569–4573 [DOI] [PubMed] [Google Scholar]
- 17.Vijgen GH, Bouvy ND, Hoeks J, Wijers S, Schrauwen P, and van Marken Lichtenbelt WD (2013) Impaired skeletal muscle mitochondrial function in morbidly obese patients is normalized one year after bariatric surgery. Surgery for Obesity and Related Diseases 9, 936–941 [DOI] [PubMed] [Google Scholar]
- 18.Coen PM, Menshikova EV, Distefano G, Zheng D, Tanner CJ, Standley RA, Helbling NL, Dubis GS, Ritov VB, and Xie H (2015) Exercise and weight loss improve muscle mitochondrial respiration, lipid partitioning, and insulin sensitivity after gastric bypass surgery. Diabetes 64, 3737–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Feuers RJ, Desai VG, Lewis SM, Duffy PH, Mayhugh MA, Cowan G Jr., and Buffington CK (2007) Surgical caloric restriction ameliorates mitochondrial electron transport dysfunction in obese females. Obesity surgery 17, 800–808 [DOI] [PubMed] [Google Scholar]
- 20.Marino S, Vooijs M, van der Gulden H, Jonkers J, and Berns A (2000) Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development 14, 994–1004 [PMC free article] [PubMed] [Google Scholar]
- 21.McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, Ley RE, Chouinard ML, and Cummings BP (2017) TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 66, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garibay D, McGavigan AK, Lee SA, Ficorilli JV, Cox AL, Michael MD, Sloop KW, and Cummings BP (2016) beta-Cell Glucagon-Like Peptide-1 Receptor Contributes to Improved Glucose Tolerance After Vertical Sleeve Gastrectomy. Endocrinology 157, 3405–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummings BP, Bettaieb A, Graham JL, Stanhope KL, Kowala M, Haj FG, Chouinard ML, and Havel PJ (2012) Vertical sleeve gastrectomy improves glucose and lipid metabolism and delays diabetes onset in UCD-T2DM rats. Endocrinology 153, 3620–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI, Seale P, and Spiegelman BM (2014) Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White P, Brestelli JE, Kaestner KH, and Greenbaum LE (2005) Identification of transcriptional networks during liver regeneration. Journal of Biological Chemistry 280, 3715–3722 [DOI] [PubMed] [Google Scholar]
- 26.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan S-T, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, and Lowe SW (2006) Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbendary AA, Cirisano FD, Evans AC Jr., Davis PL, Iglehart JD, Marks JR, and Berchuck A (1996) Relationship between p21 expression and mutation of the p53 tumor suppressor gene in normal and malignant ovarian epithelial cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2, 1571–1575 [PubMed] [Google Scholar]
- 28.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, and Gores GJ (2009) JNK1-dependent PUMA Expression Contributes to Hepatocyte Lipoapoptosis. Journal of Biological Chemistry 284, 26591–26602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, Okazaki H, Tamura Y, Iizuka Y, Inoue N, Nakagawa Y, Takeuchi Y, Ohashi K, Harada K, Gotoda T, Nagai R, Kadowaki T, Ishibashi S, Osuga J, and Yamada N (2004) p53 involvement in the pathogenesis of fatty liver disease. The Journal of biological chemistry 279, 20571–20575 [DOI] [PubMed] [Google Scholar]
- 30.Lidell ME, and Enerback S (2010) Brown adipose tissue--a new role in humans? Nat Rev Endocrinol 6, 319–325 [DOI] [PubMed] [Google Scholar]
- 31.Moulder DE, Hatoum D, Tay E, Lin Y, and McGowan EM (2018) The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cariello NF, Romach EH, Colton HM, Ni H, Yoon L, Falls JG, Casey W, Creech D, Anderson SP, Benavides GR, Hoivik DJ, Brown R, and Miller RT (2005) Gene Expression Profiling of the PPAR-alpha Agonist Ciprofibrate in the Cynomolgus Monkey Liver. Toxicological Sciences 88, 250–264 [DOI] [PubMed] [Google Scholar]
- 33.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, and Hwang PM (2006) p53 regulates mitochondrial respiration. Science (New York, N.Y.) 312, 1650–1653 [DOI] [PubMed] [Google Scholar]
- 34.Peter A, Stefan N, Cegan A, Walenta M, Wagner S, Königsrainer A, Königsrainer I, Machicao F, Schick F, Häring H-U, and Schleicher E (2011) Hepatic Glucokinase Expression Is Associated with Lipogenesis and Fatty Liver in Humans. The Journal of Clinical Endocrinology & Metabolism 96, E1126–E1130 [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Duan W, Lee WN, Zhang Y, Xiang F, Liu Q, Go VL, and Xiao GG (2016) Overexpression of p53 Improves Blood Glucose Control in an Insulin Resistant Diabetic Mouse Model. Pancreas 45, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 36.Burns DM, and Richter JD (2008) CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes & development 22, 3449–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park J-Y, Wang P. y., Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, and Kang J-G (2009) p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circulation research 105, 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hallenborg P, Fjaere E, Liaset B, Petersen RK, Murano I, Sonne SB, Falkerslev M, Winther S, Jensen BA, Ma T, Hansen JB, Cinti S, Blagoev B, Madsen L, and Kristiansen K (2016) p53 regulates expression of uncoupling protein 1 through binding and repression of PPARgamma coactivator-1alpha. American journal of physiology. Endocrinology and metabolism 310, E116–128 [DOI] [PubMed] [Google Scholar]
- 39.Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Gotoda T, Nagai R, Kimura S, Ishibashi S, Osuga J, and Yamada N (2003) p53 Activation in adipocytes of obese mice. The Journal of biological chemistry 278, 25395–25400 [DOI] [PubMed] [Google Scholar]
- 40.Derdak Z, Lang CH, Villegas KA, Tong M, Mark NM, de la Monte SM, and Wands JR (2011) Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. Journal of hepatology 54, 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Z, Mumphrey MB, Townsend RL, Morrison CD, Munzberg H, Ye J, and Berthoud HR (2016) Body Composition, Food Intake, and Energy Expenditure in a Murine Model of Roux-en-Y Gastric Bypass Surgery. Obesity surgery 26, 2173–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faria SL, Faria OP, Cardeal Mde A, de Gouvea HR, and Buffington C (2012) Diet-induced thermogenesis and respiratory quotient after Roux-en-Y gastric bypass. Surgery for obesity and related diseases : official journal of the American Society for Bariatric Surgery 8, 797–802 [DOI] [PubMed] [Google Scholar]
- 43.Werling M, Olbers T, Fandriks L, Bueter M, Lonroth H, Stenlof K, and le Roux CW (2013) Increased postprandial energy expenditure may explain superior long term weight loss after Roux-en-Y gastric bypass compared to vertical banded gastroplasty. PloS one 8, e60280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin AC, Zheng H, Townsend RL, Patterson LM, Holmes GM, and Berthoud H-R (2013) Longitudinal assessment of food intake, fecal energy loss, and energy expenditure after Roux-en-Y gastric bypass surgery in high-fat-fed obese rats. Obesity surgery 23, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nestoridi E, Kvas S, Kucharczyk J, and Stylopoulos N (2012) Resting energy expenditure and energetic cost of feeding are augmented after Roux-en-Y gastric bypass in obese mice. Endocrinology 153, 2234–2244 [DOI] [PubMed] [Google Scholar]
- 46.Berisha SZ, Serre D, Schauer P, Kashyap SR, and Smith JD (2011) Changes in Whole Blood Gene Expression in Obese Subjects with Type 2 Diabetes Following Bariatric Surgery: a Pilot Study. PloS one 6, e16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.