

Abstract
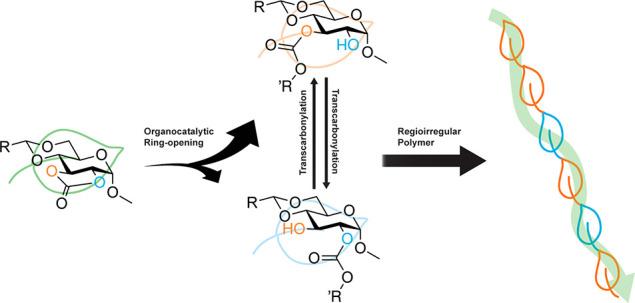
Rigorous investigations of the organobase-catalyzed ring-opening polymerizations (ROPs) of a series of five-membered cyclic carbonate monomers derived from glucose revealed that competing transcarbonylation reactions scrambled the regiochemistries of the polycarbonate backbones. Regioirregular poly(2,3-α-d-glucose carbonate) backbone connectivities were afforded by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-catalyzed ROPs of three monomers having different cyclic acetal protecting groups through the 4- and 6-positions. Small molecule studies conducted upon isolated unimers and dimers indicated a preference for Cx–O2 vs Cx–O3 bond cleavage from tetrahedral intermediates along the pathways of addition–elimination mechanisms when the reactions were performed at room temperature. Furthermore, treatment of isolated 3-unimer or 2-unimer, having the carbonate linkage in the 3- or 2-position as obtained from either Cx–O2 or Cx–O3 bond cleavage, respectively, gave the same 74:26 (3-unimer:2-unimer) ratio, confirming the occurrence of transcarbonylation reactions with a preference for 3-unimer vs. 2-unimer formation in the presence of organobase catalyst at room temperature. In contrast, unimer preparation at −78 °C favored Cx–O3 bond cleavage to afford a majority of 2-unimer, presumably due to a lack of transcarbonylation side reactions. Computational studies supported the experimental findings, enhancing fundamental understanding of the regiochemistry resulting from the ring-opening and subsequent transcarbonylation reactions during ROP of glucose carbonates. These findings are expected to guide the development of advanced carbohydrate-derived polymer materials by an initial monomer design via side chain acetal protecting groups, with the ability to evolve the properties further through later-stage structural metamorphosis.
Keywords: ring-opening polymerization, regiochemistry, transcarbonylation, glucose-derived polycarbonates, organocatalytic
Introduction
Polymeric materials constructed from renewable resources are prominent candidates toward achieving sustainability goals for the plastics market1−6 while also offering potential for rich chemical functionalities,7 stereochemical diversities,8 and regiochemical differentiation.9 The regiochemical connectivity between repeating units along the backbone is known to have substantial impacts on physical,10 chemical,11,12 and mechanical properties.13,14 Regiochemical differences may be introduced through polymer connectivities being installed at different polymerizable sites within a common substrate,15 via polymerization conducted on positional isomers,12,16 or from directional polymer backbone connectivity (head-to-head, head-to-tail, tail-to-tail).17−19
Naturally sourced sugar-based polymers are promising alternatives to petroleum-based materials for various functional and structural applications, due to their high abundance, structural diversities, and degradation potential.20−28 To date, organocatalytic ring-opening polymerizations (ROPs) of cyclic carbonate monomers have been demonstrated as low toxic, metal-free, and mild pathways to prepare sugar-based polycarbonates with predeterminable molar masses and narrow dispersities.29−37 However, there are few reports on the regioregularity between sugar monomer repeat units. Buchard and co-workers studied the 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-catalyzed ROP of six-membered d-mannose-based 4,6-cyclic carbonates.38 Initiated with 4-methylbenzyl alcohol, regioregular polycarbonates with head-to-tail carbonate linkages were obtained, as indicated by the presence of a single distinct carbonyl resonance frequency in 13C NMR spectra. Recently, we showed that the regiochemistry of TBD-catalyzed ROPs of d-glucose-based 4,6-cyclic carbonates could be tuned by varying the side-chain substituents on the 2- and 3-positions of the sugar ring.18 Density functional theory (DFT) calculations revealed that the regioselectivity differences may have resulted from intermolecular hydrogen bonding between the carbonate side-chain functionalities and TBD in the transition states. Further investigations are needed to fully understand the mechanistic origins of regiochemical outcomes, and then to translate that information into meaningful adjustments to polymer properties.
In this work, fundamental studies on ROP regioregularity were performed using methyl-α-d-glucopyranoside-derived five-membered 2,3-cyclic carbonate monomers, which contained different cyclic acetal protecting groups through the 4- and 6-positions (Scheme 1). Based upon a hypothesis that regioregularities at the initiation and early propagation stages of ROP would essentially determine the overall repeat unit connection directionalities along the backbones of resulting poly(d-glucose carbonate)s (PGC), the regioselective preferences were examined on corresponding unimers and dimers via a combination of comprehensive 1D and 2D NMR spectroscopic studies (Scheme 2). Surprisingly, however, transcarbonylation events were revealed, which upon initiation and polymer chain extension led to scrambling of the polymer backbone regiochemistry.
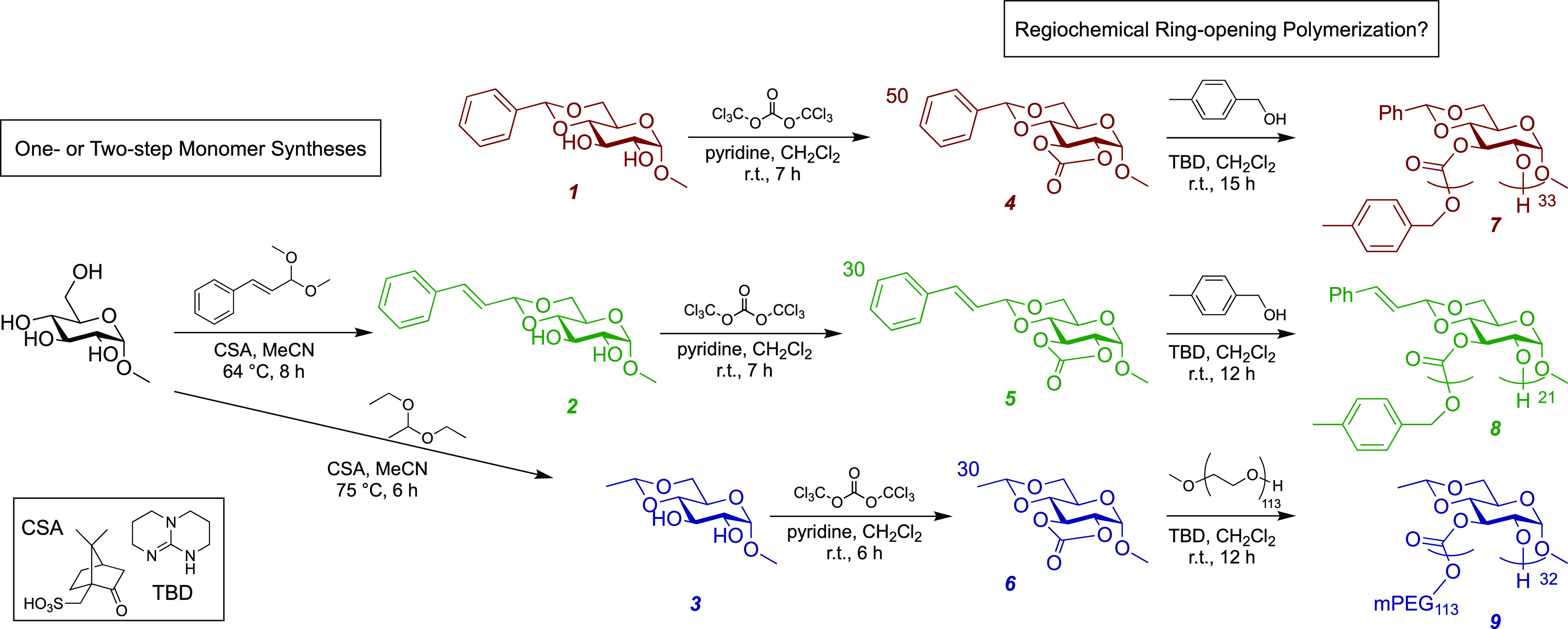
Scheme 1. Reaction Scheme for the Syntheses of the Three Five-Membered Glucose Carbonate Monomers (4–6), Followed by Their TBD-Catalyzed ROPs to Afford the Respective PGCs (7–9).
Scheme 2.

(a) Initiation of the series of 2,3-carbonate monomers (4–6) leads to two distinct unimer regiochemistries for each during TBD-catalyzed ROPs. (b) Isolated MBGC unimers (10, 11) were prepared for use as initiators, respectively, for in-depth regiochemical evaluation at the dimer stage. (c) MBGC dimer was also prepared with MBA as the initiator.
Results and Discussion
Three five-membered cyclic carbonate monomers with varying naturally sourced acetal protecting groups were prepared from commercially available methyl-α-d-glucopyranoside or methyl 4,6-O-benzylidene-α-d-glucopyranoside, 1, through two- or one-step sequences, as outlined in Scheme 1. The dimethyl acetal of cinnamaldehyde, prepared according to literature procedure,39 and the diethyl acetal of acetaldehyde, commercially available, were used for selective acetalizations at the 4- and 6-positions of methyl-α-d-glucopyranoside to afford the respective methyl-4,6-O-alkylidene-α-d-glucopyranosides, 2 and 3, in 86 and 95% yields, respectively. From each glucose derivative, having the 1-position protected by a methyl group and the 4- and 6-positions tied up into benzylidene (1, commercially available), cinnamylidene (2), or ethylidene (3) acetals, the five-membered cyclic 2,3-glucose carbonates (4–6) were synthesized through carbonylation reactions employing phosgene (generated in situ from triphosgene).36 Monomers 4–6 were named MBGC, MCGC, and MEGC based on the benzylidene, cinnamylidene, and ethylidene protecting groups at the 4- and 6-positions of the sugar ring, respectively. Installation of the carbonyls through the 2- and 3-positions was confirmed by 13C NMR spectroscopy, with the introduction of new peaks resonating at ca. 153 ppm, and by FT-IR spectroscopy, as the observance of carbonyl stretching bands at ca. 1800 cm–1, in agreement with the carbonate functionalities. The monomer structures were characterized additionally by further NMR spectroscopy studies, electrospray ionization mass spectrometry (ESI-MS), and single-crystal X-ray diffraction, the data from which can be found in the Supporting Information.
A series of three poly(2,3-α-d-glucose carbonate)s 7–9 was constructed through ROPs of 4–6 catalyzed by TBD and initiated by a primary alcohol (Scheme 1). PMBGC, 7, and PMCGC, 8, were prepared using 4-methylbenzyl alcohol (MBA) as the initiator. In contrast, MEGC (6) was initiated by a macroinitiator, mPEG113-OH, because the corresponding homopolymer was insoluble in various organic solvents when MBA was employed as the initiator, suggesting the ethylidene protecting group is of insufficient size to influence the overall polymer solubility favorably. The number-average molar masses (Mn) and dispersities (Đ) were determined using size exclusion chromatography (SEC), calibrated relative to polystyrene standards. The molar stoichiometries of monomer-to-initiator and the experimentally determined DPn values are included in the reaction sequences of Scheme 1. To investigate the polymer backbone regiochemistries, 1H NMR and 13C NMR spectra of each polymer were collected. Polymer backbone regioirregularities were suggested from indistinct and broad 1H resonances across the NMR spectra (Figure S4a), in addition to multiple broad 13C peaks for the backbone carbonate carbons at 152–155 ppm (Figure S4b), in comparison to narrow signals observed by 1H and 13C NMR for the corresponding monomers (see Figures S1–S3).
To quantitatively probe the ring-opening directionalities in the initiation step, unimers resulting from ring opening of the three five-membered glucose carbonates were synthesized, isolated, and characterized (Scheme 2a and Figures S5–S30). Each unimer was prepared by allowing 1 equiv of monomer, 2 equiv of MBA, and 0.02 equiv of TBD to undergo reaction in CH2Cl2 in a septum-sealed vial in a glovebox for 2 h before being quenched by addition of an aliquot of acetic acid. Various oligomers with repeat units of 2–4 were formed simultaneously with the desired unimers (Figure S5b); therefore, preparative SEC was employed to isolate the unimer fractions, as confirmed by high-resolution mass spectrometry (HRMS). Further characterization of the unimeric fractions showed the existence of two isomeric unimers per each composition, giving two distinctive spots by thin layer chromatography (TLC) (Figure S5c) and two sets of 1H resonances (Figures 1a and S6), suggesting regioisomers resulting from cleavage of the acyl-oxygen bond at either the 2- or 3-position from the tetrahedral intermediates during the ring-opening processes. The two isomers of each unimer fraction were produced in differing proportions. For the MBGC unimers (10, 11) and unimers derived from MCGC and MEGC (12–15), integration of 1H signals that were distinguishable as belonging to either the 3- or 2-unimer indicated, consistently, ca. 70:30 differential concentrations of the two regioisomers for each unimer type. However, the absolute quantitation and determination of configuration required in-depth 1D and 2D NMR studies. To facilitate such studies and determine their absolute configurations, they were separated by preparative TLC for further structural determination.
Figure 1.

(a) 1H NMR (500 MHz, CDCl3) spectra of a crude mixture of MBGC unimers (bottom), MBGC 3-unimer (10, middle), and MBGC 2-unimer (11, top), (b) COSY and HMBC spectra of MBGC 3-unimer (10), and (c) COSY and HMBC spectra of MBGC 2-unimer (11).
Data obtained from detailed structural characterization studies of MBGC unimers to determine the bond connectivities are presented in Figures 1 and S7–S14 and in Table S1. Homonuclear correlation spectroscopy (COSY), 1H–13C heteronuclear single quantum correlation (HSQC), and 1H–13C heteronuclear multiple-bond (HMBC) 2D-NMR analyses were employed to examine the structural details, thereby identifying bond cleavage preferences during the first ring opening step. The COSY spectrum of the high Rf isomer (Figure 1c) displayed a cross peak at (2.48, 4.21 ppm) resulting from the coupling between the protons of the hydroxyl group and C3–H, while no coupling signal was detected for the OH and proton at C2. Furthermore, a key 3JCH coupling in the HMBC spectrum was observed between the carbonate carbon and the proton connected with C2 at (4.66, 154.90 ppm), which correlated to the linkage of Cx–O2–C2. Taken together, these data agree that cleavage of the Cx–O3 bond of the monomer had occurred during the initiation process, placing the OH at position C3 and retaining attachment of the carbonyl to C2, therefore, this unimer isomer was named as the 2-unimer (11). The single-crystal X-ray structure of 11 (Table S1) further confirmed its exact configuration as the 2-unimer, showing a carbonate linkage at the 2-position of the glucose. In addition, the same characterization experiments were performed on the low Rf fraction isomer, confirming its structure as the 3-unimer (Figure 1b). Crosspeak signals resonating at (2.23, 3.71 ppm) in COSY indicated connection between the OH and C2, and at (5.14, 155.38 ppm) in the HMBC indicated connection between the carbonyl and C3–H. These 2D NMR data reflected the main structural differences between the 3-unimer and 2-unimer. The absolute crystal structure of the 3-unimer reinforced the exact bond connectivities in agreement with the NMR studies. Having identified the isomer structures and their 1H NMR resonances, the quantitative ratio of the two regioisomers of the initial fraction that contained the mixture of isolated unimers was determined to be a molar ratio of 1:2.3 (2-unimer:3-unimer), according to integrations of the C7–H signals (Figure 1a, the black dashed box with inset enlargement). The higher intensity peak of the 3-unimer resonating slightly upfield at 5.47 ppm, relative to the corresponding C7–H signal of the 2-unimer resonating at 5.55 ppm, allowed for quantitation of the extent to which Cx–O2 bond cleavage was preferred from tetrahedral intermediates along the addition–elimination mechanistic pathway during the overall initiation reaction procedure at room temperature.
The same 1D and 2D NMR analyses were applied to the unimers of MCGC and MEGC (12–15) to determine their structural configurations. In both of these cases, similar to that for the MBGC, the dominant bond cleavage during the initiation step happened at the Cx–O2 site to generate a majority fraction of 3-unimers. Interestingly, the ratios between the two unimer isomers were nearly equivalent at ca. 1:2.3 (2-unimer:3-unimer, Figure S6). Therefore, the acetal protecting group was found to have little to no effect on regioselectivities during the ring-opening initiation process with TBD as the catalyst under the conditions of room temperature in CH2Cl2.
To study the regioselectivity during the ring-opening propagation steps, MBGC dimers were synthesized using MBA as initiator at a 1:2 molar ratio of MBA:MBGC, 4, (Scheme 2c), followed by fractionation with preparative SEC. There were four different dimers existing in the dimer fraction, indicated by four distinct sets of proton resonances in the 1H NMR spectrum (Figure 2a, bottom spectrum). The dimer mixture was further separated using preparative TLC, followed by NMR analyses on each fraction (Figure S31). The lowest Rf fraction, as well as the most abundant species, was determined to be the 3,3′-dimer (16) (Figures S36–S38), involving both Cx–O2 cleavages of monomers in each ring-opening process. By comparison, the highest Rf fraction was composed of the 2,2′-dimer (18) (Figures S32–S34), involving both Cx–O3 cleavages. However, the middle Rf fraction contained an inseparable mixture of the other two dimers: 3,2′-dimer (17) and 2,3′-dimer (19) (Figure S35).
Figure 2.

(a) 1H NMR (500 MHz, CDCl3) spectra of a crude mixture of MBGC dimers (bottom curve: MBA as initiator; middle curve: MBGC 3-unimer as initiator; top curve: MBGC 2-unimer as initiator). (b) Scheme of the model reactions of 2-unimer (right) or 3-unimer (left) with TBD yielding unimer mixture with same molar ratio. 1H NMR (500 MHz, CD2Cl2) spectra of unimers and unimers with TBD after 9 min.
To further distinguish the middle two dimers, MBGC 3-unimer, 10, and MBGC 2-unimer, 11, were employed as pre-established unimer-based initiators to prepare the corresponding dimers, respectively (Scheme 2b). Ideally, it was expected that each experiment would lead to only two distinct dimers: 3,2′- and 3,3′-dimers from the 3-unimer vs 2,2′- and 2,3′-dimers from the 2-unimer. Instead, more than two sets of proton resonances were found in the 1H NMR spectra (Figure 2a, top and middle spectra), and the 3,3′-dimer was the most abundant species in both scenarios. These results suggested that other coincident reactions occurred during the ring-opening process of chain extensions from unimers to dimers.
Therefore, model studies of 2-unimer (or 3-unimer) mixed with TBD were conducted in CD2Cl2 to probe the hypothesis of a transcarbonylation mechanism (Figure 2b), especially involving intramolecular transcarbonylation. As expected, both 2-unimer and 3-unimer signals appeared in the 1H NMR spectra for both samples, although only one unimer was employed. The final molar ratio between the two unimers was ca. 26:74 in both scenarios, of which the primary component was the 3-unimer, suggesting the transcarbonylation favored the formation of 3-unimer. In consequence, the transcarbonylation reaction would be expected to scramble regioselectivies during TBD-catalyzed ring-opening reactions at room temperature.
Further complicating these experimental findings, when the unimer preparation was repeated at −78 °C, the elimination reaction from the tetrahedral intermediate favored Cx–O3 bond cleavage to form the 2-unimer and produced an opposite regiochemical molar ratio of 10:1 (2-unimer:3-unimer) (Figure S43). These differential regiochemical outcomes could be explained by either differential favoring of Cx–O2 vs Cx–O3 bond cleavage at room temperature vs −78 °C or inhibition of the transcarbonylation side reaction at the reduced temperature, or a combination of both.
Therefore, computational studies were pursued. Inspired by previous ROP mechanistic studies,18,38,40−42 four different structural arrangements of the initiator, monomer, and bifunctional catalyst TBD were calculated through a proton shift mechanism (Figure S39). The initiator attacked syn to the anomeric methoxy group in pathways a and b, while the initiator was anti to the methoxy group in pathways c and d, yielding an alcohol chain end at the 3-position (pathways a and c) or the 2-position (pathways b and d). The association of initiator was the rate-determining step in the ring-opening process, the barrier of which was accessible for all four pathways at room temperature. Similar energy levels were observed at the second transition state (TS2), resulting in possible bond cleavage at either site to produce 3-unimer or 2-unimer. Unfortunately, the differences of the Gibbs free energy barriers found between C–O2 cleavage and C–O3 cleavage were less than 2 kcal/mol, which was insufficient to make an accurate prediction of the product ratio. Noteworthy, however, the limiting energy barrier of intramolecular transcarbonylation reaction (Figure S41) found that ΔΔG was less than +12 kcal/mol for the TBD-assisted nucleophilic attack step (Figure S42), allowing the reaction to occur reversibly and readily at room temperature. The ring-opening propagation from each unimer would thereby afford four different isomeric dimers, which agreed with the experimental results.
Conclusions
The development of new synthetic routes for sustainable materials using renewable feedstocks is of critical current interest to deal with both the increasing global pressures from plastic pollution to the planet and the need to reduce dependence on petroleum-based chemicals. This work investigated the regiochemical outcomes for a series of naturally derived carbohydrate-based monomers that possess significantly greater chemical complexity than typical olefinic monomer building blocks. We intended to advance the development of glucose-based polycarbonates through determination of relationships between monomer and polymer composition, structure, reactivity, and properties.
Comprehensive 1D and 2D NMR spectroscopic analyses were used to study the regiochemical outcomes obtained from the organobase-catalyzed ring opening of glucose monomers having unique five-membered 2,3-cyclic carbonates, with variable 4,6-cyclic acetal protecting groups. The composition of the cyclic acetal protecting groups were essential to the solubility of the derived polymers, but provided little to no effect on the regioselective preference. An unexpected complication was found, in that transcarbonylation competed with ROP and led to scrambling of the backbone connectivities. Regioirregular backbone linkages were installed in the resulting 2,3-PGCs by a combination of variability in organocatalytic ring-opening directionality for five-membered cyclic glucose carbonates and coincident organocatalytic transcarbonylation side reactions. The exact regiochemical determinations within the polymer structures were complicated by signal overlaps in NMR analyses, however, small molecule model studies provided detailed information, experimentally and computationally. For each of three monomers, a certain degree of selectivity was observed during the initial ring-opening process, with the Cx–O2 bond cleavage being dominant for 3-unimer formation at room temperature. Coincident transcarbonylation reactions diminished the initial regiochemical preference. Interestingly, when the unimer study was repeated at −78 °C, the elimination reaction from the tetrahedral intermediate favored Cx–O3 bond cleavage and produced the opposite regiochemical outcome, likely due to inhibition of the transcarbonylation side reaction. Unfortunately, the polymerization at such low temperatures proceeded exceedingly slowly, so that the experimental effects of reduced temperature on regioselectivity for longer chain polymers could not be determined.
Ongoing studies continue for monomers with variable noncyclic protecting groups to pursue an in-depth understanding of composition–structure–topology–morphology–property relationships for this family of 2,3-PGCs. These abilities to tune the regiochemistry during polymerization of highly functional monomers provide hints at new chemical designs that could produce polymer structures with versatile properties and that are capable of structural metamorphosis to meet requirements of different applications. In addition, an ability to understand and tune the relative intra- vs intermolecular transcarbonylation events could be a powerful approach to innovative and adaptive materials.
Acknowledgments
This work is supported by the National Science Foundation (CHE-1610311, CHE-2003771, and DMREF-1629094), the Robert A. Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry (A-0001), and the use of the Texas A&M University Laboratory for Synthetic-Biologic Interactions. We appreciate contributions from Dr. Yohannes H. Rezenom for obtaining mass spectrometry data, and Dr. Nattamai Bhuvanesh and Dr. Joseph H. Reibenspies for providing single-crystal studies of the research reported in this article. We also thank Prof. Lei Fang for access to the preparative SEC, with experimental assistance from Dr. Xiaozhou Ji.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00545.
Experimental procedures; computational details; 1H NMR, 13C NMR, COSY, HSQC, HMBC, SEC, and X-ray data (PDF)
Crystallographic data for monomers and unimers (ZIP)
Crystallographic data for MBGC (CIF)
Crystallographic data for MBGC 2-unimer (CIF)
Crystallographic data for MBGC 3-unimer (CIF)
Crystallographic data for MCGC (CIF)
Crystallographic data for MEGC (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gandini A. Polymers from renewable resources: a challenge for the future of macromolecular materials. Macromolecules 2008, 41 (24), 9491–9504. 10.1021/ma801735u. [DOI] [Google Scholar]
- Tschan M. J.-L.; Brulé E.; Haquette P.; Thomas C. M. Synthesis of biodegradable polymers from renewable resources. Polym. Chem. 2012, 3 (4), 836–851. 10.1039/C2PY00452F. [DOI] [Google Scholar]
- Zhu Y.; Romain C.; Williams C. K. Sustainable polymers from renewable resources. Nature 2016, 540 (7633), 354–362. 10.1038/nature21001. [DOI] [PubMed] [Google Scholar]
- Kristufek S. L.; Wacker K. T.; Tsao Y.-Y. T.; Su L.; Wooley K. L. Monomer design strategies to create natural product-based polymer materials. Nat. Prod. Rep. 2017, 34 (4), 433–459. 10.1039/C6NP00112B. [DOI] [PubMed] [Google Scholar]
- Mohanty A. K.; Vivekanandhan S.; Pin J.-M.; Misra M. Composites from renewable and sustainable resources: Challenges and innovations. Science 2018, 362 (6414), 536–542. 10.1126/science.aat9072. [DOI] [PubMed] [Google Scholar]
- Llevot A.; Dannecker P. K.; von Czapiewski M.; Over L. C.; Söyler Z.; Meier M. A. Renewability is not enough: recent advances in the sustainable synthesis of biomass-derived monomers and polymers. Chem.—Eur. J. 2016, 22 (33), 11510–11521. 10.1002/chem.201602068. [DOI] [PubMed] [Google Scholar]
- Behabtu N.; Kralj S. Enzymatic Polymerization Routes to Synthetic–Natural Materials: A Review. ACS Sustain. Chem. Eng. 2020, 8 (27), 9947–9954. 10.1021/acssuschemeng.0c01664. [DOI] [Google Scholar]
- Worch J. C.; Prydderch H.; Jimaja S.; Bexis P.; Becker M. L.; Dove A. P. Stereochemical enhancement of polymer properties. Nat. Rev. Chem. 2019, 3 (9), 514–535. 10.1038/s41570-019-0117-z. [DOI] [Google Scholar]
- Fox S. C.; Li B.; Xu D.; Edgar K. J. Regioselective esterification and etherification of cellulose: a review. Biomacromolecules 2011, 12 (6), 1956–1972. 10.1021/bm200260d. [DOI] [PubMed] [Google Scholar]
- DeRosa C. A.; Luke A. M.; Anderson K.; Reineke T. M.; Tolman W. B.; Bates F. S.; Hillmyer M. A. Regioregular Polymers from Biobased (R)-1, 3-Butylene Carbonate. Macromolecules 2021, 54 (13), 5974–5984. 10.1021/acs.macromol.1c00828. [DOI] [Google Scholar]
- López-Vidal E. M.; Gregory G. L.; Kociok-Köhn G.; Buchard A. Polymers from sugars and CS 2: synthesis and ring-opening polymerisation of sulfur-containing monomers derived from 2-deoxy-d-ribose and d-xylose. Polym. Chem. 2018, 9 (13), 1577–1582. 10.1039/C8PY00119G. [DOI] [Google Scholar]
- Besset C. J.; Lonnecker A. T.; Streff J. M.; Wooley K. L. Polycarbonates from the polyhydroxy natural product quinic acid. Biomacromolecules 2011, 12 (7), 2512–2517. 10.1021/bm2003048. [DOI] [PubMed] [Google Scholar]
- Sahu P.; Bhowmick A. K.; Kali G. Terpene based elastomers: Synthesis, properties, and applications. Processes 2020, 8 (5), 553. 10.3390/pr8050553. [DOI] [Google Scholar]
- Zanchin G.; Leone G. Polyolefin thermoplastic elastomers from polymerization catalysis: Advantages, pitfalls and future challenges. Prog. Polym. Sci. 2021, 113, 101342. 10.1016/j.progpolymsci.2020.101342. [DOI] [Google Scholar]
- Sarkar P.; Bhowmick A. K. Synthesis, characterization and properties of a bio-based elastomer: polymyrcene. Rsc Advances 2014, 4 (106), 61343–61354. 10.1039/C4RA09475A. [DOI] [Google Scholar]
- Lonnecker A. T.; Lim Y. H.; Felder S. E.; Besset C. l. J.; Wooley K. L. Four different regioisomeric polycarbonates derived from one natural product, D-glucose. Macromolecules 2016, 49 (20), 7857–7867. 10.1021/acs.macromol.6b00591. [DOI] [Google Scholar]
- DiCiccio A. M.; Longo J. M.; Rodríguez-Calero G. G.; Coates G. W. Development of highly active and regioselective catalysts for the copolymerization of epoxides with cyclic anhydrides: an unanticipated effect of electronic variation. J. Am. Chem. Soc. 2016, 138 (22), 7107–7113. 10.1021/jacs.6b03113. [DOI] [PubMed] [Google Scholar]
- Song Y.; Yang X.; Shen Y.; Dong M.; Lin Y.-N.; Hall M. B.; Wooley K. L. Invoking Side-Chain Functionality for the Mediation of Regioselectivity during Ring-Opening Polymerization of Glucose Carbonates. J. Am. Chem. Soc. 2020, 142 (40), 16974–16981. 10.1021/jacs.0c05610. [DOI] [PubMed] [Google Scholar]
- Tsao Y.-Y. T.; Smith T. H.; Wooley K. L. Regioisomeric Preference in Ring-Opening Polymerization of 3′, 5′-Cyclic Phosphoesters of Functional Thymidine DNA Analogues. ACS Macro Lett. 2018, 7 (2), 153–158. 10.1021/acsmacrolett.7b00858. [DOI] [PubMed] [Google Scholar]
- Song Y.; Elsabahy M.; Collins C. A.; Khan S.; Li R.; Hreha T. N.; Shen Y.; Lin Y.-N.; Letteri R. A.; Su L.; et al. Morphologic Design of Silver-Bearing Sugar-Based Polymer Nanoparticles for Uroepithelial Cell Binding and Antimicrobial Delivery. Nano Lett. 2021, 21 (12), 4990–4998. 10.1021/acs.nanolett.1c00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L.; Faig A.; Abdelhamid D.; Uhrich K. Sugar-based amphiphilic polymers for biomedical applications: from nanocarriers to therapeutics. Acc. Chem. Res. 2014, 47 (10), 2867–2877. 10.1021/ar4003009. [DOI] [PubMed] [Google Scholar]
- Su L.; Khan S.; Fan J.; Lin Y.-N.; Wang H.; Gustafson T. P.; Zhang F.; Wooley K. L. Functional sugar-based polymers and nanostructures comprised of degradable poly (D-glucose carbonate) s. Polym. Chem. 2017, 8 (10), 1699–1707. 10.1039/C6PY01978A. [DOI] [Google Scholar]
- Gregory G. L.; Hierons E. M.; Kociok-Köhn G.; Sharma R. I.; Buchard A. CO 2-Driven stereochemical inversion of sugars to create thymidine-based polycarbonates by ring-opening polymerisation. Polym. Chem. 2017, 8 (10), 1714–1721. 10.1039/C7PY00118E. [DOI] [Google Scholar]
- Galbis J. A.; García-Martín M. d. G.; de Paz M. V.; Galbis E. Synthetic polymers from sugar-based monomers. Chem. Rev. 2016, 116 (3), 1600–1636. 10.1021/acs.chemrev.5b00242. [DOI] [PubMed] [Google Scholar]
- Dong M.; Song Y.; Wang H.; Su L.; Shen Y.; Tran D. K.; Letteri R. A.; Flores J. A.; Lin Y.-N.; Li J.; Wooley K. L. Degradable sugar-based magnetic hybrid nanoparticles for recovery of crude oil from aqueous environments. Polym. Chem. 2020, 11 (30), 4895–4903. 10.1039/D0PY00029A. [DOI] [Google Scholar]
- Xie Y.; Peng C.; Gao Y.; Liu X.; Liu T.; Joy A. Mannose-based graft polyesters with tunable binding affinity to concanavalin A. J. Polym. Sci., Part A: Polym. Chem. 2017, 55 (23), 3908–3917. 10.1002/pola.28776. [DOI] [Google Scholar]
- Ong Z. Y.; Yang C.; Gao S. J.; Ke X. Y.; Hedrick J. L.; Yan Yang Y. Galactose-Functionalized Cationic Polycarbonate Diblock Copolymer for Targeted Gene Delivery to Hepatocytes. Macromol. Rapid Commun. 2013, 34 (21), 1714–1720. 10.1002/marc.201300538. [DOI] [PubMed] [Google Scholar]
- Lin Y.-N.; Khan S.; Song Y.; Dong M.; Shen Y.; Tran D. K.; Pang C.; Zhang F.; Wooley K. L. A Tale of Drug-Carrier Optimization: Controlling Stimuli Sensitivity via Nanoparticle Hydrophobicity through Drug Loading. Nano Lett. 2020, 20 (9), 6563–6571. 10.1021/acs.nanolett.0c02319. [DOI] [PubMed] [Google Scholar]
- Song Y.; Ji X.; Dong M.; Li R.; Lin Y.-N.; Wang H.; Wooley K. L. Advancing the development of highly-functionalizable glucose-based polycarbonates by tuning of the glass transition temperature. J. Am. Chem. Soc. 2018, 140 (47), 16053–16057. 10.1021/jacs.8b10675. [DOI] [PubMed] [Google Scholar]
- Kamber N. E.; Jeong W.; Waymouth R. M.; Pratt R. C.; Lohmeijer B. G.; Hedrick J. L. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107 (12), 5813–5840. 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- Bexis P.; De Winter J.; Arno M. C.; Coulembier O.; Dove A. P. Organocatalytic Synthesis of Alkyne-Functional Aliphatic Polycarbonates via Ring-Opening Polymerization of an Eight-Membered-N-Cyclic Carbonate. Macromol. Rapid Commun. 2021, 42 (3), 2000378. 10.1002/marc.202000378. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Jones G. O.; Hedrick J. L.; Waymouth R. M. Fast and selective ring-opening polymerizations by alkoxides and thioureas. Nat. Chem. 2016, 8 (11), 1047–1053. 10.1038/nchem.2574. [DOI] [PubMed] [Google Scholar]
- Wei J.; Meng H.; Guo B.; Zhong Z.; Meng F. Organocatalytic ring-opening copolymerization of trimethylene carbonate and dithiolane trimethylene carbonate: Impact of organocatalysts on copolymerization kinetics and copolymer microstructures. Biomacromolecules 2018, 19 (6), 2294–2301. 10.1021/acs.biomac.8b00415. [DOI] [PubMed] [Google Scholar]
- Dove A. P. Organic catalysis for ring-opening polymerization. ACS Macro Lett. 2012, 1, 1409. 10.1021/mz3005956. [DOI] [PubMed] [Google Scholar]
- Buchard A.; Carbery D. R.; Davidson M. G.; Ivanova P. K.; Jeffery B. J.; Kociok-Köhn G. I.; Lowe J. P. Preparation of Stereoregular Isotactic Poly (mandelic acid) through Organocatalytic Ring-Opening Polymerization of a Cyclic O-Carboxyanhydride. Angew. Chem. 2014, 53 (50), 13858–13861. 10.1002/anie.201407525. [DOI] [PubMed] [Google Scholar]
- Felder S. E.; Redding M. J.; Noel A.; Grayson S. M.; Wooley K. L. Organocatalyzed ROP of a glucopyranoside derived five-membered cyclic carbonate. Macromolecules 2018, 51 (5), 1787–1797. 10.1021/acs.macromol.7b01785. [DOI] [Google Scholar]
- Gregory G. L.; Kociok-Köhn G.; Buchard A. Polymers from sugars and CO 2: Ring-opening polymerisation and copolymerisation of cyclic carbonates derived from 2-deoxy-d-ribose. Polym. Chem. 2017, 8 (13), 2093–2104. 10.1039/C7PY00236J. [DOI] [Google Scholar]
- Gregory G. L.; Jenisch L. M.; Charles B.; Kociok-Köhn G.; Buchard A. Polymers from sugars and CO2: synthesis and polymerization of a D-mannose-based cyclic carbonate. Macromolecules 2016, 49 (19), 7165–7169. 10.1021/acs.macromol.6b01492. [DOI] [Google Scholar]
- Dong J.-L.; Yu L.-S.-H.; Xie J.-W. A simple and versatile method for the formation of acetals/ketals using trace conventional acids. ACS omega 2018, 3 (5), 4974–4985. 10.1021/acsomega.8b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt R. C.; Lohmeijer B. G.; Long D. A.; Waymouth R. M.; Hedrick J. L. Triazabicyclodecene: a simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2006, 128 (14), 4556–4557. 10.1021/ja060662+. [DOI] [PubMed] [Google Scholar]
- Simon L.; Goodman J. M. The mechanism of TBD-catalyzed ring-opening polymerization of cyclic esters. J. Org. Chem. 2007, 72 (25), 9656–9662. 10.1021/jo702088c. [DOI] [PubMed] [Google Scholar]
- Chuma A.; Horn H. W.; Swope W. C.; Pratt R. C.; Zhang L.; Lohmeijer B. G.; Wade C. G.; Waymouth R. M.; Hedrick J. L.; Rice J. E. The reaction mechanism for the organocatalytic ring-opening polymerization of l-lactide using a guanidine-based catalyst: hydrogen-bonded or covalently bound?. J. Am. Chem. Soc. 2008, 130 (21), 6749–6754. 10.1021/ja0764411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.