

Abstract
Mulberrin (Mul) is a key component of the traditional Chinese medicine Romulus Mori with various biological functions. However, the effects of Mul on liver fibrosis have not been addressed, and thus were investigated in our present study, as well as the underlying mechanisms. Here, we found that Mul administration significantly ameliorated carbon tetrachloride (CCl4)-induced liver injury and dysfunction in mice. Furthermore, CCl4-triggerd collagen deposition and liver fibrosis were remarkably attenuated in mice with Mul supplementation through suppressing transforming growth factor β1 (TGF-β1)/SMAD2/3 signaling pathway. Additionally, Mul treatments strongly restrained the hepatic inflammation in CCl4-challenged mice via blocking nuclear factor-κB (NF-κB) signaling. Importantly, we found that Mul markedly increased liver TRIM31 expression in CCl4-treated mice, accompanied with the inactivation of NOD-like receptor protein 3 (NLRP3) inflammasome. CCl4-triggered hepatic oxidative stress was also efficiently mitigated by Mul consumption via improving nuclear factor E2-related factor 2 (Nrf2) activation. Our in vitro studies confirmed that Mul reduced the activation of human and mouse primary hepatic stellate cells (HSCs) stimulated by TGF-β1. Consistently, Mul remarkably retarded the inflammatory response and reactive oxygen species (ROS) accumulation both in human and murine hepatocytes. More importantly, by using hepatocyte-specific TRIM31 knockout mice (TRIM31Hep-cKO) and mouse primary hepatocytes with Nrf2-knockout (Nrf2KO), we identified that the anti-fibrotic and hepatic protective effects of Mul were TRIM31/Nrf2 signaling-dependent, relieving HSCs activation and liver fibrosis. Therefore, Mul-ameliorated hepatocyte injury contributed to the suppression of HSCs activation by improving TRIM31/Nrf2 axis, thus providing a novel therapeutic strategy for hepatic fibrosis treatment.
Keywords: Liver fibrosis, Mulberrin (Mul), TRIM31-Nrf2 axis, Hepatocyte injury, HSCs activation
Graphical abstract
1. Introduction
Liver fibrosis occurs in many types of liver disease, such as hepatitis and chronic alcoholism, and leads to scarring and injury to the liver [1]. Liver fibrosis is characterized by excessive accumulation of extracellular matrix (ECM) proteins, hepatocyte damage, distortion of the hepatic lobules, and changes in the vascular architecture [2,3]. HSCs activation plays an essential role in the pathogenesis of liver fibrosis. Activated HSCs can transform to myofibroblast-like cells, expressing α-SMA and secreting ECM that consists of various proteoglycans and proteins [4]. Presently, the most effective therapeutic approach for hepatic cirrhosis is liver transplantation, but its clinical application is limited due to the shortage of donor material, the prerequisite for expert technical support, and the high hospital costs [5]. If left untreated or without effective treatment, liver fibrosis can progress to cirrhosis or even hepatocellular carcinoma (HCC) [6]. Herein, exploring the mechanisms and finding promising therapeutic strategies are urgently necessary for liver fibrosis management.
Hepatic fibrosis is generally prolonged by chronic inflammatory response, and persistent inflammation is involved in hepatic fibrosis progression and cirrhosis development [7]. Continuous hepatic injury leads to inflammation pathology and inflammatory cells infiltration, such as macrophages and lymphocytes [8]. A key factor of hepatocyte-driven liver fibrosis is the activation of the pro-inflammatory NF-κB signaling in hepatocytes. The family of NF-κB transcription factors are crucial regulators of inflammatory processes [9]. NF-κB activation in injured hepatocytes results in the secretion of various pro-inflammatory cytokines and chemokines including interleukin 1β (IL-1β), tumor necrosis factor-α (TNF-ɑ), IL-6, and monocyte chemoattractant protein-1 (MCP-1) [10]. Chronic hepatic inflammation, derived from liver injury or infection, is a major driving force for liver fibrosis. Although previous work has reported that activated NF-κB is involved in fibrosis development, the exact contribution process still remains enigmatic.
The tripartite motif (TRIM) family consists of a RING domain, one or two B-box domains, and a coiled-coil domain, which contributes to a wide range of biological processes [11]. TRIM31, a member of the TRIM protein family, can mediate various pathological conditions including inflammatory diseases, viral infection and tumor progression [12]. Recently, TRIM31 was shown to attenuate NLRP3 inflammasome activation, subsequently accelerating IL-1β releases and alum-induced peritonitis in vivo [13]. More recently, TRIM31 over-expression was reported to reduce the effects of oxidized low-density lipoprotein (ox-LDL) on NLRP3 expression, pyroptosis, and inflammatory cytokine levels in vitro [14]. Under stimuli conditions, NLRP3 assembles into a large cytoplasmic complex through recruiting apoptosis associated speck-like protein (ASC) and Caspase-1, thereafter, causing the cleavage of pro-IL-1β, which enables its maturation and releases from cells depending on NF-κB activation [15]. Increasing studies have demonstrated that excessive NLRP3 inflammasome activation plays an essential role in the regulation of liver inflammation and fibrosis [16,17]. Therefore, we hypothesized that TRIM31/NLRP3 signaling pathway might be involved in liver fibrosis. However, as for this, little has been investigated and reported, and thus was explored in our study to disclose whether it could be a therapeutic target for liver fibrosis treatment.
Oxidative stress is another predominant pro-fibrogenic factor involved in liver fibrosis progression [18]. Liver fibrosis-associated oxidative stress is largely attributed to the abundant ROS production and weakened antioxidant capacity. Nrf2, as a key transcription factor, crucially protects cells against oxidative stress by promoting the expression of numerous antioxidative genes, such as heme oxygenase-1 (HO-1), NAD(P)H: quinone oxidoreductase (NQO1), glutamate-cysteine ligase modifier subunit (GCLM) and glutamate-cysteine ligase catalytic subunit (GCLC) [19,20]. Nrf2 signaling inactivation is intimately associated with the progression of liver fibrosis [21]. Compounds or approaches that can better Nrf2 activation have been reported to efficiently ameliorate hepatic fibrosis development [20,22,23]. Mulberrin (Mul) is a key component of the traditional Chinese medicine Romulus Mori and has been demonstrated to exert anti-inflammatory and antioxidant biological activities [24]. Mul could meliorate spinal cord injury by suppressing neuroinflammation, oxidative stress and neuronal death in vivo and in vitro [25]. Recently, Mul was shown to attenuate 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson's disease through inhibiting microglial activation and inflammatory response [26]. Given these effects of Mul, we supposed that Mul might have therapeutic potential against liver fibrosis.
In the present work, we found that Mul treatments significantly ameliorated CCl4-induced hepatic fibrosis in mice via depressing inflammation and oxidative stress. The anti-fibrotic, anti-inflammatory and antioxidant biofunctions of Mul were validated in HSCs and hepatocytes in vitro. An interaction between TRIM31 and Nrf2 was detected. Importantly, we identified that the protective effects of Mul against hepatic injury were TRIM31/Nrf2 signaling-dependent in hepatocytes, contributing to the suppression of HSCs activation and liver fibrosis. Together, Mul may be a promising therapeutic agent for the management of liver fibrosis.
2. Materials and methods
For extended Material and Methods, see online Supplementary materials and methods.
2.1. Animals and treatments
The male wild type (WT) C57BL/6 N mice (6- to 8-week-old; 22–25 g body weight) used in the current study were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All mice were housed in a constant temperature, humidity (controlled by GREE central air-conditioner, #GMV-Pd250W/NaB-N1, China) and pathogen-free-controlled environment (23 ± 25 °C, 50–60%) cage with a standard 12 h light/12 h dark cycle, plenty of water and food (pathogen-free) in their cages. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee in Chongqing Key Laboratory of Medicinal Resources in the Three Gorges Reservoir Region, School of Biological and Chemical Engineering, Chongqing University of Education (Chongqing, China). Mice received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH) in 1996.
2.1.1. Animal model 1#
WT mice were allowed to adapt to their living environment for 1 week before all experiment's proper starts. All mice were then randomly divided into 6 groups, including the Oil/Veh group (n = 20), Oil/Mul-H (high dosage 60 mg/kg; n = 20) group, CCl4/Veh group (n = 25), CCl4/Mul-L (low dosage 15 mg/kg; n = 25) group, CCl4/Mul-M (moderate dosage 30 mg/kg; n = 25) group, and CCl4/Mul-H (high dosage 60 mg/kg; n = 20) group.
2.1.2. Animal model 2#
To generate mice with a conditional knockout allele of TRIM31, the TRIM31Flox/Flox mice with C57BL/6 N background were constructed using CRISPR/Cas9-regulated genome engineering system. The exon 4/5 of TRIM31 was selected as conditional knockout region (cKO). Briefly, the selected exons of TRIM31 were flanked by two loxP sites, and therefore two single guide RNAs (gRNA1# and gRNA2#) targeting TRIM31 introns were designed. The targeting vector containing TRIM31 exon 4/5 flanked by two loxP sites and the two homology arms was used as the template. The targeting vector, gRNA1# and gRNA2#, and together with Cas9 were co-injected into fertilized eggs for cKO mouse production. The obtained mice, which had exon 4/5 flanked by two loxP sites on one allele, were used to establish TRIM31Flox/Flox mice. Hepatocyte-specific TRIM31 deletion (TRIM31Hep-cKO) mice were produced by mating TRIM31Flox/Flox mice with albumin-Cre (Alb-Cre) mice (Jackson Laboratory, Bar Harbor, Maine, USA). A simple schematic diagram has been indicated in Supplementary Fig. S13A. TRIM31Flox/Flox (Flox) mice littermates were used in the work as controls for the obtained TRIM31Hep-cKO mice. All TRIM31Flox/Flox and TRIM31Hep-cKO mice were then randomly divided into three subgroups, including the Oil/Veh group (n = 15), CCl4/Veh group (n = 20), and CCl4/Mul-H (high dosage 60 mg/kg; n = 15) group.
2.1.3. Establishments of hepatic fibrosis murine model in vivo
Toxic liver fibrosis was established by intraperitoneal (i.p.) injections of CCl4 (1 ml/kg, dissolved in corn oil at a ratio of 1:4) twice a week for 6 weeks. The mice were sacrificed 24 h after the final CCl4 (Aladdin, Shanghai, China) injection. Same volume of corn oil was subjected to the normal control group. Mul (Catalog number: #S-119; HPLC>98%; Chengdu Herbpurify CO., LTD, Chengdu, China) at 15, 30 or 60 mg/kg was administered to each group of mice by gavage daily. The dosages of Mul for animal treatment were referred to previous studies and our preliminary experiments [25,26]. Vehicle groups of mice received 0.9% saline. Animal experimental protocols were presented in Fig. 1A. During the treatments, body weights of mice were recorded weekly. Survival rates of mice were measured. Blood samples were taken either from the tail vein or via the cardiac puncture. The sampled blood was collected into EDTA-coated tubes through retro-orbital bleeding, and centrifuged at 5000 rpm for 10 min at 4 °C. The supernatant was then collected and stored at −80 °C for further biological analysis. The liver samples were either immediately frozen in liquid nitrogen and kept at −80 °C or fixed with 4% paraformaldehyde for histological analysis.
Fig. 1.

Mulberrin ameliorates hepatic dysfunction in CCl4-induced mice. (A) The experiment design scheme. (B) Body weights of each group of mice. (C) Survival rate of mice from all groups was quantified. Serum (D) TBIL, (E) ALT, AST, ALP, and (F) LDH levels were examined. Representative data were expressed as mean ± SEM (n = 10 per group). **P < 0.01 and ***P < 0.001 vs the Oil/Veh group; +P < 0.05 and ++P < 0.01 vs the CCl4/Veh group.
2.2. In vitro experiments
2.2.1. Cells treatment
Human hepatocyte cell line L02 was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Human hepatic stellate cell (HSC) line LX2 was obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). All cells were maintained in Dulbecco's Modified Eagle Medium (DMEM; Gibco, USA) with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin in a 5% CO2 incubator at 37 °C. To imitate the in vivo hepatic injury, cells were incubated with recombinant TGF-β1 (#240-B-002 and #7666-MB; R&D system, USA), recombinant TNF-α (#210-TA; R&D system), recombinant IL-1β (#201-LB; R&D system), LPS (derived from Escherichia coli (055:B5); #L2880, Sigma-Aldrich, St. Louis, USA) or H2O2 (Sigma-Aldrich) as demonstrated in figure legends to investigate the effects of Mul on liver fibrosis in vitro.
2.2.2. Primary hepatocytes isolation and culture
Mouse primary hepatocytes used in the study were isolated from wild type or Nrf2-knockout (Nrf2KO) mice with C57BL/6J background (Jackson Laboratory, Bar Harbor, ME) using liver perfusion method as described previously [27,28]. Briefly, mice abdominal cavity was opened under a painless anesthesia condition. Thereafter, the liver tissue was carefully perfused with 1×liver perfusion medium (#17701-038, Gibco™) and 1×liver digest medium (#17703-034, Gibco™) via the portal vein. Subsequently, 100 μm steel mesh was used to grind and filter the digested liver samples. The mice primary hepatocytes were then collected through centrifuging the filter liquor at 800 rpm, 4 °C for 5 min, and were further purified using 50% percoll solution (#17-0891-01, GE Healthcare Life Sciences). The obtained hepatocytes were maintained in DMEM medium containing 10% FBS and 1% penicillin-streptomycin and cultured at 37 °C in a cell incubator with 5% CO2.
2.2.3. Primary HSCs isolation
Primary HSCs were isolated from wild type C57BL/6 N mice. Briefly, HSCs were isolated through collagenase-pronase perfusion of livers as described previously with the minor modifications [29,30]. Liver of mice was digested using Collagenase IV (Sigma-Aldrich, USA) and Pronase E (Sigma-Aldrich) dissolved in PB buffer. Suspension of dispersed cells was layered through gradient centrifugation in Nycodenz (Sigma-Aldrich) according to manufacturer's instructions. Isolated HSCs were cultured in DMEM (Gibco) containing 10% FBS and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The medium was changed 24 h after seeding to remove dead cells and debris.
2.2.4. Vectors establishment and transfection
For in vitro transfection, TRIM31 si-RNAs (siTRIM31), Nrf2 siRNAs (siNrf2), and the corresponding negative control siRNAs (Ctrl/siRNAs) were obtained from Generay Biotechnology (Shanghai, China). Transfection for TRIM31 or Nrf2 knockdown was performed using Lipofectamine 3000 reagent (Invitrogen Life Technologies, Carlsbad, USA) according to the provider's instructions. To overexpress TRIM31, the entire coding region of human TRIM31 was introduced to a replication-defective adenoviral vector under the control of the cytomegalovirus promoter. Recombinant adenoviruses expressing Flag tagged TRIM31 protein were purified. Adenovirus was infected at a multiplicity of infection of 50 in cells for 24 h.
2.3. Histological and immunohistochemical (IHC) analysis
To explore histopathologic changes, the liver tissues were fixed with 10% neutral formalin, embedded in paraffin, and then sectioned transversely (5-μm-thick). The thin liver tissue sections were then stained with hematoxylin and eosin (H&E). The deposition of collagen fibers in hepatic sections was measured using Masson staining and Sirius red staining by the conventional protocols. Fibrosis stage was assessed according to the Ishak score [31]. As for IHC staining, embedded liver sections were dewaxed, and antigens were retrieved via sodium citrate heating. Endogenous peroxidase was removed by adding 30% H2O2, and an immunohistochemical pen was used to draw a circle around the tissue. Then, 5% goat serum (#C0265, Beyotime Biotechnology) was added to block the liver tissues. The liver sections were then incubated with primary antibodies (Supplementary Table 1) at 4 °C overnight. Sections were then washed, followed by incubation with secondary antibodies (Supplementary Table 1) for 1 h at room temperature. IHC staining was observed using 3,3′-diaminobenzidine (DAB) substrate kit (#ab64238, Abcam, USA) and were counterstained with hematoxylin. All images were captured under a microscope. The percentage of collagen areas or positive signal expression fields was measured by Image Pro Plus software (Media Cybernetics, USA).
2.4. Examination for oxidative stress markers
Commercial assay kits for the examination of hepatic malondialdehyde (MDA; #A003-1-2), catalase (CAT; #A007-1-1), superoxide dismutase (SOD; #A001-3-2) and glutathione (GSH; #A006-2-1) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Hydrogen peroxide (H2O2) levels in liver were tested using commercially available kit (#S0038; Beyotime Biotechnology) in line with the provider suggested. Hepatic lipid peroxidation was examined by thiobarbituric acid reactive substances (TBARS) formation [32]. TBARS Assay Kit (#801192, ZeptoMetrix Corporation, USA) was used for the measurements of liver TBARS contents following the manufacturer's protocols.
2.5. Immunofluorescence (IF) staining
For IF analysis, the frozen liver sections or cells after treatments were washed with PBS and were then blocked in 10% goat serum (#C0265, Beyotime Biotechnology) containing 0.3% Triton X-100 (#ST797, Beyotime Biotechnology) for 1 h at room temperature and incubated with primary antibodies (Supplementary Table 1) at 4 °C overnight. Samples were then washed, and secondary fluorescent antibodies (Supplementary Table 1) were prepared for incubation at room temperature in dark for 45 min. After washing, 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride solution (DAPI; #C1006, Beyotime Biotechnology) was added to the samples for nuclei staining. Images were captured under a fluorescence microscopy.
2.6. Statistical analysis
Statistical analysis was conducted using GraphPad Prism 8.0 (San Diego, CA, USA). Data represented as mean ± standard error of the mean (SEM) unless otherwise indicated. All analysis were repeated independently with similar results at least three times. Differences between two groups were analyzed using Student's t-test. One-way analysis of variance (ANOVA) with Tukey's post hoc tests were performed for comparisons between multiple groups. P value < 0.05 was considered indicative of statistical significance. The experimenters were blinded to the animal grouping information.
3. Results
3.1. Mulberrin ameliorates hepatic dysfunction and fibrosis in CCl4-induced mice
In the present study, a mouse model with hepatic fibrosis was established using CCl4 to explore the regulatory effects of Mul on liver injury (Fig. 1A). As displayed in Fig. 1B, CCl4 injection significantly reduced the body weights of mice, but were moderately rescued by Mul at higher dosages (30 and 60 mg/kg). CCl4 treatment led to poorer survival rates of mice over the course of the experiment compared with Oil/Veh group, while Mul treatments reduced the mortality of CCl4-challenged mice (Fig. 1C). CCl4 injection significantly induced the hepatic dysfunction and injury in mice, as evidenced by the increased serum TBIL, ALT, AST, ALP and LDH contents; however, all these results caused by CCl4 were efficiently reversed by Mul treatments in a dose-dependent manner (Fig. 1D–F). These findings initially revealed the protective effects of Mul against CCl4-triggered hepatic damage. Furthermore, we showed that under normal physiological conditions, Mul at 60 mg/kg had no significant influences on the changes of body weights, animal survival and hepatic functions, indicating the safe use of Mul in vivo.
3.2. Mulberrin inhibits CCl4-induced activation of TGF-β1/SMADs signaling in liver of mice
H&E staining showed that CCl4 led to evident histological changes in liver tissues, confirming the hepatic injury. Sirius red and Masson's Trichrome staining demonstrated that CCl4-challenged mice exhibited severer collagen deposition and fibrosis in liver sections than that of the Oil/Veh group. Notably, these histological alterations and hepatic fibrosis were significantly mitigated by Mul treatments via a concentration-dependent fashion (Fig. 2A–D). Furthermore, the levels of three key liver fibrosis hallmarks including hyaluronic acid, PC III and Collagen IV were strongly promoted by CCl4, while being strongly ameliorated in Mul-treated mice compared with the model group (Fig. 2E–G). These results demonstrated that Mul could mitigate liver fibrosis progression in vivo.
Fig. 2.

Mulberrin reduces liver fibrosis in CCl4-treated mice. (A) H&E, Sirius red and Masson's Trichrome staining of liver sections. (B) Quantification for fibrosis score. (C,D) Quantification for fibrotic areas by Sirius red and Masson's Trichrome staining, respectively. Serum contents of (E) hyaluronic acid, (F) procollagen III (PC III) (G) and Collagen IV were measured. Representative data were expressed as mean ± SEM (n = 5 for histological analysis, or 10 for biological analysis per group). Magnification: × 100. **P < 0.01 and ***P < 0.001 vs the Oil/Veh group; +P < 0.05, ++P < 0.01 and +++P < 0.001 vs the CCl4/Veh group. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further reveal the protective effect of Mul in the mouse model with liver fibrosis, α-SMA, Col-I and TGF-β1/SMADs signaling that plays a contributory role in fibrosis, were then explored. IF staining showed that Mul significantly reduced the positive expression of α-SMA and Col-I in liver samples of CCl4-challenged mice compared with CCl4/Veh group (Fig. 3A–D). Consistently, CCl4-enhanced mRNA expression levels of fibrotic genes including α-SMA, collagen types I (Col1a1), collagen types III (Col3a1) and TGF-β1, were markedly reversed by Mul treatments in a dose-dependent manner (Fig. 3E). Western blotting demonstrated that the protein expression levels of TGF-β1, p-SMAD2/3 and α-SMA in liver were strongly down-regulated by Mul in CCl4-treated mice compared with the model group (Fig. 3F). Therefore, Mul could restrain TGF-β1/SMADs signaling to meliorate hepatic fibrosis.
Fig. 3.

Mulberrin inhibits CCl4-induced activation of TGF-β1/SMADs signaling in liver of mice. IF staining for (A) α-SMA and (B) Col-I expression in liver sections from each group of mice. Quantification for positive fluorescent intensity of (C) α-SMA and (D) Col-I following IF assays. (E) RT-qPCR results for fibrogenic genes including α-SMA, Col1a1, Col3a1 and TGF-β1 in liver of all groups of mice. (F) Western blotting analysis for TGF-β1, p-SMAD2/3 and α-SMA protein expression in liver tissues of all groups of mice. Representative data were expressed as mean ± SEM (n = 3 per group). Magnification: × 200. ***P < 0.001 vs the Oil/Veh group; +P < 0.05 and ++P < 0.01 vs the CCl4/Veh group.
3.3. Mulberrin ameliorates hepatic inflammation in CCl4-challenged mice
Inflammatory response was then investigated to reveal whether it might be a therapeutic target for Mul to perform its protective function. F4/80 and CD68 are macrophage markers and indicate the infiltration of macrophages, which is a sign of inflammation [33]. IHC staining showed that CCl4 injection significantly up-regulated the positive expression of F4/80 and CD68 in liver sections, and these effects were remarkably ameliorated in mice co-treated with Mul (Fig. 4A–C). By ELISA analysis, we found that Mul treatments strongly reduced the systematic inflammatory cytokines and chemokine in CCl4-challenged mice, as proved by the decreased serum IL-1β, TNF-α, IL-6 and CXCL-10 contents (Fig. 4D). Consistently, CCl4-elevated expression levels of inflammatory genes such as IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 were significantly abolished in mice with Mul administration (Fig. 4E). NF-κB signaling is a classic pathway for inflammatory response induction through promoting the releases of pro-inflammatory factors [9]. Western blotting analysis subsequently showed that CCl4 strongly promoted the phosphorylation of IKKα, IκBα, and NF-κB/P65 in liver tissues, which were, however, efficiently abolished by Mul treatments. Meanwhile, Mul restored total IκBα expression in liver of CCl4-treated mice (Fig. 4F), indicating the suppressed activation of NF-κB signaling pathway. Together, these findings illustrated that Mul could repress inflammatory response induced by CCl4 through retarding NF-κB signaling.
Fig. 4.

Mulberrin ameliorates hepatic inflammation in CCl4-challenged mice. (A) IHC staining for F4/80 and CD68 expression in hepatic sections. (B) F4/80- and (C) CD68-positive areas were quantified after IHC assays. (D) ELISA analysis for serum contents of IL-1β, TNF-α, IL-6 and CXCL-10 from the shown groups of mice. (E) RT-qPCR analysis for inflammatory factors including IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 in liver tissues from the shown groups of mice. (F) Western blotting analysis for hepatic p-IKKα, p-IκBα, IκBα and p-NF-κB/p65 protein expression levels. Representative data were expressed as mean ± SEM (n = 3 for IHC, RT-qPCR and western blotting assays, or 8 for biological analysis per group). Magnification: ×200. **P < 0.01 and ***P < 0.001 vs the Oil/Veh group; +P < 0.05 and ++P < 0.01 vs the CCl4/Veh group.
3.4. Mulberrin mediates TRIM31/NLRP3 signaling pathway in liver of CCl4-treated mice
TRIM31/NLRP3 signaling is involved in the mediation of inflammation under various stimuli [13,14], and was previously reported to regulate NF-κB pathway to subsequently control various cellular events, particularly inflammatory response [9]. Here, we notably found that CCl4 challenge significantly reduced the expression of TRIM31 in liver samples, evidenced by the weakened fluorescent intensity; however, such effect was greatly abolished by Mul treatments (Fig. 5A and B). Consistently, RT-qPCR results confirmed that Mul administration markedly up-regulated TRIM31 expression levels in liver of CCl4-challenged mice. On the contrary, hepatic NLRP3 and its down-streaming signal ASC stimulated by CCl4 were strongly decreased by Mul (Fig. 5C). As expected, the protein expression levels of TRIM31 restrained by CCl4 were efficiently restored in mice with Mul administration. However, the expression levels of NLRP3 and its associated inflammasome were greatly diminished by Mul in liver of CCl4-challenged mice, as evidenced by the markedly decreased NLRP3, ASC, Caspase-1, mIL-1β and mIL-18, which were all comparable with the fibrosis model group (Fig. 5D and E). These findings indicated that Mul could mediate TRIM31/NLRP3 signaling to mitigate hepatic fibrosis.
Fig. 5.

Mulberrin mediates TRIM31/NLRP3 signaling pathway in liver of CCl4-treated mice. (A) IF staining for TRIM31 in hepatic sections from all groups of mice. (B) TRIM31-positive fluorescent intensity was quantified following IF staining. (C) RT-qPCR analysis for hepatic TRIM31, NLRP3 and ASC gene expression levels. (D,E) Western blotting analysis for TRIM31, NLRP3, ASC, Caspase-1, mature IL-1β (mIL-1β) and mature IL-18 (mIL-18) protein expression levels in liver tissues of all groups of mice. Representative data were expressed as mean ± SEM (n = 3 per group). Magnification: × 200. **P < 0.01 and ***P < 0.001 vs the Oil/Veh group; +P < 0.05, ++P < 0.01 and +++P < 0.001 vs the CCl4/Veh group.
3.5. Mulberrin restrains hepatic oxidative stress in CCl4-challenged mice
Oxidative stress is another key for the progression of liver fibrosis [18] and was thus investigated. As shown in Fig. 6A–C, CCl4 challenge significantly increased the oxidative stress hallmarks including H2O2, MDA and TBARS in liver of fibrotic mice, while being dose-dependently reduced by Mul treatments. In contrast, antioxidants such as CAT, GSH and SOD were strongly restored in liver of Mul-treated mice after CCl4 challenge (Fig. 6D–F). IF staining confirmed that hepatic lipid peroxidation product 4-HNE was highly induced by CCl4, whereas being greatly abrogated in mice with Mul supplementation (Fig. 6G and H). What's more, compared with CCl4/Veh group, mice co-treated with Mul exhibited higher expression levels of antioxidant genes including HO-1, NQO1, GCLC and GCLM (Fig. 6I). NAD(P)H oxidase is a key source of ROS production and plays an essential role in liver injury and fibrosis [34]. The mRNA expression levels of NAD(P)H oxidase subunits including NOX1, NOX2, NOX4 and p22phox were highly up-regulated in liver of CCl4-challenged mice, which were, however, efficiently mitigated by Mul treatments (Fig. 6J). IHC and western blotting assays showed that CCl4 injection clearly reduced Nrf2 nuclear translocation in liver tissues compared with the Oil/Veh group. Of note, Mul markedly rescued nuclear Nrf2 expression in hepatic samples compared with CCl4/Veh group (Fig. 6K-M), indicating the improved activation of Nrf2 signaling. Cytochrome P450 2E1 (CYP2E1) aggravates oxidative stress and ROS generation, leading to various kinds of liver cell damage and death and fibrotic liver progression [35]. As expected, we found that CCl4-treated mice exhibited higher CYP2E1 expression levels than that of the Oil/Veh group, while being significantly abrogated by Mul consumption via a dose-dependent manner (Fig. 6N). These results elucidated that Mul could facilitate Nrf2 activation to attenuate hepatic oxidative stress under fibrotic pressure.
Fig. 6.

Mulberrin restrains hepatic oxidative stress in CCl4-challenged mice. Examination for (A) H2O2, (B) MDA, (C) TBARS, (D) CAT, (E) GSH and (F) SOD in liver tissues of all groups of mice. (G) IF staining for 4-HNE expression in hepatic sections. (H) Positive fluorescent intensity of 4-HNE was quantified after IF analysis. (I) RT-qPCR analysis for antioxidants including HO-1, NQO1, GCLC and GCLM in liver tissues. (J) RT-qPCR results for oxidative stress markers NOX1, NOX2, NOX4 and p22phox gene expression levels in liver samples. (K) IHC staining for hepatic Nrf2 expression levels. (L) Nrf2-positive nucleus was quantified following IHC assay. (M) Nuclear and cytoplastic Nrf2 protein expression levels were measured using western blotting analysis. (N) Western blotting results for hepatic CYP2E1 protein expression levels. Representative data were expressed as mean ± SEM (n = 3 for IHC, IF, RT-qPCR and western blotting assays, or 8 for biological analysis per group). Magnification: × 200. **P < 0.01 and ***P < 0.001 vs the Oil/Veh group; +P < 0.05 and ++P < 0.01 vs the CCl4/Veh group.
3.6. Mulberrin reduces the expression of fibrosis markers in TGF-β1-activated LX-2 cells
Our in vivo results indicated the protective effects of Mul against fibrosis, inflammation and oxidative stress in CCl4-treated mice. To confirm the function of Mul, in vitro experiments were subsequently performed using human or mouse hepatocyte and HSC cell lines. At first, CCK-8 analysis was used to examine the safe use of Mul. As displayed in Supplementary Figs. 1A and B, Mul treatments at various concentrations had no significant influences on the survival of human HSC cell line LX-2 and hepatocyte L02, respectively, indicating the non-cytotoxicity of Mul. Considering that TGF-β1 plays an important role in hepatic fibrosis and is an inducer of the fibrotic response [36], TGF-β1 was then exposed to HSCs for its stimulation. RT-qPCR results showed that the expression levels of fibrosis markers α-SMA, Col1a1, Col3a1 and Fibronectin were markedly up-regulated by TGF-β1 in LX-2 cells, which were significantly and dose-dependently decreased by Mul, particularly starting from 10 μM (Fig. 7A). Thereafter, 25 μM (low dosage), 50 μM (moderate concentration) and 100 μM (high dosage) of Mul were used for further in vitro studies. CCK-8 results confirmed the safe use of 100 μM Mul treatment from 0 to 72 h both in LX-2 and L02 cells (Supplementary Figs. 1C and D). IF analysis then validated that Mul treatments reduced α-SMA expression in LX-2 cells after TGF-β1 stimulation (Fig. 7B and C). Finally, western blotting results demonstrated that compared with TGF-β1/Veh group, Mul-treated cells exhibited lower protein expression levels of α-SMA and p-SMAD2/3 in LX-2 cells following TGF-β1 stimulation (Fig. 7D). To verify the suppressive effects of Mul on fibrosis in vitro, primary mouse HSCs were isolated and employed. Morphology of the isolated primary mouse HSCs was displayed in Supplementary Fig. 1E. CCK-8 results confirmed the safety of Mul (100 μM) exposure to primary HSCs for 24 h (Supplementary Figs. 1F and G). As expected, TGF-β1 stimulation significantly increased the expression of α-SMA, Col1a1, Col3a1 and Fibronectin in primary HSCs, while being strongly abolished upon Mul co-culture (Supplementary Fig. 2A). IF staining and western blotting results confirmed that TGF-β1-induced elevation of α-SMA and p-SMAD2/3 in primary mouse HSCs was highly eliminated after Mul incubation (Supplementary Figs. 2B–D).
Fig. 7.

Mulberrin reduces the expression of fibrosis markers in TGF-β1-activated LX-2 cells. (A) After stimulation with TGF-β1 (10 ng/ml) for 24 h in the absence or presence of the indicated concentrations of Mul, LX-2 cells were collected for RT-qPCR analysis of α-SMA, Col1a1, Col3a1 and Fibronectin. (B–G) LX-2 cells were subjected to TGF-β1 (10 ng/ml) treatment for 24 h with or without Mul treatments (25, 50 or 100 μM). Then, all cells were harvested for studies as follows. (B) IF staining for α-SMA expression in LX-2 cells. (C) Quantification for α-SMA positive fluorescent intensity was exhibited. (D) Western blotting results for α-SMA, p-SMAD2/3 protein expression levels in LX-2 cells. (E) RT-qPCR analysis for inflammatory factors including TNF-α, IL-1β, IL-6, IL-18, MCP-1 and CXCL10, and (F) oxidative stress related molecules such as HO-1, NQO1, NOX2 and NOX4 in LX-2 cells. (G) Western blotting results for nuclear and cytoplastic Nrf2 protein expression levels in LX-2 cells. Representative data were expressed as mean ± SEM (n = 3 per group). Scale bar, 10 μm. ***P < 0.001 vs the Ctrl/Veh group; +P < 0.05 and ++P < 0.01 vs the TGF-β1/Veh group. Mul-L, 25 μM; Mul-M, 50 μM; and Mul-H, 100 μM.
Inflammatory response and oxidative stress in HSCs are involved in fibrosis progression [37,38], and thus were investigated in LX-2 cells or primary HSCs under TGF-β1 stimuli. As shown in Fig. 7E, TGF-β1 stimulation led to inflammatory response in LX-2 cells, proved by the markedly enhanced gene expression levels of TNF-α, IL-1β, IL-6, IL-18, MCP-1 and CXCL10; however, such event caused by TGF-β1 was significantly abrogated following Mul exposure. Such anti-inflammatory effects mediated by Mul were validated in mouse primary HSCs upon TGF-β1 stimulation (Supplementary Fig. 2E). Moreover, HO-1 and NQO1 gene expression levels were highly decreased in LX-2 cells and mouse primary HSCs stimulated by TGF-β1, while being efficiently reversed upon Mul co-treatment. In contrast, NOX2 and NOX4 expression levels were greatly abolished by Mul in a dose-dependent fashion (Fig. 7F and Supplementary Fig. 2F). In response to TGF-β1, nuclear Nrf2 protein expression levels were significantly weakened, whereas being markedly rescued after Mul exposure. Opposite profile was detected in the expression of cytoplastic Nrf2 both in LX-2 and primary mouse HSCs (Fig. 7G and Supplementary Fig. 2G). These in vitro findings suggested that Mul could restrain the activation, inflammation and oxidative stress in HSCs.
Given the crucial role of inflammatory response in the modulation of HSCs activation, LX-2 cells were then exposed to recombinant human TNF-α (rTNF-α) and IL-1β (rIL-1β) to further examine the effects of Mul on inflammation-caused fibrosis in vitro. As shown in Supplementary Figs. 3A–D, we found that rTNF-α and rIL-1β treatment significantly increased α-SMA expression in LX-2 cells, proved by the stronger fluorescent intensity, which were, however, markedly abolished after Mul treatment. Consistently, in response to rTNF-α and rIL-1β stimulation, fibrosis markers α-SMA, Col1a1, Col3a1 and Fibronectin were greatly induced, whereas being remarkably diminished in LX-2 cells with Mul exposure (Supplementary Figs. 3E and F). As expected, the protein expression levels of α-SMA, p-SMAD2 and p-SMAD3 enhanced by rTNF-α and rIL-1β were dramatically ameliorated upon Mul treatment (Supplementary Figs. 3G and H). These in vitro data suggested that inflammatory factors contributed to HSCs activation, which could be attenuated by Mul.
3.7. Mulberrin mitigates inflammatory response through mediating TRIM31/NLRP3 signaling in L02 cells
Inflammatory response in hepatocytes participates in HSCs activation and liver fibrosis [3,17,39], and thus was explored by the use of L02 cells and the isolated primary mouse hepatocytes (Supplementary Fig. 4A). CCK-8 results suggested that 100 μM of Mul treatment for 24 h was safe for primary mouse hepatocytes culture (Supplementary Figs. 4B and C). We found that TGF-β1 stimulation significantly increased the expression of IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 in L02 cells and the mouse primary hepatocytes, and such effect was remarkably abolished upon Mul co-incubation (Fig. 8A and Supplementary Fig. 5A). Consistently, lower contents of IL-1β and TNF-α in supernatants were detected collected from Mul-exposed L02 cells and the mouse primary hepatocytes after TGF-β1 treatment, which was comparable with TGF-β1/Veh group (Fig. 8B and Supplementary Fig. 5B). LPS is frequently used to induce inflammatory response [40]. As expected, LPS-enhanced IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 in L02 cells or supernatants were considerably mitigated by Mul (Fig. 8C and D), confirming the anti-inflammatory bioactivity of Mul in vitro. We also found that Mul-treated L02 cells exhibited markedly decreased TGF-β1 both in the collected supernatants and cells compared with the LPS group (Supplementary Figs. 6A and B). IF results showed that TRIM31-positive expression was markedly restrained by TGF-β1 or LPS in L02 cells and the isolated primary murine hepatocytes, which were, however, strongly restored by Mul treatments (Fig. 8E–G, and Supplementary Figs. 5C and D). In line with in vivo results, RT-qPCR and western blotting analysis verified that in response to TGF-β1 or LPS, Mul treatments significantly improved TRIM31 and total IκBα expression levels, while decreased NLRP3, ASC, Caspase-1, p-IκBα, p-NF-κB, mIL-1β and mIL-18 expression levels in L02 cells and mouse primary hepatocytes (Fig. 8H and I, and Supplementary Figs. 5E–G). These results illustrated that Mul suppressed inflammatory response by mediating TRIM31/NLRP3 signaling in vitro.
Fig. 8.

Mulberrin mitigates inflammatory response through mediating TRIM31/NLRP3 signaling in L02 cells. L02 cells were incubated with TGF-β1 (10 ng/ml) or LPS (100 ng/ml) for 24 h in the presence or absence of Mul (25, 50 or 100 μM). Subsequently, all cells and supernatants were harvested for experiments as follows. (A) RT-qPCR analysis for inflammatory factors including TNF-α, IL-1β, IL-6, IL-18, MCP-1 and CXCL10 in L02 cells. (B) ELISA analysis for TNF-α and IL-1β in the collected supernatants. (C) Inflammatory gene markers as shown were measured using RT-qPCR assay. (D) TNF-α and IL-1β contents in the collected supernatants were examined using ELISA analysis. (E) TRIM31 expression in L02 cells by IF staining. (F) TRIM31-positive fluorescent intensity was quantified following IF assay. Rt-qPCR results for (G) TRIM31, (H) NLRP3 and ASC gene expression levels. (I) Western blotting results for the protein expression levels of TRIM31, NLRP3, ASC, Caspase-1, p-IκBα, IκBα, p-NF-κB/P65, mIL-1β and mIL-18 in L02 cells. Representative data were expressed as mean ± SEM (n = 3 for IF, RT-qPCR and western blotting assays, or 6 for ELISA analysis per group). Scale bar, 10 μm. **P < 0.01 and ***P < 0.001 vs the Ctrl/Veh group; +P < 0.05 and ++P < 0.01 vs the TGF-β1/Veh or LPS/Veh group; #P < 0.05, ##P < 0.01 and ###P < 0.001.
To explore whether improving TRIM31 was required for Mul to suppress inflammation, TRIM31 was then silenced in L02 cells by transfecting with its siRNAs. As shown in Supplementary Figs. 7A and B, transfection efficacy of siTRIM31 was confirmed by RT-qPCR and western blotting assays. Because siTRIM31-1# exerted the strongest inhibitory effect on TRIM31, and thus was selected for subsequent in vitro analysis. RT-qPCR results indicated that TGF-β1-increased expression of IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 was further accelerated upon TRIM31 knockdown. Importantly, Mul-ameliorated expression of these inflammatory factors was completely abolished by siTRIM31 (Supplementary Fig. 8A). IF and western blotting results confirmed that after TGF-β1 stimulation, Mul-improved TRIM31 expression was strongly abolished in L02 cells with TRIM31 silence (Supplementary Figs. 8B–D). As expected, under TGF-β1 stimuli, TRIM31 knockdown significantly abrogated the capacity of Mul to suppress NLRP3, ASC, Caspase-1, p-NF-κB, mIL-1β and mIL-18 protein expression levels in L02 cells (Supplementary Fig. 8D). These supporting data demonstrated that Mul-inhibited hepatocyte inflammation was largely through increasing TRIM31 expression.
3.8. Mulberrin meliorates oxidative stress via improving Nrf2 signaling in L02 cells
The regulatory role of Mul on oxidative stress and Nrf2 signaling was further explored in human hepatocytes and the isolated mouse primary hepatocytes. DCF-DA staining showed that TGF-β1 exposure clearly elevated ROS production in L02 cells and the mouse primary hepatocytes, which was strongly abrogated by Mul treatments. Furthermore, H2O2-induced ROS generation was also diminished by Mul (Fig. 9A and Supplementary Figs. 9A and B), confirming the antioxidant bioactivity of Mul. Moreover, Mul-incubated L02 cells and primary murine hepatocytes showed lower H2O2 and MDA contents after TGF-β1 or H2O2 stimulation (Fig. 9B and C, and Supplementary Figs. 9C and D). On the contrary, SOD, CAT and GSH activities were greatly reduced in TGF-β1- or H2O2-exposed L02 cells and primary murine hepatocytes, whereas being efficiently rescued by Mul (Fig. 9D–F, and Supplementary Figs. 9E–G). HO-1, NQO1, GCLC and GCLM gene expression levels decreased by TGF-β1 or H2O2 were considerably abolished by Mul co-incubation in L02 cells or primary murine hepatocytes (Fig. 9G and Supplementary Fig. 9H). Nevertheless, NOX1, NOX2, NOX4 and p22phox stimulated by TGF-β1 or H2O2 were highly down-regulated by Mul in L02 cells and primary murine hepatocytes (Fig. 9H and Supplementary Fig. 9I). IF staining thereafter showed that nuclear Nrf2 was highly decreased in L02 cells and primary murine hepatocytes after TGF-β1 or H2O2 exposure, which was reversed upon Mul co-treatment (Fig. 9I and J, and Supplementary Figs. 9J and K). Western blotting results confirmed the capacity of Mul to improve Nrf2 nuclear translocation both in L02 cells and primary murine hepatocytes under fibrotic or oxidative stresses (Fig. 9K and Supplementary fig. 9L).
Fig. 9.

Mulberrin meliorates oxidative stress via improving Nrf2 signaling in L02 cells. L02 cells were subjected to 24 h of TGF-β1 (10 ng/ml) or H2O2 (100 μM) incubation combined with or without Mul (25, 50 or 100 μM). Next, all cells were harvested for studies as follows. (A) DCF-DA staining for ROS production in L02 cells. Scale bar, 50 μm. Cellular (B) H2O2, (C) MDA, (D) SOD, (E) CAT and (F) GSH contents or activities were examined. RT-qPCR results for (G) antioxidants including HO-1, NQO1, GCLC and GCLM, and (H) oxidative stress hallmarks NOX1, NOX2, NOX4 and p22phox gene expression levels in L02 cells. (I) IF staining for Nrf2 expression in L02 cells. Scale bar, 10 μm. (J) Quantification for nuclear Nrf2-positive fluorescent intensity was exhibited. (K) Western blotting analysis for nuclear and cytoplastic Nrf2 protein expression levels in L02 cells. Representative data were expressed as mean ± SEM (n = 3 for DCF-DA, IF, RT-qPCR and western blotting assays, or 6 for biological analysis per group). **P < 0.01 and ***P < 0.001 vs the Ctrl/Veh group; #P < 0.05 and ##P < 0.01.
To further investigate whether Nrf2 was required for Mul to perform its antioxidant function, Nrf2 was then knocked down in L02 cells. RT-qPCR and western blotting results showed that siNrf2-2# showed the highest transfection efficacy (Supplementary Figs. 7C and D), and thus was chosen for subsequent analysis. DCF-DA staining indicated that in TGF-β1-stimulated L02 cells, Mul-ameliorated ROS generation was significantly diminished upon Nrf2 silence (Supplementary Fig. 10A). Consistently, H2O2 and MDA levels restrained by Mul were also restrengthened by siNrf2 in TGF-β1-exposed L02 cells (Supplementary Figs. 10B and C). In contrast, siNrf2 markedly eliminated the capacity of Mul to improve SOD and GSH levels in L02 cells under TGF-β1 stimuli (Supplementary Figs. 10D and E), along with decreased HO-1 and NQO1 expression levels (Supplementary Fig. 10F). In hepatocytes with TGF-β1 exposure, Mul-suppressed NOX2 and NOX4 gene expression levels were almost abolished upon Nrf2 deletion (Supplementary Fig. 10G). Western blotting confirmed that after TGF-β1 stimulation, Nrf2 was almost undetectable both in nucleus and cytoplasm either with or without Mul exposure (Supplementary Fig. 10H). Taken together, all these data depicted that Mul exerted antioxidant function mainly through improving Nrf2 signaling.
3.9. Effects of mulberrin on fibrosis markers of LX-2 cells cultured in conditional medium from TGF-β1-treated hepatocytes
Given that the inflammatory response and oxidative stress in hepatocytes are crucial for HSCs activation and fibrosis, conditional medium (CM) derived from L02 cells was collected and subjected to LX-2 culture to further explore the underlying mechanisms (Fig. 10A). CM from TGF-β1-treated L02 cells markedly increased the expression of α-SMA, Col1a1 and Col3a1 in LX-2 cells, which was significantly ameliorated by Mul (Fig. 10B). IF staining by α-SMA indicated that CM derived from TGF-β1-exposed L02 cells led to HSCs activation, while being greatly ameliorated upon Mul treatment (Fig. 10C and D). CM obtained from TGF-β1-stimulated L02 cells significantly promoted the TGF-β1/SMADs signaling in LX-2 cells, which was almost diminished after Mul treatment, as indicated by the decreased expression of TGF-β1, p-SMAD2/3 and α-SMA (Fig. 10E). These findings elucidated that under fibrotic stresses, hepatocyte damage contributed to HSCs activation, and such effect could be abolished by Mul.
Fig. 10.

Effects of mulberrin on fibrosis markers of LX-2 cells cultured in conditional medium from TGF-β1-treated hepatocytes. L02 cells were incubated with or without TGF-β1 (10 ng/ml) for 24 h. Then, the culture media was collected and mixed with fresh media at 1:1 ratio, which was served as the conditional medium (CM). Next, LX-2 cells were incubated in the CM with or without Mul-H (100 μM) treatment for another 24 h. Finally, all LX-2 cells were collected for studies as follows. (A) Scheme for the in vitro experimental design. (B) RT-qPCR results for fibrosis markers including α-SMA, Col1a1 and Col3a1. (C,D) IF staining for α-SMA expression in LX-2 cells. Positive α-SMA fluorescent intensity was quantified. (E) Western blotting analysis for TGF-β1, α-SMA, p-SMAD2/3 protein expression levels. Representative data were expressed as mean ± SEM (n = 3 per group). Scale bar, 10 μm. **P < 0.01 and ***P < 0.001 vs the CM-Ctrl/Veh group; ++P < 0.01 vs the CM-TGF-β1/Veh group.
3.10. Mulberrin inhibits fibrosis via the improvements of TRIM31 and Nrf2 signaling pathways
Persistent inflammation can promote HSCs activation and liver fibrosis [7]. We therefore explored the effect of Mul-mediated hepatocyte inflammation on HSCs activation. To study the cell- to- cell crosstalk between hepatocytes and HSCs, we established an in vitro CM system derived from L02 cells, which was then collected for LX-2 cell culture [40]. We found that CM derived from TGF-β1-incubated L02 cells indeed promoted α-SMA, Col1a1, Col3a1 and Fibronectin expression levels in LX-2 cells, which were, however, accelerated upon TRIM31 and Nrf2 deletion. Additionally, in LX-2 cells exposed to CM from TGF-β1-stimulated L02 cells, Mul-inhibited expression of these fibrotic genes was considerably abolished by siTRIM31 and siNrf2 (Fig. 11A). Activation of LX-2 cells provoked by CM-TGF-β1 was further aggravated when TRIM31 and Nrf2 was ablated in L02 cells, as proved by the increased expression of α-SMA in LX-2 cells. Meanwhile, siTRIM31 and siNrf2 significantly eliminated the inhibitory effect of Mul on α-SMA expression in LX-2 cells cultured in CM derived from TGF-β1-treated L02 cells (Fig. 11B and C). Under fibrotic pressure, CM derived from siTRIM31 and siNrf2 L02 cells markedly exacerbated TGF-β1, p-SMAD2/3 and α-SMA expression in LX-2 cells. What's more, CM obtained from TGF-β1-incubated L02 cells with TRIM31 or Nrf2 deletion significantly abolished the capacity of Mul to reduce the expression of TGF-β1, p-SMAD2/3 and α-SMA in LX-2 cells (Fig. 11D and E). Collectively, these findings demonstrated that the TRIM31-Nrf2 axis in hepatocytes was crucial for Mul to restrain HSCs activation in vitro.
Fig. 11.

Mulberrin inhibits fibrosis via the improvements of TRIM31 and Nrf2 signaling pathways. L02 cells were transfected with siTRIM31-1# or siNRf2-2# for 24 h for the knockdown of TRIM31 and Nrf2, respectively, and were then stimulated by TGF-β1 (10 ng/ml) for another 24 h in the absence or presence of Mul-H (100 μM). The cultured medium was then collected, and mixed with fresh medium at 1:1 ratio, served as CM. The CM was then subjected to the culture of LX-2 cells. After 24 h, LX-2 cells and the cultured supernatants were harvested for the following studies. (A) RT-qPCR results for fibrosis markers including α-SMA, Col1a1, Col3a1 and Fibronectin. (B,C) IF staining for α-SMA expression in LX-2 cells. Quantification for positive α-SMA fluorescent intensity was shown. (D,E) Western blotting results for TGF-β1, α-SMA, p-SMAD2/3 protein expression levels in LX-2 cells. (F) ELISA analysis for IL-1β, TNF-α, IL-18 and CXCL-10 in the collected supernatants. (G) Western blotting analysis for protein expression levels of cellular TRIM31, NLRP3, ASC, Caspase-1 and p-NF-κB/P65, and nuclear Nrf2. Representative data were expressed as mean ± SEM (n = 3 per group). Scale bar, 10 μm. **P < 0.01 and ***P < 0.001 vs the CM-Ctrl group; +P < 0.05 and ++P < 0.01 vs the CM-TGF-β1 group; #P < 0.05 and ##P < 0.01.
Moreover, CM derived from TGF-β1-incubated L02 cells led to higher contents of IL-1β, TNF-α, IL-18 and CXCL-10 in supernatants, which were significantly accelerated upon TRIM31 and Nrf2 knockdown. Similarly, Mul-reduced concentrations of these inflammatory factors were markedly restrengthened by siTRIM31 or siNrf2 (Fig. 11F). Western blotting suggested that CM-TGF-β1 exposure strongly reduced TRIM31 and nuclear Nrf2 protein expression levels in LX-2 cells, and such effects were further weakened upon TRIM31 and Nrf2 deletion. Mul-improved expression of TRIM31 and Nrf2 was also significantly abolished when TRIM31 and Nrf2 were ablated. In contrast, NLRP3, ASC, Caspase-1 and p-NF-κB protein expression levels potentiated by CM-TGF-β1 were further exacerbated by siTRIM31 and siNrf2. Consistently, Mul-inhibited expression of these signals was strongly restored upon TRIM31 and Nrf2 silence (Fig. 11G). The expression changes of TRIM31/NLRP3 and Nrf2 signaling in LX-2 cells might be attributed to the severer inflammatory conditions.
3.11. Anti-fibrosis effects of mulberrin are TRIM31-dependent in CCl4-treated mice
To confirm the interaction between hepatocytes and HSCs mediated by Mul under fibrotic stresses in vivo, the hepatocyte-specific TRIM31 knockout (TRIM31Hep-cKO) mice were generated (Supplementary Fig. 11A). Western blotting results confirmed that TRIM31 was not detected in liver of TRIM31Hep-cKO mice (Supplementary Figs. 11B and C). H&E staining demonstrated that there were no evident histological changes in liver sections between TRIM31Flox and TRIM31Hep-cKO groups of mice (Supplementary Fig. 11D). IF staining validated the successful TRIM31 deletion in liver of TRIM31Hep-cKO mice (Supplementary Fig. 11E). In response to CCl4, Mul improved the body weights of mice, which was, however, abolished in TRIM31Hep-cKO mice (Fig. 12A). As shown in Fig. 12B–D, Mul failed to mitigate the elevation of serum TBIL, LDH, ALT, AST and ALP in the CCl4/TRIM31Hep-cKO mice. Likewise, the effects of Mul to reduce fibrotic parameters including hyaluronic acid, PC III and Collagen IV levels were impeded in TRIM31Hep-cKO mice after CCl4 challenge (Fig. 12E–G). Furthermore, histological staining suggested that Mul treatment failed to restrain the collagen deposition in liver of TRIM31Hep-cKO mice induced by CCl4, along with restored fibrosis scores (Fig. 13A–D). IF staining confirmed that after CCl4 injection, the function of Mul to reduce α-SMA and Col-I accumulation in liver sections was almost diminished upon hepatocyte-specific TRIM31 knockout (Fig. 14A–D). Consistently, Mul failed to repress α-SMA, Col1a1, Col3a1 and TGF-β1 gene expression levels in liver of CCl4-treated TRIM31Hep-cKO mice (Fig. 14E). Similarly, the effects of Mul to decrease TGF-β1, p-SMAD2/3 and α-SMA protein expression levels were also completely diminished in CCl4/TRIM31Hep-cKO mice (Fig. 14F). Together, these findings elucidated that Mul might exert its anti-fibrotic effects through improvement of hepatocyte TRIM31 signaling.
Fig. 12.

Hepatic protective effects of mulberrin are TRIM31-dependent in vivo. (A) Body weights of TRIM31Flox or TRIM31Hep-cKO mice were recorded. Serum (B) TBIL, (C) LDH levels, (D) ALT, AST, ALP contents, (E) hyaluronic acid, (F) PC III, and (G) Collagen IV levels were examined. Representative data were expressed as mean ± SEM (n = 9 per group). #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference.
Fig. 13.

Anti-fibrosis effects of mulberrin are TRIM31-dependent in CCl4-treated mice. (A) H&E, Sirius red and Masson's Trichrome staining of liver sections from TRIM31Flox or TRIM31Hep-cKO mice. Quantification results for (B) fibrosis scores, (C) Sirius red- and (D) Masson's Trichrome-positive staining areas were exhibited. Representative data were expressed as mean ± SEM (n = 6 or 9 per group). Magnification: × 100. #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 14.

Mulberrin suppresses collagen deposition by improving TRIM31 in CCl4-challenged mice. IF staining for (A) α-SMA and (B) Col-I expression in hepatic sections from TRIM31Flox or TRIM31Hep-cKO mice. Quantification for positive fluorescent intensity of (C) α-SMA and (D) Col-I following IF assays. (E) RT-qPCR results for α-SMA, Col1a1, Col3a1 and TGF-β1 in liver of all groups of mice. (F) Western blotting analysis for TGF-β1, p-SMAD2/3 and α-SMA protein expression in liver tissues of all groups of mice. Representative data were expressed as mean ± SEM (n = 4 per group). Magnification: × 200. #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference.
3.12. Anti-inflammatory effects of mulberrin are TRIM31-dependent in CCl4-treated mice
In this regard, IHC staining suggested that in response to CCl4 stimuli, Mul failed to mitigate F4/80 and CD68 positive expression in liver sections of TRIM31Hep-cKO mice (Fig. 15A and B). TRIM31Hep-cKO also abolished the capacity of Mul to reduce serum IL-1β, TNF-α and IL-6 contents in CCl4-challenged mice (Fig. 15C). Consistently, after CCl4 injection, hepatic IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 gene expression levels were also failed to be diminished by Mul upon hepatocyte-specific TRIM31 deletion (Fig. 15D). As expected, Mul lost its inhibitory effects on NLRP3, ASC, Caspase-1, p-IKKα, p-IκBα, p-NF-κB, mIL-1β and mIL-18 protein expression levels in liver of CCl4/TRIM31Hep-cKO mice (Fig. 15E). These findings suggested that hepatocyte TRIM31 expression was required for Mul to perform its anti-inflammatory role.
Fig. 15.

Anti-inflammatory actions of mulberrin in liver fibrosis mice regulated by TRIM31 in vivo. (A) IHC staining for F4/80 and CD68 expression in hepatic sections from TRIM31Flox or TRIM31Hep-cKO mice. (B) F4/80- and CD68-positive areas were quantified after IHC assays. (C) ELISA analysis for serum contents of IL-1β, TNF-α and IL-6 from the shown groups of mice. (E) RT-qPCR analysis for IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10 in liver tissues from the shown groups of mice. (F) Western blotting analysis for hepatic NLRP3, ASC, Caspase-1, p-IKKα, p-IκBα, IκBα, p-NF-κB/p65, mIL-1β and mIL-18 protein expression levels. Representative data were expressed as mean ± SEM (n = 4 for IHC, RT-qPCR and western blotting assays, or 9 for biological analysis per group). Magnification: × 200. #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference.
3.13. Anti-fibrotic effect of mulberrin is Nrf2-dependent in the primary HSCs
To further explore whether Nrf2 in hepatocytes was necessary for Mul to control HSCs activation, primary hepatocytes were isolated from the wild type (WT) and Nrf2-knockout (KO) mice and were employed in vitro. Western blotting and IF staining assays confirmed the successful knockout of Nrf2 in the extracted primary mouse hepatocytes (Supplementary Figs. 12A and B). We then established an in vitro CM system derived from Nrf2WT or Nrf2KO primary hepatocytes with or without TGF-β1 stimuli, which was then collected for mouse primary HSCs culture in the presence or absence of Mul treatment (Fig. 16A). Firstly, we showed that Mul remarkably improved nucleus Nrf2 expression, but decreased cytoplastic Nrf2 in primary Nrf2WT hepatocytes after TGF-β1 stimulation. As expected, Nrf2 was undetectable in Nrf2KO hepatocytes (Fig. 16B). We then found that Mul failed to reduce α-SMA, Col1a1, Col3a1 and Fibronectin expression levels in primary HSCs cultured in CM derived from TGF-β1-incubated Nrf2KO hepatocytes (Fig. 16C–E). Consistently, the effects of Mul to down-regulate TGF-β1, p-SMAD2/3 and α-SMA protein expression levels were also significantly diminished in mouse primary HSCs cultured in CM obtained from Nrf2KO hepatocytes under TGF-β1 stimuli (Fig. 16F). These data elucidated that the anti-fibrotic effects of Mul were at least in part attributed to hepatocyte Nrf2 expression.
Fig. 16.

Anti-fibrotic effect of mulberrin is Nrf2-dependent in the primary HSCs. Primary hepatocytes isolated from Nrf2WT or Nrf2KO mice were stimulated by TGF-β1 (10 ng/ml) for 24 h in the absence or presence of Mul-H (100 μM). The cultured medium was then collected, and mixed with fresh medium at 1:1 ratio, served as CM. The CM was then subjected to the culture of primary HSCs. After 24 h, these primary HSCs were collected for the following studies. (A) Scheme for the in vitro experimental design. (B) Western blotting analysis for nuclear and cytoplastic Nrf2 protein expression levels in the treated primary hepatocytes. (C) IF staining for α-SMA expression in the treated primary HSCs. (D) Positive fluorescent intensity of α-SMA expression was quantified. (E) RT-qPCR results for α-SMA, Col1a1, Col3a1 and Fibronectin in primary HSCs. (F) Western blotting analysis for TGF-β1, p-SMAD2/3 and α-SMA protein expression levels in primary HSCs. Representative data were expressed as mean ± SEM (n = 4 per group). Scale bar, 50 μm. #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference.
Finally, to further explore the underlying molecular mechanisms, we examined whether there might be a possible interaction between TRIM31 and Nrf2. Endogenous immunoprecipitation and western blotting assays confirmed the interaction between TRIM31 and Nrf2 in human hepatocytes (Fig. 17A and B). Additionally, the interaction between TRIM31 and Nrf2 in L02 cells was weakened upon TGF-β1 stimulation (Fig. 17C). The interaction between TRIM31 and Nrf2 was verified in human HSCs (Fig. 17D and E), and was receded under TGF-β1 stress (Fig. 17F). Moreover, knockdown of TRIM31 expression by siRNA further reduced Nrf2 nuclear translocation after TGF-β1 treatment in L02 and LX2 cells (Fig. 17G). On the contrary, Nrf2 nuclear expression was up-regulated by TRIM31 in a concentration-dependent manner (Fig. 17H). As expected, IHC staining showed that Mul-rescued expression of nuclear Nrf2 in liver of CCl4-challenged mice was completely abrogated upon hepatic-specific TRIM31 knockout (Fig. 17I and J), which was validated by western blotting assay (Fig. 17K), partially demonstrating the requirement of TRIM31 for Mul to improve Nrf2 signaling. Together, these findings suggested that there might be an interaction between TRIM31 and Nrf2.
Fig. 17.

Exploration of the interaction between TRIM31 and Nrf2. (A,B) The interaction of endogenous TRIM31 with Nrf2 in L02 cells was examined by immunoprecipitation assay using the indicated antibodies. (C) After TGF-β1 (10 ng/ml) incubation for 24 h, interaction between TRIM31 and Nrf2 in L02 cells was assessed by immunoprecipitation analysis using anti-TRIM31 antibody and by immunoblotting assay with anti-Nrf2 antibody. (D,E) The interaction of endogenous TRIM31 with Nrf2 in LX2 cells was explored with immunoprecipitation assay using the shown antibodies. (F) LX2 cells were exposed to TGF-β1 (10 ng/ml) culture for 24 h and were then harvested to explore the interaction between TRIM31 and Nrf2 by immunoprecipitation analysis using anti-TRIM31 antibody and by immunoblotting assay with anti-Nrf2 antibody. (G) After siTRIM31 transfection for 24 h, L02 and LX2 cells were stimulated by TGF-β1 (10 ng/ml) for another 24 h. Then, all cells were collected for western blotting analysis of TRIM31 protein expression levels in whole cells, and Nrf2 expression in nucleus and cytoplasm, respectively; siCtrl was used as a control. (H) Representative western blot analysis of cellular TRIM31, nuclear and cytoplastic Nrf2 protein expression levels in L02 and LX2 cells infected with Ad-Flag-TRIM31 for 24 h, followed by an additional 24 h of TGF-β1 (10 ng/ml) exposure. (I) IHC staining for Nrf2 positive expression in hepatic sections from TRIM31Flox or TRIM31Hep-cKO mice. (J) Quantification for Nrf2-positive nucleus was shown. (K) Western blotting analysis for nuclear and cytoplastic Nrf2 protein expression levels in liver of the indicated groups of mice. Representative data were expressed as mean ± SEM (n = 4 per group). Magnification: × 200. #P < 0.05, ##P < 0.01 and ###P < 0.001; ns, no significant difference.
4. Discussion
Hepatic inflammatory response and oxidative stress contribute to fibrosis progression [2,4,7,8,18]. Active TGF-β promotes hepatocyte injury and the activation of HSCs and fibroblast, leading to collagen formation [41]. TGF-β1 binds to its receptor and contributes to the phosphorylation of mothers against decapentaplegic homolog SMAD2/3, which is required for their nuclear translocation and transcriptional modulation of their fibrotic target genes including α-SMA, Col1a1, Col3a1 and Fibronectin, ultimately resulting in collagen deposition and fibrosis [2,4,7,42]. Unfortunately, liver fibrosis is still an intractable disease without effective therapeutic options due to its intricate pathogenesis [3,5]. Approaches to restrain inflammation, ROS production and collagen deposition are pivotal and promising for the treatment of liver fibrosis [10,20,22]. Mulberrin (Mul), as a key component of Chinese medicine Romulus Mori, has anti-inflammatory and antioxidant properties to meliorate tissue injury [13,14].
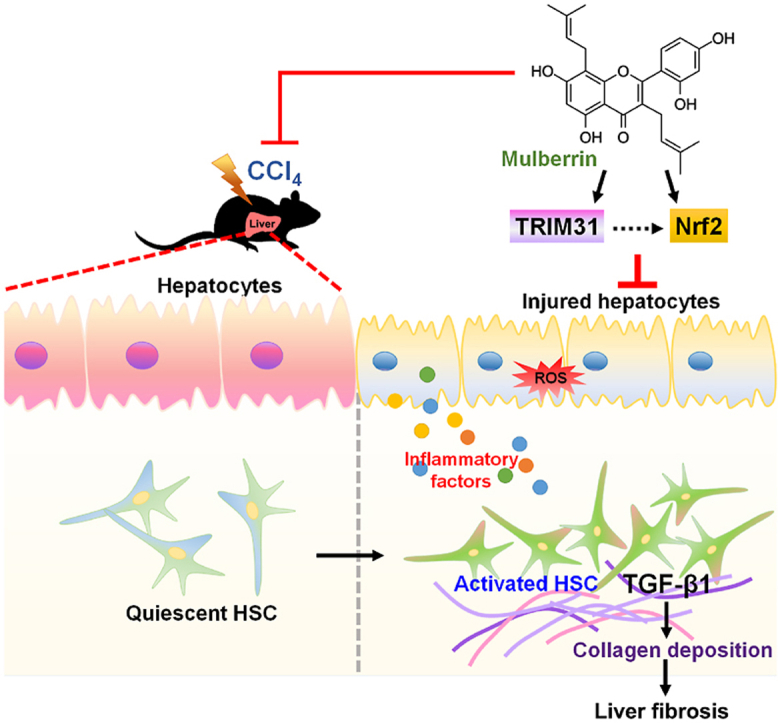
Here in our present study, we for the first time demonstrated that Mul exerted protective effects against liver fibrosis both in vivo and in vitro. Briefly, our findings showed that Mul administration significantly reduced hepatic dysfunction and toxicity in CCl4-challenged mice, proved by the reduced serum AST, ALT, AKP, TBIL and LDH levels. We then revealed that CCl4-indcued hepatic collagen deposition was considerably ameliorated by Mul supplementation through suppressing TGF-β1/SMAD2/3 signaling, thereby decreasing the expression of fibrotic genes, such as α-SMA, Co1a1 and Col3a1. More studies indicated that Mul remarkably suppressed inflammatory response in liver of CCl4-injected mice, evidenced by the down-regulated expression and releases of pro-inflammatory cytokines or chemokines through blocking NF-κB signaling pathway. We surprisingly found that CCl4 injection significantly reduced TRIM31, while promoted NLRP3 inflammasome in liver samples; however, these effects were dramatically reversed by Mul treatments. Moreover, CCl4-triggered hepatic oxidative stress was strongly ameliorated in mice with Mul administration through improving Nrf2 signaling. Our in vitro studies subsequently confirmed that Mul could suppress TGF-β1-induced HSCs activation by decreasing α-SMA expression. In line with in vivo findings, TGF-β1- or LPS-induced inflammatory response in L02 cells and primary mouse hepatocytes was remarkably alleviated by Mul through restraining NF-κB and NLRP3 signaling pathways, and such anti-inflammatory property of Mul was largely TRIM31-dependent. Additionally, ROS production induced by TGF-β1 or H2O2 was also strongly repressed by Mul exposure both in human and mouse hepatocytes through improving Nrf2 activation. Importantly, we found that CM derived from TGF-β1-stimulated L02 cells led to LX-2 cell activation, whereas being restrained by Mul. Of note, these anti-fibrotic effects mediated by Mul were significantly abolished upon TRIM31 and Nrf2 knockdown in L02 cells. More in vivo and in vitro studies by using TRIM31Hep-cKO mice and Nrf2KO hepatocytes supported that the anti-fibrotic and liver protective effects of Mul were mainly dependent on the expression of TRIM31 and Nrf2 in hepatocytes with considerably ameliorated inflammatory response and oxidative stress. Immunoprecipitation analysis surprisingly showed that there might be an interaction between TRIM31 and Nrf2. Mul-improved Nrf2 nuclear translocation in liver was almost eliminated upon TRIM31Hep-cKO under fibrotic stress. Taken together, all our findings demonstrated that inflammation and oxidative stress in hepatocytes contributed to HSCs activation and collagen deposition, which could be mitigated by Mul through improving TRIM31-regulated Nrf2 signaling pathways, consequently ameliorating hepatic fibrosis (Fig. 18). Therefore, Mul may be a promising therapeutic strategy for the treatment of liver fibrosis.
Fig. 18.

Proposed mechanisms indicate the effects of mulberrin on liver fibrosis. In brief, under fibrotic stimuli, TRIM31-mediated activation of Nrf2 signaling pathway was restrained, resulting in inflammatory response and oxidative stress in hepatocytes, which contributed to the HSC activation and hepatic fibrosis. Notably, mulberrin treatment could improve TRIM31/Nrf2 signaling to ameliorate hepatic inflammation and ROS production, consequently attenuating liver fibrosis.
Inflammasome activation plays a key role in the progression of liver disease. NLRP3 is the most well-studied inflammasome and is a multiprotein cytoplasmic complex that consists of ASC and the effector molecule pro-Caspase-1 [[15], [16], [17]]. Under stimulation by pathogen-associated molecular patterns and damage-related molecular patterns, NLRP3 inflammasome activation mediates the cleavage of Capase-1, leading to the maturation and extracellular releases of IL-1β and IL-18 [43,44]. IL-1β and IL-18 are synthesized as inactive precursor forms (pro-IL-1β and pro-IL-18), which is dependent on the activation of NF-κB under various stimuli conditions [10,15]. The activation of NLRP3 inflammasome can result in severe liver inflammation, hepatocyte pyroptotic cell death, and hepatic fibrosis in mice [45], whereas depressing NLRP3 inflammasome ameliorates liver fibrosis. For instance, in an animal model of non-alcoholic steatohepatitis (NASH), NLRP3 inflammasome activation was indispensable in fibrotic response, and blockage of NLRP3 by an NLRP3 selective inhibitor attenuated nonalcoholic steatohepatitis (NASH) profiles and fibrosis [16,46]. Additionally, NLRP3 suppression by MCC950, a selective NLRP3 inhibitor, could markedly ameliorate liver injury and retard the progression of bile-duct-ligation (BDL)-induced hepatic fibrosis in mice [47]. Herein, suppressing NLRP3 inflammasome activation may be a promising therapeutic option to effectively depress liver injury and fibrosis. TRIM31, a member of the TRIM protein family, has been recognized to play important roles in regulating tumor growth and immuno-inflammatory diseases [12,48]. TRIM31 restrained the activation NLRP3 inflammasome through promoting proteasomal degradation of NLRP3, consequently maintaining immune homeostasis [13]. A recent study revealed that TRIM31 could promote NLRP3 ubiquitination, thereafter, suppressing NLRP3 inflammasome and pyroptosis in human retinal pigment epithelial (RPE) cells, accompanied by decreased expression of mature IL-1β [14]. In pancreatic cancer, TRIM31 promoted NF-κB/p65 nuclear translocation through catalyzing the K63-linked polyubiquitination of TRAF2 and thus facilitated NF-κB activation to accelerate tumor growth consequently [49]. TRIM31 also promoted colorectal cancer cell proliferation and invasion by activating NF-κB signaling pathway [50]. Here in our present study, we found that CCl4 treatment significantly reduced TRIM31 expression levels in liver of mice, and promoted NLRP3 inflammasome activation, as indicated by the increased expression of NLRP3, ASC and Caspase-1, which was along with up-regulated expression of mIL-1β and mIL-18. Consistent with previous studies [9,51], NF-κB signaling pathway in liver was highly stimulated by CCl4, proved by the elevated expression of p-IKKα, p-IκBα and p-NF-κB/p65, resulting in the high expression of pro-inflammatory factors including IL-1β, TNF-α, IL-6, IL-18, MCP-1 and CXCL-10. Notably, Mul treatments considerably improved TRIM31 expression, and reduced the activation of NLRP3 inflammasome and NF-κB signaling, thereby mitigating hepatic inflammatory response. These regulatory effects of Mul on TRIM31/NLRP3 and NF-κB signaling were validated both in L02 cells and mouse primary hepatocytes in vitro under TGF-β1 or LPS irritating status. Our more in vitro studies showed that in response to TGF-β1 stimulus, TRIM31 knockdown accelerated NLRP3 inflammasome and NF-κB activation in L02 cells, thereby aggravating inflammatory response. In line with previous research [13,14], TRIM31 exerted inhibitory effect on NLRP3 inflammasome activation. However, the negative regulatory effect of TRIM31 on NF-κB/p65 here we observed was different with previous studies [49,50]. We hypothesized that it might be associated with the different types of cells. As for this, more studies are still required to explore the correlation between TRIM31 and NF-κB under different physiological status. More importantly, we found that siTRIM31 significantly abolished the function of Mul to retard inflammation via restrengthening NLRP3 inflammasome and NF-κB activation in TGF-β1-stimulated L02 cells. In vivo studies confirmed that Mul failed to suppress NLRP3 inflammasome activation and inflammatory response in liver of CCl4-challened mice with hepatocyte-specific TRIM31 knockout, disclosing the necessity of TRIM31 for Mul to perform its anti-inflammatory capacities. Collectively, both our in vivo and in vitro findings demonstrated that Mul could restrain hepatic inflammation by suppressing NLRP3 and NF-κB signaling pathways through the improvement of TRIM31 signaling (Fig. 18).
Nrf2 pathway plays an essential role in defending against oxidative stress. As a transcription factor, Nrf2 induces the expression of numerous cytoprotective and detoxifying genes, including HO-1 and NQO1 [18,19,21]. As reported, Nrf2 activation could increase the expression of its down-streaming antioxidant factors HO-1 and NQO1 to meliorate hepatic fibrosis [[20], [21], [22], [23]]. GCLC and GCLM are two crucial enzymes during the synthesis of GSH. As the major components of the Nrf2 pathway, GCLC and GCLM increases are involved in the suppression of liver fibrosis [52]. NOXs are a major source of ROS in liver and modulate fibrogenic responses induced by angiotensin II, platelet derived growth factor (PDGF), and TGF-β in cells [34,36,53,54]. The function of Mul to improve Nrf2 signaling to subsequently ameliorate spinal cord injury has been recognized [25]. In the current study, we consistently found that Mul treatments restored the expression of HO-1, NQO1, GCLC, and GCLM in liver of CCl4-challenged mice, and such effect was concomitant with the up-regulated expression of nuclear Nrf2. Therefore, Mul exerted its antioxidant effect through modifying Nrf2 signaling. Meanwhile, NOX1, NOX2, NOX4 and p22phox stimulated by CCl4 were strongly restrained by Mul supplements in liver of mice. These regulatory effects of Mul on Nrf2 were validated in TGF-β1- or H2O2-stimulated L02 cells and mouse primary hepatocytes. Intriguingly, we found that siNrf2 markedly diminished the effects of Mul against ROS production and oxidative stress in TGF-β1-treated L02 cells, and accelerated TGF-β1-induced oxidative damage in vitro. Herein, we concluded that Mul-suppressed oxidative stress in liver was Nrf2-dependent, contributing to the amelioration of hepatic injury (Fig. 18).
Hepatic inflammation is a pan-etiology driver of hepatic damage and liver fibrosis. Inflammatory factors such as IL-1β and IL-6 released by hepatocytes are important mediators of the inflammatory response which initiate and perpetuate an abnormal wound-healing response and facilitate the progression of hepatic fibrosis through promoting HSCs activation [55]. Therefore, blockade of inflammation in hepatocytes emerges as a novel therapeutic target to reduce liver inflammation and fibrosis in different types of hepatic diseases such as NASH [56]. NLRP3 inflammasome and inflammatory response in hepatocytes led to the HSC activation, which is then responsible for collagen deposition and fibrosis [16,17,46,47]. The profibrogenic effects of ROS are compounded by the fact that NOX4 up-regulation in hepatocytes results in the cell damage, further inducing the cascade of cellular events that lead to HSCs activation and cirrhosis [52,57]. Similarly, here in our present study, we found that after exposure to CM derived from TGF-β1-stimulated L02 cells containing higher levels of inflammatory factors and oxidative markers, HSCs activation of LX-2 cells was promoted, which might be mainly attributed to the elevated inflammatory and oxidative microenvironment; however, these phenomena were strongly abolished by Mul. Notably, the effects of Mul to suppress LX-2 cell activation induced by CM collected from TGF-β1-stimulated L02 cells were almost abrogated upon TRIM31 or Nrf2 deletion. More animal studies using TRIM31Hep-cKO mice demonstrated that Mul failed to ameliorate liver dysfunction and collagen deposition, revealing that the anti-fibrotic and liver protective effects of Mul were mainly TRIM31-dependent in hepatocytes with reduced inflammatory factors. Similarly, Mul lost its function to reduce the activation of primary mouse HSCs when cultured in CM derived from Nrf2KO hepatocytes under stimuli. Therefore, hepatocytes played the main role in restraining HSCs activation and liver fibrosis through regulating Nrf2 signaling by Mul treatment. All these in vitro and in vivo studies elucidated that TRIM31 and Nrf2 expression in hepatocytes was necessary for Mul to perform its suppressive effects on HSCs activation and hepatic fibrosis. Growing studies have reported that TRIMs family members such as TRIM16 and TRIM25 regulate Nrf2 signaling and its activation to control numerous cellular events, including oxidative stress, ECM process and inflammatory response by governing the protein quality control [58,59]. Here in our study, we found that Mul-improved expression of nuclear Nrf2 was considerably abrogated by hepatocyte-specific knockout of TRIM31 in liver of CCl4-treated mice, partially revealing the beneficial effect of TRIM31 on Nrf2 activation. Notably, we initially showed that there might be an interaction between TRIM31 and Nrf2 by immunoprecipitation analysis. Nevertheless, how TRIM31 regulates Nrf2 signaling deserves further attention. As reported, there are numerous types of cells in liver tissues, such as Kupffer cells, Dendritic cells (DCs) and NK cells, which play crucial roles in mediating inflammation and fibrosis [60,61]. Given that the inflammation and macrophage activation at different stages of liver injury is a pivotal character to regulate fibrosis progression [62], more studies are still warranted to explore whether and how Mul performs its biological functions in macrophage to subsequently control hepatic damage. In addition, if TRIM31 expression changes in HSCs could govern hepatic fibrosis, further studies are required to address this issue.
In conclusion, we established a link between hepatocyte TRIM31 and liver fibrosis in mouse models and revealed the therapeutic potential of Mul on the disease. Specifically, we found that Mul treatments significantly ameliorated HSCs activation and collagen deposition both in CCl4-induced mouse models and TGF-β1-stimulated HSCs. Mul mainly mitigated inflammatory response and oxidative stress in hepatocytes through improving TRIM31/Nrf2 signaling pathway, contributing to the blockage of HSCs activation and ameliorating liver fibrosis consequently (Fig. 18). Therefore, Mul can be considered as a promising therapeutic strategy for liver fibrosis management through improving TRIM31/Nrf2 axis.
Declaration of competing interest
The authors see no conflict of interest.
Acknowledgments
This work was supported by (1) National Natural Science Foundation of China (NSFC Grant No.: 81703527); (2) Chongqing Research Program of Basic Research and Frontier Technology (Grant No. cstc2018jcyjAX0393, cstc2018jcyjAX0811, cstc2018jcyjA3533); (3) Science and Technology Research Program of Chongqing Education Commission of China (Grant No.: KJQN201901608, KJQN201901615, KJZD-M201801601, KJZD-K202001603); (4) Chongqing Professional Talents Plan for Innovation and Entrepreneurship Demonstration Team (CQCY201903258, cstc2021ycjh-bgzxm0202); (5) School-level Research Program of Chongqing University of Education (Grant No.: 2019BSRC001); (6) Advanced Programs of Post-doctor of Chongqing (Grant No.: 2017LY39); (7) Supported by Youth Project of Science and Technology Research Program of Chongqing Education Commission of China (Grant No.: KJQN201901606).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102274.
Contributor Information
Jun Tan, Email: tanjun@cque.edu.cn.
Bochu Wang, Email: wangbc2000@126.com.
Minxuan Xu, Email: minxuanxu@foxmail.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Parola M., Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol. Aspect. Med. 2019;65:37–55. doi: 10.1016/j.mam.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Kawai H., Osawa Y., Matsuda M., et al. Sphingosine-1-phosphate promotes tumor development and liver fibrosis in mouse model of congestive hepatopathy. Hepatology. 2021 doi: 10.1002/hep.32256. [DOI] [PubMed] [Google Scholar]
- 3.Roehlen N., Crouchet E., Baumert T.F. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4):875. doi: 10.3390/cells9040875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kisseleva T., Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021;18(3):151–166. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 5.Altamirano-Barrera A., Barranco-Fragoso B., Méndez-Sánchez N. Management strategies for liver fibrosis. Ann. Hepatol. 2017;16(1):48–56. doi: 10.5604/16652681.1226814. [DOI] [PubMed] [Google Scholar]
- 6.Schuppan D., Ashfaq-Khan M., Yang A.T., et al. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68:435–451. doi: 10.1016/j.matbio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Koyama Y., Brenner D.A. Liver inflammation and fibrosis. J. Clin. Invest. 2017;127(1):55–64. doi: 10.1172/JCI88881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner D.A. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 2009;120:361. [PMC free article] [PubMed] [Google Scholar]
- 9.Elsharkawy A.M., Mann D.A. Nuclear factor‐κB and the hepatic inflammation‐fibrosis‐cancer axis. Hepatology. 2007;46(2):590–597. doi: 10.1002/hep.21802. [DOI] [PubMed] [Google Scholar]
- 10.Muriel P. NF‐κB in liver diseases: a target for drug therapy. J. Appl. Toxicol. 2009;29(2):91–100. doi: 10.1002/jat.1393. [DOI] [PubMed] [Google Scholar]
- 11.Nisole S., Stoye J.P., Saïb A. TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005;3(10):799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- 12.Sugiura T., Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J. Cell. Biochem. 2008;105(4):1081–1091. doi: 10.1002/jcb.21908. [DOI] [PubMed] [Google Scholar]
- 13.Song H., Liu B., Huai W., et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016;7(1):1–11. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang P., Liu W., Chen J., et al. TRIM31 inhibits NLRP3 inflammasome and pyroptosis of retinal pigment epithelial cells through ubiquitination of NLRP3. Cell Biol. Int. 2020;44(11):2213–2219. doi: 10.1002/cbin.11429. [DOI] [PubMed] [Google Scholar]
- 15.Chen S., Tang C., Ding H., et al. Maf1 ameliorates sepsis-associated encephalopathy by suppressing the NF-kB/NLRP3 inflammasome signaling pathway. Front. Immunol. 2020;11:3310. doi: 10.3389/fimmu.2020.594071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mridha A.R., Wree A., Robertson A.A.B., et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017;66(5):1037–1046. doi: 10.1016/j.jhep.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C.Y., Deng Y., Li P., et al. Prediction of biochemical nonresolution in patients with chronic drug-induced liver injury: a large multicenter study. Hepatology. 2021 doi: 10.1002/hep.32283. [DOI] [PubMed] [Google Scholar]
- 18.Sánchez-Valle V., C Chavez-Tapia N., Uribe M., et al. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr. Med. Chem. 2012;19(28):4850–4860. doi: 10.2174/092986712803341520. [DOI] [PubMed] [Google Scholar]
- 19.Ge C., Tan J., Zhong S., et al. Nrf2 mitigates prolonged pm2. 5 exposure-triggered liver inflammation by positively regulating sike activity: protection by juglanin. Redox Biol. 2020;36:101645. doi: 10.1016/j.redox.2020.101645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Q., Zhang H., Cao Y., et al. Schisandrin B attenuates CCl4-induced liver fibrosis in rats by regulation of Nrf2-ARE and TGF-β/Smad signaling pathways. Drug Des. Dev. Ther. 2017;11:2179. doi: 10.2147/DDDT.S137507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin S.M., Yang J.H., Ki S.H. Role of the Nrf2-ARE pathway in liver diseases. Oxid. Med. Cell. Longev. 2013;2013:763257. doi: 10.1155/2013/763257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J.Q., Ding J., Zhang L., et al. Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin. Res. Hepatol. Gastroenterol. 2015;39(2):188–197. doi: 10.1016/j.clinre.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Lyu H., Wang H., Li L., et al. Hepatocyte-specific deficiency of Nrf2 exacerbates carbon tetrachloride-induced liver fibrosis via aggravated hepatocyte injury and subsequent inflammatory and fibrogenic responses. Free Radic. Biol. Med. 2020;150:136–147. doi: 10.1016/j.freeradbiomed.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Jing H., Wang S., Wang M., et al. Isobavachalcone attenuates MPTP-induced Parkinson's disease in mice by inhibition of microglial activation through NF-κB pathway. PLoS One. 2017;12(1) doi: 10.1371/journal.pone.0169560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia P., Gao X., Duan L., et al. Mulberrin (Mul) reduces spinal cord injury (SCI)-induced apoptosis, inflammation and oxidative stress in rats via miroRNA-337 by targeting Nrf-2. Biomed. Pharmacother. 2018;107:1480–1487. doi: 10.1016/j.biopha.2018.07.082. [DOI] [PubMed] [Google Scholar]
- 26.Cao W., Dong Y., Zhao W., et al. Mulberrin attenuates 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced Parkinson's disease by promoting Wnt/β-catenin signaling pathway. J. Chem. Neuroanat. 2019;98:63–70. doi: 10.1016/j.jchemneu.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Ji Y.X., Huang Z., Yang X., et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med. 2018;24(2):213–223. doi: 10.1038/nm.4461. [DOI] [PubMed] [Google Scholar]
- 28.Liu D., Zhang P., Zhou J., et al. TNFAIP3 interacting protein 3 overexpression suppresses nonalcoholic steatohepatitis by blocking TAK1 activation. Cell Metabol. 2020;31(4):726–740. doi: 10.1016/j.cmet.2020.03.007. e8. [DOI] [PubMed] [Google Scholar]
- 29.Cai S.P., Cheng X.Y., Chen P.J., et al. Transmembrane protein 88 attenuates liver fibrosis by promoting apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2016;80:58–67. doi: 10.1016/j.molimm.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Guimarães E.L.M., Empsen C., Geerts A., et al. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J. Hepatol. 2010;52(3):389–397. doi: 10.1016/j.jhep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Polasek M., Fuchs B.C., Uppal R., et al. Molecular MR imaging of liver fibrosis: a feasibility study using rat and mouse models. J. Hepatol. 2012;57(3):549–555. doi: 10.1016/j.jhep.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghatak S., Biswas A., Dhali G.K., et al. Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol. Appl. Pharmacol. 2011;251(1):59–69. doi: 10.1016/j.taap.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X., Wu Z., Xu Y., et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell. Mol. Immunol. 2019;16(11):878–886. doi: 10.1038/s41423-018-0032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan T., Kisseleva T., Brenner D.A. Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J., Ma H.Y., Liang S., et al. The role of human cytochrome P450 2E1 in liver inflammation and fibrosis. Hepatol. Commun. 2017;1(10):1043–1057. doi: 10.1002/hep4.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su J., Morgani S.M., David C.J., et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. 2020;577(7791):566–571. doi: 10.1038/s41586-019-1897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y., Zeng Z., Shen X., et al. MicroRNA-146a-5p negatively regulates pro-inflammatory cytokine secretion and cell activation in lipopolysaccharide stimulated human hepatic stellate cells through inhibition of toll-like receptor 4 signaling pathways. Int. J. Mol. Sci. 2016;17(7):1076. doi: 10.3390/ijms17071076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong M., Chen X., Lv F., et al. Serum response factor (SRF) promotes ROS generation and hepatic stellate cell activation by epigenetically stimulating NCF1/2 transcription. Redox Biol. 2019;26:101302. doi: 10.1016/j.redox.2019.101302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida K., Matsuzaki K., Murata M., et al. Clinico-Pathological importance of TGF-β/phospho-smad signaling during human hepatic fibrocarcinogenesis. Cancers. 2018;10(6):183. doi: 10.3390/cancers10060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie G., Jiang R., Wang X., et al. Conjugated secondary 12α-hydroxylated bile acids promote liver fibrogenesis. EBioMedicine. 2021;66:103290. doi: 10.1016/j.ebiom.2021.103290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J., Zhao Y., Wang Y., et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 42.Xu F., Liu C., Zhou D., et al. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem. Cytochem. 2016;64(3):157–167. doi: 10.1369/0022155415627681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chenxu G., Minxuan X., Yuting Q., et al. Loss of RIP3 initiates annihilation of high-fat diet initialized nonalcoholic hepatosteatosis: a mechanism involving Toll-like receptor 4 and oxidative stress. Free Radic. Biol. Med. 2019;134:23–41. doi: 10.1016/j.freeradbiomed.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 45.Al Mamun A., Akter A., Hossain S., et al. Role of NLRP3 inflammasome in liver disease. J. Digest. Dis. 2020;21(8):430–436. doi: 10.1111/1751-2980.12918. [DOI] [PubMed] [Google Scholar]
- 46.Dong Z., Zhuang Q., Ning M., et al. Palmitic acid stimulates NLRP3 inflammasome activation through TLR4-NF-κB signal pathway in hepatic stellate cells. Ann. Transl. Med. 2020;8(5):168. doi: 10.21037/atm.2020.02.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu J., Yuan Z., Wang G., et al. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharm. 2019;70:147–155. doi: 10.1016/j.intimp.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 48.Ra E.A., Lee T.A., Kim S.W., et al. TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat. Commun. 2016;7(1):1–15. doi: 10.1038/ncomms11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu C., Chen S., Guo Y., et al. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics. 2018;8(12):3224. doi: 10.7150/thno.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H., Yao L., Gong Y., et al. TRIM31 regulates chronic inflammation via NF-κB signal pathway to promote invasion and metastasis in colorectal cancer. Am. J. Tourism Res. 2018;10(4):1247. [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R., Yu X.Y., Guo Z.Y., et al. Inhibitory effects of salvianolic acid B on CCl4-induced hepatic fibrosis through regulating NF-κB/IκBα signaling. J. Ethnopharmacol. 2012;144(3):592–598. doi: 10.1016/j.jep.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 52.Mortezaee K. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and liver fibrosis: a review. Cell Biochem. Funct. 2018;36(6):292–302. doi: 10.1002/cbf.3351. [DOI] [PubMed] [Google Scholar]
- 53.Liang S., Kisseleva T., Brenner D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016;7:17. doi: 10.3389/fphys.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crosas-Molist E., Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015;6:106–111. doi: 10.1016/j.redox.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Del Campo J.A., Gallego P., Grande L. Role of inflammatory response in liver diseases: therapeutic strategies. World J. Hepatol. 2018;10(1):1. doi: 10.4254/wjh.v10.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y., Wen H., Fu J., et al. Hepatocyte TNF receptor–associated factor 6 aggravates hepatic inflammation and fibrosis by promoting lysine 6–linked polyubiquitination of apoptosis signal‐regulating kinase 1. Hepatology. 2020;71(1):93–111. doi: 10.1002/hep.30822. [DOI] [PubMed] [Google Scholar]
- 57.Luangmonkong T., Suriguga S., Mutsaers H.A.M., et al. Targeting oxidative stress for the treatment of liver fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018;175:71–102. doi: 10.1007/112_2018_10. [DOI] [PubMed] [Google Scholar]
- 58.Jena K.K., Mehto S., Kolapalli S.P., et al. TRIM16 employs NRF2, ubiquitin system and aggrephagy for safe disposal of stress-induced misfolded proteins. Cell Stress. 2018;2(12):365. doi: 10.15698/cst2018.12.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y., Tao S., Liao L., et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020;11(1):1–13. doi: 10.1038/s41467-019-14190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu M., Ge C., Zhu L., et al. iRhom2 promotes hepatic steatosis by activating MAP3K7‐dependent pathway. Hepatology. 2021;73(4):1346–1364. doi: 10.1002/hep.31436. [DOI] [PubMed] [Google Scholar]
- 61.Weiskirchen R., Tacke F. Liver fibrosis: which mechanisms matter? Clin. Liver Dis. 2016;8(4):94–99. doi: 10.1002/cld.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fallowfield J., Hayes P. Pathogenesis and treatment of hepatic fibrosis: is cirrhosis reversible? Clin. Med. 2011;11(2):179. doi: 10.7861/clinmedicine.11-2-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.