

Abstract
Aire allows medullary thymic epithelial cells (mTECs) to express and present a large number of self‐antigens for central tolerance. Although mTECs express a high diversity of self‐antigen splice isoforms, the extent and regulation of alternative splicing events (ASEs) in their transcripts, notably in those induced by Aire, is unknown. In contrast to Aire‐neutral genes, we find that transcripts of Aire‐sensitive genes show only a low number of ASEs in mTECs, with about a quarter present in peripheral tissues excluded from the thymus. We identify Raver2, as a splicing‐related factor overexpressed in mTECs and dependent on H3K36me3 marks, that promotes ASEs in transcripts of Aire‐neutral genes, leaving Aire‐sensitive ones unaffected. H3K36me3 profiling reveals its depletion at Aire‐sensitive genes and supports a mechanism that is preceding Aire expression leading to transcripts of Aire‐sensitive genes with low ASEs that escape Raver2‐induced alternative splicing. The lack of ASEs in Aire‐induced transcripts would result in an incomplete Aire‐dependent negative selection of autoreactive T cells, thus highlighting the need of complementary tolerance mechanisms to prevent activation of these cells in the periphery.
Keywords: Aire, alternative splicing, central tolerance, Raver2, thymic epithelial cells
Subject Categories: Chromatin, Transcription & Genomics; Immunology; RNA Biology
Genes in the thymic epithelium sensitive to Aire expression are characterized by low levels of H3K36me3 marks and show few alternative splicing events (ASEs) in their transcripts, as they escape the splicing‐related factor Raver2.
Introduction
Tolerance against self‐tissues is an essential feature of the immune system. It is established in the thymus following the presentation of self‐antigen peptides to developing T cells. T cells recognizing their cognate antigen either undergo negative selection by apoptosis, preventing the release of autoreactive T cells, or develop into regulatory T cells (Tregs) able to suppress potential autoreactive T cells in the periphery (Goodnow et al, 2005; Cowan et al, 2013; Klein et al, 2014). The subset of medullary thymic epithelial cells characterized by high levels of major histocompatibility complex class II (MHC II) molecules (mTEChi) is essential to the presentation of self‐antigens to developing T cells. Indeed, mTEChi have the unique ability to load, onto MHC II molecules, antigenic peptides originated from a wide array of endogenously expressed self‐antigens (Sansom et al, 2014; Danan‐Gotthold et al, 2016), including those controlled by the autoimmune regulator (Aire) and restricted to specific peripheral tissues (Derbinski et al, 2001; Sansom et al, 2014). In mice, invalidation of the Aire gene results in a drop of Aire‐sensitive gene expression in mTEChi, and the presence of autoantibodies and immune infiltrates directed at multiple peripheral tissues due to impaired negative selection of autoreactive T cells and their harmful activation in the periphery (Anderson et al, 2002). Mutations in the human AIRE gene also cause the multi‐organ devastating autoimmune disorder named Autoimmune Polyendocrine Syndrome type 1 (APS1) (Nagamine et al, 1997; Peterson et al, 2004).
The breadth of the repertoire of self‐peptides presented by mTEChi does not only rely on the expression of a high number of self‐antigen genes but also on the high diversity of their transcript isoforms whose translation produces multiple protein variants as a result of high rates of alternative splicing (Keane et al, 2015; Danan‐Gotthold et al, 2016). Hence, a variety of alternative splicing events (ASEs) are expected to be spliced‐in in mTEChi and the resulting processed peptides presented to developing T cells, thereby ensuring the efficient elimination of T cells capable to elicit autoreactive responses against peptides derived from these ASEs in the periphery. Although the expression of a wide range of transcript isoforms has been unambiguously established in mTEChi, the impact and the regulation mechanisms of alternative splicing on the genes controlled by Aire remain to be determined. Notably, it is unknown whether they encode a high transcript‐isoform diversity similarly to total mTEChi, or whether the ASEs included in their transcripts equal the diversity of spliced‐in ASEs detected for the same genes in their tissues of expression.
We therefore investigated the pattern of ASE inclusion for transcripts of Aire‐sensitive genes in WT and Aire‐KO mTEChi, as well as in peripheral tissues. Comparison of ASE inclusion for transcripts of Aire‐sensitive versus neutral genes in mTEChi revealed an unexpected conservation of alternative splicing regulation between Aire‐positive and Aire‐negative subsets. Differences in the pattern of ASE inclusion with peripheral tissues provided clues on the relative importance of the role of negative selection, in comparison to peripheral mechanisms, for the establishment and maintenance of immunological tolerance against Aire‐dependent self‐antigens. Finally, we identified an epigenetic mechanism, involving the splicing‐related factor Raver2 that sustains the regulation of alternative splicing in mTECs and explains the patterns of ASE inclusion for transcripts of Aire‐sensitive and neutral genes in these cells.
Results
Aire‐sensitive genes encode a low diversity of transcript isoforms in mTEChi
To characterize the alternative splicing complexity of Aire‐sensitive genes in mTEChi sorted following the outline strategy (Appendix Fig S1 and S2), we determined the diversity of Aire‐induced transcript isoforms that result from alternative splicing through the calculation of the median splicing entropy of these genes (Ritchie et al, 2008). For a given gene, splicing entropy was calculated using the levels of transcript‐isoform expression obtained by assignment of paired‐end sequencing reads to the RefSeq mRNA isoform annotations (Fig 1A). The more diverse the transcript isoforms, the greater the associated splicing entropy. Since a sufficient number of mapped junction‐spanning reads is necessary for accurate characterization of transcript isoforms, we looked for the minimum expression value below which transcript isoform diversity could not be reliably captured in our mTEChi RNA‐seq data. For that purpose, we binned the genes based on expression and calculated their median splicing entropy (Fig 1B). We found stable values of splicing entropy for genes with expression levels over 1 FPKM and highly degraded values for genes showing lower expression levels. This prevented the detection of minor transcript isoforms and therefore prompted us to exclude weakly expressed genes for subsequent splicing analyses. Next, we selected the Aire‐sensitive genes characterized by a twofold expression increase in WT versus Aire‐KO mTEChi (Fig 1C) and found that their median splicing entropy was significantly lower than those of Aire‐neutral genes and of all genes taken together (Fig 1D). This finding thus revealed that Aire‐sensitive genes encode a lower diversity of transcript isoforms in mTEChi, denoting lower rates of alternative splicing at these genes.
Figure 1. Low splicing entropy of Aire‐sensitive genes in mTEChi.
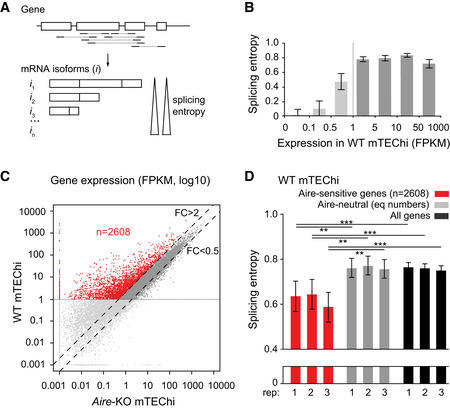
- Schematic representation of a hypothetical gene with mapped paired‐end sequencing reads identifying transcript isoforms. The isoform diversity of this gene is evaluated by calculation of its splicing entropy.
- Median splicing entropy of genes binned according to their expression levels. FPKM of 1 corresponds to the threshold over which the transcript isoform diversity can be accurately characterized in our RNA‐seq dataset. (three mTEChi biological replicates [combined]; error bars show the 95% confidence interval of the medians).
- Identification of Aire‐sensitive genes upregulated by Aire in WT versus Aire‐KO mTEChi (FC > 2) and matching the threshold of 1 FPKM in WT mTEChi (red dots, n = 2,608). Aire‐neutral genes (0.5 < FC < 2) with expression levels over 1 FPKM in WT mTEChi are represented by dark gray dots between the dashed lines. (n = 3 WT and three Aire‐KO mTEChi biological replicates).
- Median splicing entropy of Aire‐sensitive genes, Aire‐neutral genes (equal numbers), and all genes in each of the three mTEChi biological replicates (rep). Error bars show the 95% confidence interval of the medians. **P < 10−3, ***P < 10−4 (Wilcoxon test, n = 2,608, performed in each of the three biological replicates).
Aire‐induced transcripts show low ASE inclusion in mTEChi
We next sought to determine whether the low diversity of transcript isoforms induced by Aire was sustained by a biased inclusion of ASE. To this end, we first identified all ASEs from the RefSeq mRNA annotation database in parsing its content using rMATS and considered the types of ASEs whose inclusion is susceptible to add amino acid content to the encoded protein isoforms without removing some, that is, skipped exon (SE), alternative 5′ splicing site (5SS), alternative 3′ splicing site (3SS), and intron retention (IR) events. We then computed their percent splicing inclusion (PSI), that is, the relative expression of transcript isoforms spliced in versus spliced in or out for each ASE (Fig 2A). Comparison of PSI values for Aire‐sensitive versus neutral genes in mTEChi revealed a significant imbalance toward lower values (< 0.1), therefore showing a reduced inclusion of ASEs in the transcripts induced by Aire(Aire‐sensitive: PSI < 0.1 [n = 163], PSI > 0.1 [n = 265]; Aire‐neutral: PSI < 0.1 [n = 93], PSI > 0.1 [n = 332]; Chisq = 3.6 × 10−7; Fig 2B). We then calculated, for Aire‐sensitive and neutral genes, the levels of ASE inclusion imbalance, that is, the number ratio of ASEs showing some level of active inclusion (PSI > 0.1) to ASEs showing no or background inclusion (PSI < 0.1), and found similar reduced levels for Aire‐sensitive genes across three replicates (Fig 2C, Left). Comparison of the levels of ASE inclusion imbalance for Aire‐neutral genes between mTEChi and peripheral tissues for which we collected RNA‐seq datasets (Li et al, 2017) revealed higher levels in mTEChi, which is in line with reports showing higher rates of alternative splicing in mTEChi (Keane et al, 2015; Danan‐Gotthold et al, 2016). In addition, the breadth of the reduction of ASE inclusion between transcripts of Aire‐neutral and Aire‐sensitive genes in mTEChi is striking since it is much wider than the reduction observed for Aire‐neutral genes between mTEChi and each peripheral tissue (Fig 2C, Right). Since alternative splicing was reported to be influenced by gene expression in some systems (Kornblihtt et al, 2013), we calculated the median expression level of neutral genes in each mTEChi replicate and peripheral tissues. We found no significant correlation with the levels of ASE inclusion imbalance, therefore ruling out gene expression as a primary factor responsible for variation of ASE inclusion in our tested samples (Appendix Fig S3).
Figure 2. Low levels of ASE inclusion imbalance for Aire‐sensitive genes in mTEChi.
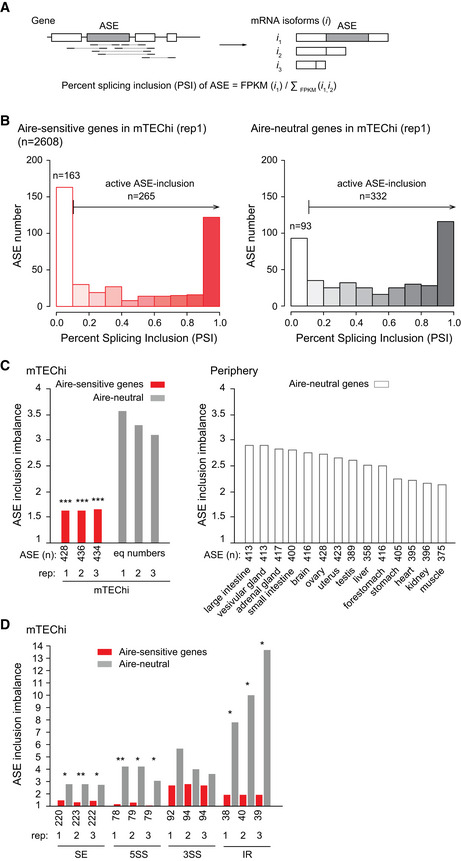
- Schematic representation exemplifying the characterization of two transcript isoforms defined by a spliced in (i1) and a spliced out (i2) ASE, respectively, as well as, of an additional isoform (i3) unrelated to the ASE. The PSI value of the ASE is calculated using the specific expression of the transcript isoforms having the ASE spliced in or out.
- Distribution of ASEs according to their PSI values for Aire‐sensitive (Left) and neutral genes (Right) in mTEChi. ASEs with PSI < 0.1 are considered as excluded, whereas ASEs with PSI > 0.1 as present, with some level of active inclusion.
- Levels of ASE inclusion imbalance for Aire‐sensitive and neutral genes (equal numbers) in each of the three mTEChi biological replicates (rep; Left) and for Aire‐neutral genes in peripheral tissues (unique samples; Right).
- Inclusion imbalance of each type of ASE in each of the three mTEChi biological replicates (rep).
Data information: In (C, D) *** P < 10−4, **P < 10−3, *P < 0.05 (chi‐squared test).
Next, we calculated in mTEChi, the inclusion imbalance of the different types of considered ASEs, and found decreased levels for each type of ASE, with SE, 5SS, and IR events reaching statistical significance (Fig 2D).
Finally, to address whether the transcripts induced by Aire also show reduced inclusion of ASEs in human mTEChi, we isolated mTEChi from human thymic tissues obtained during pediatric cardiac surgery and performed RNA‐seq experiments. We identified the minimum expression values over which transcript isoforms could be accurately characterized in these samples, as shown by the example of two individuals (Fig EV1A). We then identified all ASEs from human RefSeq mRNA annotations and selected human Aire‐sensitive and neutral ortholog genes for which we calculated the PSI values of their associated ASEs. Comparison of ASE inclusion imbalance for the Aire‐sensitive and neutral genes in one male (indiv 1) and four female (indiv 2–5) human individuals showed a marked reduction for Aire‐sensitive genes similar to what we found in mice, therefore revealing conservation of ASE inclusion in mTEChi between mice and humans (Fig EV1B).
Figure EV1. Low levels of ASE inclusion imbalance for Aire‐sensitive genes in human mTEChi.
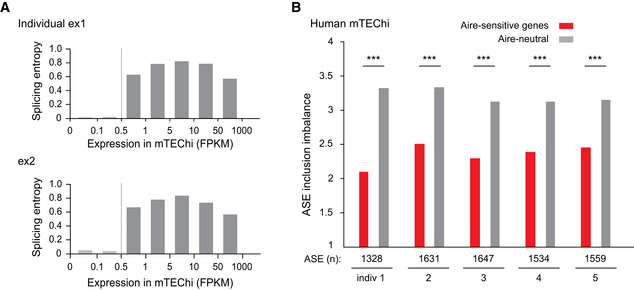
- Median splicing entropy of genes binned according to their expression values. FPKM of 0.5 corresponds to the threshold over which the transcript isoform diversity can be accurately characterized in our RNA‐seq dataset. Example of two individuals is shown.
- Levels of ASE inclusion imbalance for Aire‐sensitive and neutral genes in mTEChi of five different individuals, *** P < 10−4 (chi‐squared test).
Together, these findings revealed that the transcripts induced by Aire in mTEChi show conserved low ASEs in contrast to those that are neutral to Aire.
Low ASE inclusion in transcripts of Aire‐sensitive genes is a general feature of TECs
To discriminate whether the reduced inclusion of ASEs in the transcripts induced by Aire was directly associated with the Aire’s induction of gene expression or was also observed in the absence of Aire, we selected the genes with twofold increased expression in WT versus Aire‐KO mTEChi and with levels of expression over 1 FPKM in both WT and Aire‐KO mTEChi (Fig 3A). We noted that meeting the threshold of 1 FPKM in Aire‐KO mTEChi shrank the selection of Aire‐sensitive genes since most of Aire‐sensitive genes are inactive or expressed at very low level in the absence of Aire.
Figure 3. Low levels of ASE inclusion imbalance for Aire‐sensitive genes in Aire‐negative TECs.
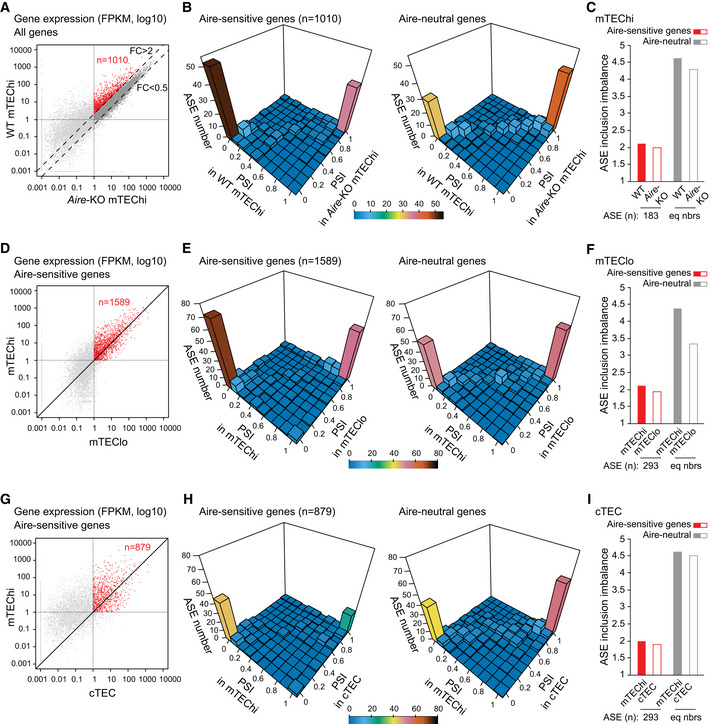
- Identification of Aire‐sensitive genes upregulated by Aire in WT versus Aire‐KO mTEChi (FC > 2) and matching the threshold of 1 FPKM in WT and Aire‐KO mTEChi (red dots, n = 1,010). (three WT and three Aire‐KO mTEChi biological replicates).
- 3D representation of the distribution of ASEs of Aire‐sensitive (Left) and neutral genes (Right) according to their PSI values calculated from three WT (combined) and three Aire‐KO mTEChi replicates (combined).
- Levels of ASE inclusion imbalance for Aire‐sensitive and neutral genes (equal numbers) based on PSI values calculated from three WT (combined) and three Aire‐KO mTEChi replicates (combined).
- Differential gene expression of Aire‐sensitive genes between mTEChi and mTEClo. Red dots show the Aire‐sensitive genes with FPKM > 1 in mTEChi and mTEClo (n = 1,589). (three mTEChi biological replicates and one mTEClo dataset).
- 3D representation of the distribution of ASEs of Aire‐sensitive (Left) and neutral genes (Right) according to their PSI values calculated from three mTEChi replicates (combined) and one mTEClo dataset.
- Levels of ASE inclusion imbalance for Aire‐sensitive and neutral genes (equal numbers) based on PSI values calculated from three mTEChi replicates (combined) and one mTEClo dataset.
- Differential gene expression of Aire‐sensitive genes between mTEChi and cTEC. Red dots show the Aire‐sensitive genes with FPKM > 1 in mTEChi and cTEC (n = 879). (three mTEChi and three cTEC biological replicates).
- 3D representation of the distribution of ASEs of Aire‐sensitive (Left) and neutral genes (Right) according to their PSI values calculated from three mTEChi replicates (combined) and three cTEC replicates (combined).
- Levels of ASE inclusion imbalance for Aire‐sensitive and neutral genes (equal numbers) based on PSI values calculated from three mTEChi replicates (combined) and three cTEC replicates (combined).
We calculated the PSI values of ASEs of the selected Aire‐sensitive and neutral genes and compared their distributions in WT and Aire‐KO mTEChi using three‐dimensional representations (Fig 3B). We observed similar PSI values for Aire‐sensitive genes in WT and Aire‐KO mTEChi, as well as for Aire‐neutral genes. We then calculated the values of ASE inclusion imbalance and identified similar low levels for Aire‐sensitive genes in mTEChi from WT and Aire‐KO mice, showing that the reduced inclusion of ASEs in the transcripts of Aire‐sensitive genes was independent of Aire expression (Fig 3C).
Since mTEChi correspond to a stage of differentiation derived from precursor cells in mTEClo that lack Aire expression (Gäbler et al, 2007; Hamazaki et al, 2007; Dhalla et al, 2020), we asked whether the low ASE inclusion in transcripts of Aire‐sensitive genes could be a feature already present in mTEClo. To this end, we selected the genes with expression values over 1 FPKM in mTEChi and mTEClo for calculation of PSI values of their associated ASEs (Fig 3D). As for the WT versus Aire‐KO mTEChi comparison, we observed similar PSI values for Aire‐sensitive genes in mTEChi and mTEClo, as well as for Aire‐neutral genes (Fig 3E). Finally, calculation of the values of ASE inclusion imbalance revealed similar low levels for Aire‐sensitive genes in mTEChi and mTEClo (Fig 3F). This finding thus shows that the low number of ASEs in the transcripts of Aire‐sensitive genes in mTEChi is also a feature of mTEClo.
Next, we sought to definitively confirm this observation in analyzing an independent RNA‐seq dataset of mTEChi/lo sorted as CD45−EpCAM+UEA1+Ly51−CD80highMHCIIhigh/CD80lowMHCIIlow (St‐Pierre et al, 2015). We first showed that the level of expression of the markers used to sort these cells and of a set of genes specific to mTEChi and mTEClo were identical or very close to those measured in our mTEChi and mTEClo data (Appendix Fig S4A and B). Then, we analyzed the extent of ASE inclusion in the independent mTEChi/lo dataset and confirmed similar low levels of ASE inclusion imbalance for Aire‐sensitive genes in mTEChi and mTEClo (Appendix Fig S4C–E).
To evaluate the levels of ASE inclusion in cTECs, we analyzed RNA‐seq data of cTECs that were also generated in (St‐Pierre et al, 2015) and showed that the low ASE inclusion in transcripts of Aire‐sensitive genes was also a feature of cTECs (Fig 3G–I), suggesting that it could stem from a common thymic epithelial progenitor.
Together these findings revealed that the low inclusion of ASEs in the transcripts of Aire‐sensitive genes is a general feature of TECs, independent of Aire expression and conserved upon maturation of mTEClo into mTEChi.
Transcripts of Aire‐sensitive genes show enhanced ASE inclusion in those tissues where they are expressed
Since transcripts of Aire‐sensitive genes are subject to low ASE inclusion in mTEChi, we asked whether they could exhibit a stronger inclusion when their expression is driven by tissue‐specific transcriptional mechanisms in the periphery. To address this question, we selected for each tissue in our dataset, the Aire‐sensitive genes showing a specific or selective expression by using the Specificity Measurement (SPM) method (Pan et al, 2013) as in Guyon et al (2020), and determined the levels of ASE inclusion imbalance. Comparison with mTEChi revealed overall stronger ASE inclusion in peripheral tissues (Fig 4A), indicating that ASE inclusion for transcripts of Aire‐sensitive genes is differentially regulated in the periphery. We further identified for each ASE, its PSI values in mTEChi and in the peripheral tissue(s) of specific/selective expression of its corresponding Aire‐sensitive gene (Fig EV2A). We selected the ASEs with PSI values < 0.1 in mTEChi and > 0.1 in the periphery, as exemplified in (Figs EV2A–C), and found that nearly a quarter of ASEs present in the tissues of specific Aire‐sensitive gene expression, were excluded in mTEChi (Fig 4B, Left). This exclusion of ASEs in mTEChi would therefore increase the risk of release of autoreactive T cells. Conversely, a significantly lower percentage of ASEs present in mTEChi were excluded in the tissues of specific Aire‐sensitive gene expression (Fig 4B, Right). We noted that among the latter ASEs, only a minority (about 2%) showed a full inclusion in mTEChi (PSI > 0.9 in mTEChi and < 0.1 in the periphery; Fig EV2A), indicating that only few autoreactive T cells specific for the antigenic epitopes generated upon the exclusion of these ASEs, would leave the thymus and contribute to the risk for autoimmunity.
Figure 4. Higher levels of ASE inclusion imbalance for Aire‐sensitive genes in their tissues of expression.
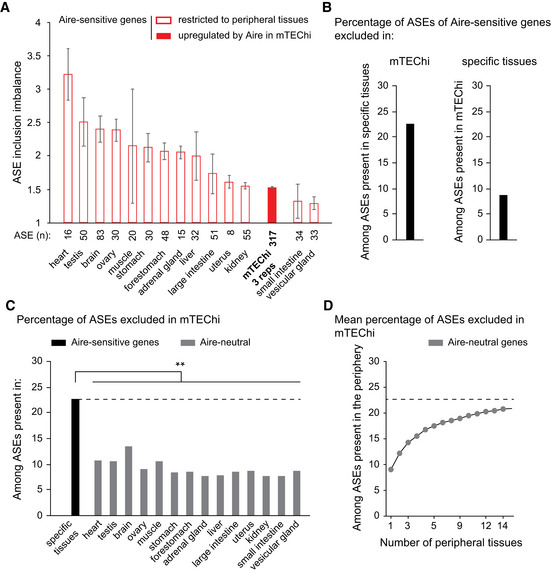
-
ALevels of ASE inclusion imbalance for Aire‐sensitive genes in their tissues of expression. Open red bars are for particular peripheral tissues, whereas the solid red bar is for mTEChi. The ASE inclusion imbalance is calculated for each of the three mTEChi biological replicates on an identical set of ASEs (n = 3, error bars show mean ± STD).
-
BPercentage of ASEs of Aire‐sensitive genes that are excluded in mTEChi among ASEs showing some level of inclusion in the tissues of specific expression (Left). The percentage of ASEs excluded in specific tissues among ASEs present in mTEChi is shown (Right).
-
C, DPercentage of ASE exclusion for Aire‐neutral genes in mTEChi among ASEs present in each peripheral tissue (C) or (D) in all 1–14 permutations of the 14 peripheral tissues in our database, as a mean percentage. The dashed line represents the percentage of ASE exclusion shown in (B, Left) and (C), **P < 10−3 (pnorm (cumulative distribution function) to the normal distribution defined by the mean and STD of ASE exclusion percentages in the 14 peripheral tissues).
Figure EV2. PSI values of ASEs of Aire‐sensitive genes in mTEChi and in their tissues of expression.
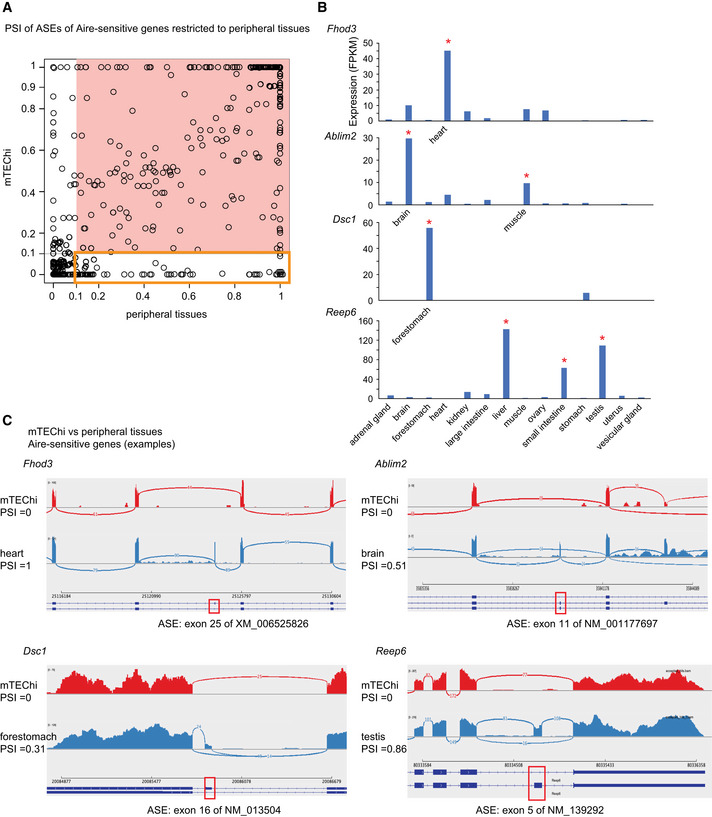
- Each ASE is represented by a circle. ASEs excluded in mTEChi or in the periphery (PSI < 0.1) are shown on a white background, otherwise on a salmon‐colored background. The ASEs present in the periphery (PSI > 0.1) and excluded in mTEChi (PSI < 0.1) are framed by an orange square.
- Level of expression of Fhod3, Ablim2, Dsc1, and Reep6 (taken as examples of Aire‐sensitive genes with a specific or selective peripheral expression) from RNA‐seq data of mTEChi and 14 mouse tissues. Asterisks indicate the tissue(s) of specific/selective expression: Fhod3 is specific to the heart, Dsc1 to the forestomach; Ablim2 is selective to the brain and muscle, Reep6 to the liver, small intestine, and testis.
- Shashimi plots of the above genes show ASE exclusion (PSI < 0.1) in mTEChi (red) and some level of inclusion (PSI > 0.1) in the tissues of expression (blue). Arcs representing splice junctions connect exons. Red boxes indicate alternative spliced exons present or absent in the transcript isoforms of Fhod3, Ablim2, Dsc1, and Reep6.
We next compared for Aire‐sensitive and neutral genes the proportion of ASEs excluded in mTEChi among those expressed in peripheral tissues and found significantly lower proportions for neutral genes (Fig 4C). Since Aire‐neutral genes can be expressed in multiple tissues, increasing their likelihood of undergoing ASE inclusion, we sought to determine for their transcripts, the proportion of exclusion in mTEChi of their ASEs present in the periphery, considering all tissues together. Hence, we considered all 1–14 permutations of the 14 tissues in our dataset and calculated for each set of permutations of the same number of tissues, the mean proportion of ASEs exclusively excluded in mTEChi (Fig 4D). We observed that the proportion of exclusion tended to reach, with the increasing number of considered tissues, the proportion of exclusion detected for Aire‐sensitive genes in mTEChi (Fig 4D and B, Left).
Together these findings revealed that transcripts of Aire‐sensitive genes show stronger ASE inclusion in their tissues of expression than in mTEChi and that an important proportion of these ASEs expressed in the periphery are excluded from mTEChi, similarly to the ASEs of Aire‐neutral genes.
Aire‐sensitive genes escape Raver2‐dependent promotion of ASE inclusion in mTECs
To get insights into the molecular mechanisms that underlie the differential inclusion of ASEs between Aire‐sensitive and neutral genes in mTEChi, we searched for the preferential or deprived gene expression of a full set of RNA‐binding proteins including factors known to influence RNA splicing, in mTEChi versus peripheral tissues. We identified a handful of RNA‐binding proteins showing statistically significant differential expression in mTEChi, including a single factor reported to modulate alternative splicing, namely Raver2 (Bartoletti‐Stella et al, 2015) (Fig 5A and Appendix Fig S5). We further showed that Raver2 expression is over‐represented in mTEChi, mTEClo, or cTECs (Fig 5B). Then, to determine whether the higher expression of Raver2 is specific to the thymic epithelium or shared with epithelia from other tissues, we collected public RNA‐seq datasets of epithelia isolated from a variety of peripheral tissues (ENCODE Project Consortium, 2012; St‐Pierre et al, 2013; Tan et al, 2020; Marincola Smith et al, 2021; Pal et al, 2021; Wiles et al, 2021) and calculated in each sample the expression of Raver2. This comparison revealed a preferential expression of Raver2 in the thymic epithelium supporting an important role for Raver2 in the thymus (Fig EV3).
Figure 5. Effect of Raver2 and Setd2 on the level of ASE inclusion imbalance for Aire‐sensitive and neutral genes in mTECs.
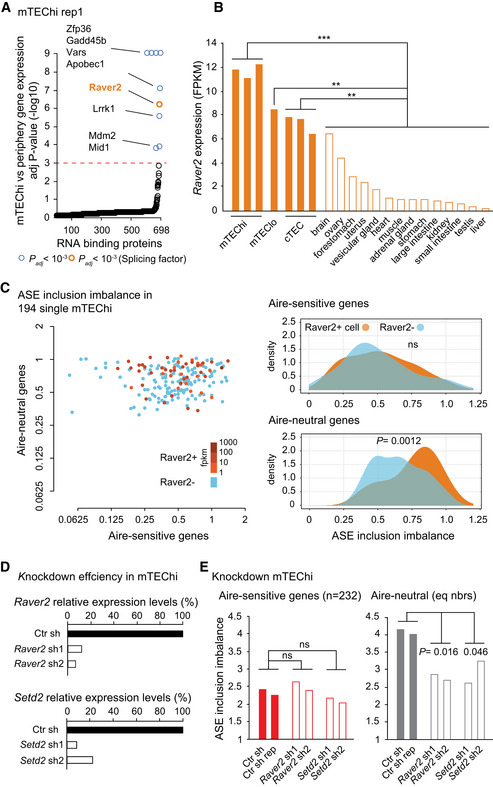
- Differential expression of genes coding for RNA‐binding proteins in mTEChi vs peripheral tissues. For each gene, the expression in mTEChi is compared to the normal distribution estimated based on the levels of expression in the periphery. The red dashed line represents the threshold for statistical significance (Benjamini–Hochberg adjusted P < 0.001). RNA‐binding proteins showing significant differential gene expression are represented by colored circles, in orange if the RNA‐binding proteins have been reported to be involved in splicing, in blue otherwise.
- Raver2 expression levels in mTEChi, mTEClo, cTEC, and across peripheral tissues (open bars). Significance of Raver2 over‐representation in mTEChi, in mTEClo and in cTEC versus the peripheral tissues is shown. *** P < 10−7, **P < 10−3 (pnorm (cumulative distribution function) to the normal distribution defined by the mean and STD of Raver2 expression in the 14 peripheral tissues).
- Levels of ASE inclusion imbalance (Left) and their distributions (Right) are shown for Aire‐neutral and Aire‐sensitive genes in 194 single mTEChi. Blue is for cells negative for Raver2, whereas orange shades are for Raver2‐positive cells. Statistical significance assessed by a Student's test.
- Relative expression levels of Raver2 (Top) and Setd2 (Bottom) in mTEChi infected by lentiviruses containing Raver2, Setd2, or the Ctr (LacZ) shRNAs.
- Levels of ASE inclusion for transcripts of Aire‐sensitive and neutral genes (equal numbers) in mTEChi infected by lentiviruses targeting Raver2, Setd2 (open bars), or the Ctr (LacZ; solid bars). Statistical significance of cumulative sh1 and sh2 assessed by a chi‐squared test (two Ctr and two sh‐treated biological replicates).
Figure EV3. Comparison of Raver2 expression levels across TECs and epithelial cells from a variety of peripheral tissues.
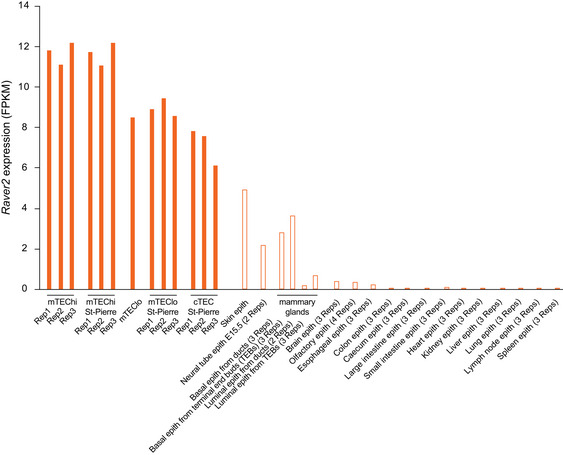
Closed bars are for TECs (mTEChi, mTEClo, and cTECs) including our dataset and St‐Pierre dataset for replication. Open bars correspond to Raver2 expression calculated by re‐analysis of public datasets of epithelial cells isolated from peripheral tissues.
To determine whether Raver2 was able to exert an effect on Aire‐sensitive or neutral genes, we first sought to correlate its expression to variation of ASE inclusion imbalance across individual mTEChi. To this end, we collected full‐length single‐cell RNA‐seq data enabling splicing analyses that were generated in mTEChi (Handel et al, 2018). We calculated, for each cell, the PSI values of ASEs of Aire‐sensitive and neutral genes, as well as their levels of ASE inclusion imbalance. Note that the comparison of the distribution of the levels of ASE inclusion imbalance across the individual mTEChi and between Aire‐sensitive and neutral genes showed significantly lower levels for Aire‐sensitive genes (Appendix Fig S6A), therefore confirming, at the single‐cell level, the difference that we identified at the cell population level in (Figs 2C and EV1B). The absence of Aire effect on the low inclusion of ASEs in transcripts of Aire‐sensitive genes was confirmed by the lack of significant correlation between the levels of ASE inclusion imbalance and those of Aire expression across the individual mTEChi (Appendix Fig S6B). We then identified which cells expressed Raver2 (threshold > 1 FPKM; Fig 5C, Left) and compared the distribution of the levels of ASE inclusion imbalance between the Raver2‐positive and Raver2‐negative cells. In contrast to Aire‐sensitive genes, we observed a significant association between the presence of Raver2 and the high levels of ASE inclusion imbalance for Aire‐neutral genes, strongly suggesting that Raver2 promotes the inclusion of ASEs specifically in transcripts of Aire‐neutral genes (Fig 5C, Right).
Then, to evaluate the specific impact of Raver2 on ASE inclusion in mTECs, we used a 3D organotypic culture system adapted from Ref Pinto et al (2013) in which we seed medullary‐ and MHCII‐enriched TECs (mTEChi: ~ 62%; mTEClo: ~ 7%; Appendix Fig S7), and retrieve, after 5 days in culture, pure mTECs showing a relative increase of mTEClo (mTEChi: ~ 70%; mTEClo: ~ 25%; Appendix Fig S8A). This shift toward mTEClo is dramatically enhanced in the absence of the addition of RankL, suggesting that it is caused by an increased mTEChi mortality and/or de‐differentiation (Appendix Fig S8B). In addition, we observe a strong expression of Aire and Ins2 (prototypic self‐antigen gene controlled by Aire) in the seeded and retrieved cells, with higher levels (2‐fold) at the beginning of the culture (Appendix Fig S9). This observation is consistent with the transitory nature of Aire expression and the fact that it does not seem to be balanced out by new Aire‐expressing mTEChi arising from the differentiation/maturation of cells from the mTEClo pool. Right after the seeding of mTECs onto the 3D system, we performed infection with concentrated lentiviruses encoding shRNAs targeting Raver2 (shRNA 1 or 2) or the control LacZ, cloned into a vector expressing GFP as a marker of transduction (Fig EV4). Three days later, we cell‐sorted GFP+ mTECs and quantified Raver2 expression level. We found a strong knockdown efficiency for each of the two Raver2 shRNAs with 10–20% remaining expression in comparison to control knockdown samples (Fig 5D, Top). To ensure that the newly transcribed isoforms, resulting from potential alternative splicing alterations promoted by Raver2 knockdown, stabilize and accumulate to be accurately detected, we isolated GFP+ mTECs 5 days after infection. RNA‐seq experiments on these cells revealed a significant reduction of the levels of ASE inclusion imbalance for Aire‐neutral genes (Fig 5E, Right), whereas no effect was detected on Aire‐sensitive genes (Fig 5E, Left). This finding demonstrated that Raver2 promotes the inclusion of ASEs in transcripts of Aire‐neutral genes and that those of Aire‐sensitive genes escape its effect.
Figure EV4. Schematic of the knockdown strategy of primary mTECs ex vivo .
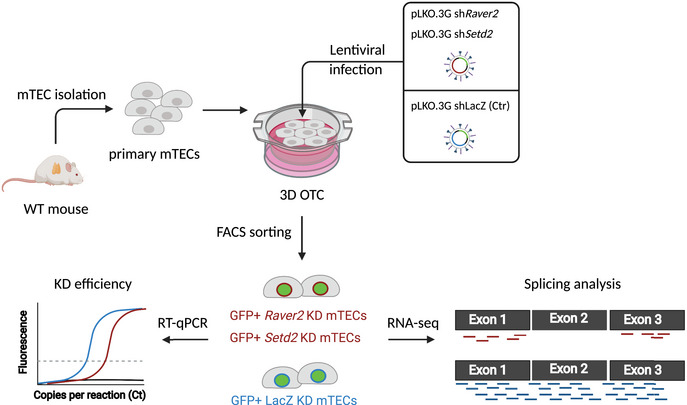
Primary mTECs were isolated from thymi of WT mice and seeded along with shRNA‐containing lentiviruses on a 3D organotypic culture system (3D OTC). Three days or 5 days after infection, GFP+ mTECs expressing the lentigenes were isolated for knockdown efficiency by qPCR or for splicing analyses by RNA‐seq experiments. This figure was created with BioRender (biorender.com).
Raver2 is a heterogeneous nuclear ribonucleoprotein (Hnrnp) that has been reported to bind to the polypyrimidine track‐binding protein (Ptb) (Kleinhenz et al, 2005; Henneberg et al, 2010) which regulates alternative splicing at regions enriched in H3K36me3 through interaction with H3K36me3 adaptor proteins (Luco et al, 2010, 2011). To evaluate the effect of H3K36 methylation on ASE inclusion in mTECs, we performed knockdown of Setd2 that is recognized as the only enzyme able to methylate H3K36 into H3K36me3 in somatic cells (Husmann & Gozani, 2019). We first validated the efficient knockdown of two shRNAs targeting Setd2 (Fig 5D, Bottom) and then performed RNA‐seq experiments to determine the levels of ASE inclusion imbalance in the knockdown samples. As for Raver2, Setd2 knockdown resulted in a significant reduction of ASE inclusion for the transcripts of Aire‐neutral genes, whereas no effect was detected for those of Aire‐sensitive genes (Fig 5E).
Together these findings showed that Aire‐sensitive genes escape Raver2’s effect promoting ASE inclusion in mTECs and suggested that the effect on Aire‐neutral genes require H3K36me3 as an anchor for this factor.
Aire‐sensitive genes are devoid of H3K36me3 and H3K36me3‐associated ASE inclusion in mTEClo
Since the low ASE inclusion in the transcripts induced by Aire in mTEChi is already present in mTEClo (Fig 3D–F), we sought to determine whether a potential escape of Raver2’s effect due to a lack of H3K36me3 could apply for such a mechanism in these cells. We looked for H3K36me3 enrichment by ChIP‐seq experiments and found very low levels of H3K36me3 at Aire‐sensitive genes in comparison to all genes (Figs 6A and EV5A). We then selected the genes with expression values over 1 FPKM and the presence of H3K36me3 marks within the last third of their gene bodies and compared the levels of ASE inclusion imbalance to those of the counterpart genes lacking H3K36me3 (Fig 6B). We observed a significant increase of ASE inclusion in transcripts of genes harboring H3K36me3 marks, very likely reflecting the functional link that we detected between increased H3K36me3 and stronger ASE inclusion in mTECs (Fig 5E, Right). Thus, these findings supported the assumption that the low ASE inclusion observed for transcripts of Aire‐sensitive genes in mTEClo was due, at least in part, to the lack of H3K36me3 deposition and the subsequent impairment of Raver2 recruitment.
Figure 6. Aire‐sensitive genes show low levels of H3K36me3 correlated with low levels of ASE inclusion imbalance in mTEClo.
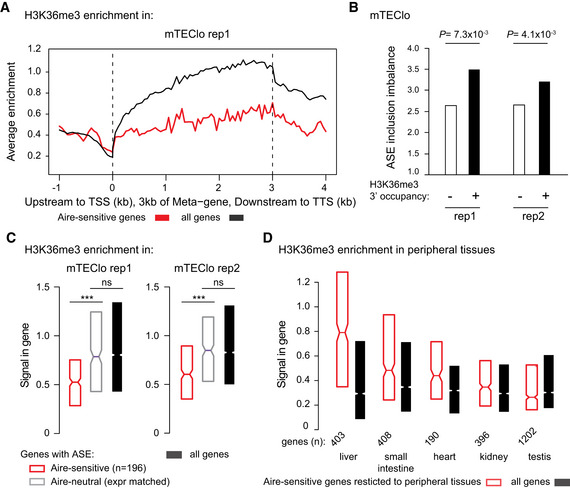
- Metagene profiles of the average normalized enrichment of H3K36me3 for Aire‐sensitive genes (red) and all genes (black) in mTEClo.
- Levels of ASE inclusion imbalance for genes with and without H3K36me3 in the last third of their gene bodies, in each biological replicate (rep1 and 2). The statistical significance is assessed by a chi‐squared test performed in each of the two replicates.
- Median enrichment of H3K36me3 for Aire‐sensitive genes and neutral genes (expression matched), as well as all genes in each biological replicate. *** P < 10−13 (Wilcoxon test).
- Median enrichment of H3K36me3 for Aire‐sensitive genes in their tissues of expression and for all genes.
Data information: In (C) and (D), notches represent the 95% confidence interval of the medians, and the limits of the upper and lower boxes the 75th and 25th percentile of H3K36me3 enrichment, respectively.
Figure EV5. Reduced H3K36me3 deposition at Aire‐sensitive genes in mTEClo in comparison to their tissues of expression.
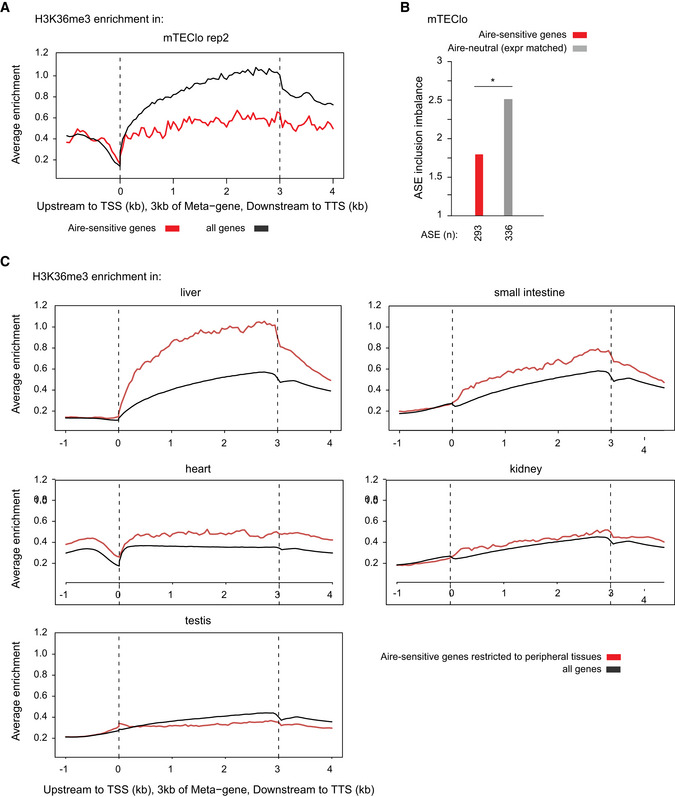
- Metagene profiles of the average normalized enrichment of H3K36me3 for Aire‐sensitive genes (red) in mTEClo; all genes are shown in (black).
- Levels of ASE inclusion imbalance for Aire‐sensitive genes and neutral genes (expression matched), *P < 0.05 (chi‐squared test performed in one mTEClo dataset).
- Metagene profiles of the average normalized enrichment of H3K36me3 for Aire‐sensitive genes (red) in their tissues of expression; all genes are shown in (black).
However, since H3K36me3 is involved in transcription (Vojnic et al, 2006; de Almeida & Carmo‐Fonseca, 2012), we asked whether the lack of H3K36me3 at Aire‐sensitive genes was specific to these genes or associated with their low expression. To address this question, we selected a set of Aire‐neutral genes with expression levels matching those of Aire‐sensitive genes and measured their enrichment for H3K36me3. We found significantly lower levels of H3K36me3 at Aire‐sensitive genes, therefore showing that the lack of H3K36me3 was specific to these genes and unrelated to their low expression (Fig 6C). In addition, a comparison of the levels of ASE inclusion imbalance for the same Aire‐sensitive and expression‐matched neutral genes showed a clear reduction of the former (Fig EV5B), confirming that the weak ASE inclusion for transcripts of Aire‐sensitive genes is not a reflection of their low expression levels.
Finally, to address whether the lack of H3K36me3 deposition at Aire‐sensitive genes was a differential feature between mTEClo and peripheral tissues, we collected H3K36me3 ChIP‐seq data generated from mouse tissues, as part of the ENCODE project (ENCODE Project Consortium et al, 2007). For each tissue, we measured the enrichment of H3K36me3 at all genes and found that Aire‐sensitive genes with a specific or selective expression showed similar to higher levels of H3K36me3 in comparison to all genes (Figs 6D and EV5C). This finding is consistent with the observation that Aire‐sensitive genes exhibit enhanced ASE inclusion, globally, in the periphery, but showed a poor correlation with ASE inclusion in each individual tested tissue (Fig 4A), indicating that H3K36me3 may be permissive to tissue‐specific mechanisms of alternative splicing without orchestrating a coordinated tissue‐independent pattern of the latter.
Discussion
In our present study, we demonstrated that, while mTEChi express a wide range of self‐antigen splice isoforms, Aire‐upregulated transcripts exhibit a low diversity and a weak inclusion of ASEs. We showed that this low ASE inclusion was independent of Aire's action on gene expression and resulted from a mechanism that was sustained by the specific depletion of H3K36me3. The linked mechanism between the decrease of H3K36me3 and the reduction of ASE inclusion that we propose was consistent with the report showing that alternative exons, subjected to exclusion or partial inclusion, have lower H3K36me3 signals than constitutive exons in Caenorhabditis elegans (Kolasinska‐Zwierz et al, 2009). In addition, a recent report showed that high inclusion levels of skipped exon (SE) ASEs correlate with the strong enrichment of H3K36me3 in the exon flanking regions in mice (Hu et al, 2020).
Unlike Aire‐sensitive genes, we showed that the genes neutral to the effect of Aire in mTECs were characterized by an enrichment of H3K36me3 and a strong enhancement of ASE inclusion. Identification of Raver2 as a splicing‐related factor overrepresented in mTECs and able to promote ASE inclusion led us to propose a mechanism by which ASE inclusion for Aire‐neutral genes is driven by the coordinated action of Raver2 and the splicing factor Ptb that is specifically recruited at H3K36me3‐rich regions. This mechanism was supported by the observation that Ptb is not only able to repress alternatively spliced exons (Wagner & Garcia‐Blanco, 2002) but also enhances the inclusion of a large number of ASEs, as shown by the effect of Ptb knockdown on ASE inclusion at the genomic scale (Luco et al, 2010). In addition, Raver2 was characterized as a potent modulator of the splicing activity of Ptb (Bartoletti‐Stella et al, 2015), therefore opening the possibility that Raver2 may function through interaction with Ptb to enhance ASE inclusion for transcripts of genes with H3K36me3 enrichment. The role of Raver2 in mTECs is further strengthened by the higher expression of Ptb in mTEChi (105 FPKM) and mTEClo (91 FPKM) than in peripheral tissues (median expression: 31 FPKM).
Hence, through depletion of H3K36me3, the self‐antigen genes controlled by Aire likely escape the effect of Raver2/Ptb and thereby generate transcripts with fewer ASEs. This weak ASE inclusion indicated that the Aire‐induced transcripts do not include all the ASEs that are spliced‐in in the periphery. Lack of presentation of Aire‐dependent antigenic peptides corresponding to ASEs absent from mTEChi, is likely to result in a number of autoreactive T cells leaving the thymus and potentially able to elicit autoimmune reactions. The formal identification and characterization of such autoreactive T cells by the use of MHC tetramers carrying peptides from thymus‐excluded ASEs of Aire‐dependent genes will be necessary to evaluate the direct impact of the described weak ASE inclusion on central tolerance. In addition, thanks to the progress of the Knockout Mouse Project (https://www.mmrrc.org) with the recent availability of Raver2 KO cryopreserved embryos, the effect of the deletion of Raver2 on the diversity of autoreactive T cells will be assessable, as well as on the susceptibility to autoimmunity in the next future.
The negative selection triggered by Aire may then be not as comprehensive as originally thought and complementary peripheral mechanisms would be likely necessary to maintain tolerance. As a possible mechanism, Tregs selected against peptides derived from constitutive exons of Aire‐sensitive genes in mTEChi would suppress the autoimmune responses triggered against immunogenic ASE‐derived peptides. This is in line with previous studies showing the importance of Aire‐induced self‐antigen expression in the selection and development of suppressive Tregs (Aschenbrenner et al, 2007; Pomié et al, 2011; Malchow et al, 2013). In humans, the study of APS1 patients revealed a role for AIRE in the development of Tregs (Ryan et al, 2005; Kekäläinen et al, 2007), as well as in the enforcement of immune tolerance by directing differentiation of autoreactive T cells into the Treg lineage (Malchow et al, 2016). This latter mechanism would thus provide the possibility for autoreactive T cells directed against immunogenic ASE‐derived peptides, to convert into Tregs, therefore preventing potential autoimmune reactions and favoring Treg‐associated suppressive responses.
Our study also revealed that the proportion of ASEs selectively excluded in mTEChi is similar between Aire‐sensitive and neutral genes. This indicated that the lack of H3K36me3 at Aire‐sensitive genes, and therefore the absence of Raver2 recruitment, would result in a certain degree of autoreactivity that Tregs may control similarly to autoreactive reactions against immunogenic peptides derived from ASEs of Aire‐neutral genes. Hence, the weak ASE inclusion for Aire‐sensitive genes in mTECs may be sufficient to impose, with the support of Tregs, immune tolerance against the full set of Aire‐dependent self‐antigens. If Aire‐mediated immunological tolerance was entirely under the control of the sole mTEChi‐dependent negative selection, which happens not to be the case, the full set of ASEs of Aire‐sensitive genes would be present in mTEChi with the full diversity of tissue‐specific splicing mechanisms activated. Such a broaden activation would certainly conflict with the proper function and physiology of mTEChi and represent a tremendous energetic cost, most probably unreachable.
Materials and Methods
Mice
Aire‐deficient (C57BL/6) mice, previously obtained by D. Mathis and C. Benoist (Harvard Medical School, Boston, MA), were bred and maintained at Cochin Institute. Wild‐type B6 mice were purchased from Charles River laboratory. Animal housing and experiments were conducted in specific‐pathogen‐free conditions according to the protocols and guidelines of the French Veterinary Department under procedures approved by the Paris‐Descartes Ethical Committee for Animal Experimentation (decision CEEA34.MG.021.11).
Isolation of medullary thymic epithelial cells
Thymi of 4‐ to 6‐week‐old mice were trimmed of fat, cut into pieces, and digested by mechanic coercion to release thymocytes. Enzymatic digestion was first performed with collagenase D (1 mg/ml; Roche) and DNase I (1 mg/ml; Sigma) for 30 min at 37°C, then with Collagenase/Dispase (2 mg/ml; Roche) and DNase I (2 mg/ml) at 37°C up to the obtention a single‐cell fraction. The cells were then filtered through a 70‐μm cell strainer and resuspended in PBS containing 1% FBS and 5 mM of EDTA. mTECs were then isolated from the obtained cell fraction using the following methods:
Isolation of mTEChi and mTEClo
Thymic stromal cells from pools of 4 thymi were enriched by CD45 depletion of thymocytes using magnetic CD45 MicroBeads (Miltenyi Biotec) and the magnetic cell sorter (autoMACS, Miltenyi Biotec). The depleted cell fraction was then stained for 20 min at 4°C with a cocktail of fluorophore‐labeled antibodies CD45‐PerCPCy5.5 (1:50; Biolegend, ref: 103131), Ly51‐PE (1:800; Biolegend, ref: 108307) and I‐A/E‐APC (MHCII; 1:1,200; eBioscience, ref: 17–5321) for sorting of mTEChi/lo (CD45−Ly51−I‐A/Ehigh/low) on a FACSAria III instrument (BD Bioscience), as previously described (Giraud et al, 2014; Guyon et al, 2020). The phenotypic validation of the isolated mTEChi and mTEClo was performed using the additional antibodies EpCAM‐PE‐Dazzle 549 (1:200; Biolegend, ref: 118235), CD80‐PE‐Cy7 (1:200; Biolegend, ref: 104733), and the lectin UEA‐1‐FITC (1:200; Thermo Fisher, L32476).
Isolation of total mTECs for 3D organotypic culture
As above, a CD45 depletion of thymocytes using magnetic beads was first performed. The depleted cell fraction was then stained for 20 min at 4°C with the CD45‐PerCPCy5.5 antibody (1:50; Biolegend) and the EpCAM‐FITC antibody (G8.8 clone; 1:500; Biolegend, ref: 118207) for FACS sorting of CD45−EpCAM+ cells. The sorted fraction was sequentially stained for 20 min at 4°C with the Ly51‐PE antibody (1:800; Biolegend) and depleted of Ly51‐positive cells using anti‐PE Microbeads (Miltenyi Biotec), then stained with the I‐A/E‐APC antibody (MHCII; 1:1,200; eBioscience) and positively selected for I‐A/E‐positive cells using anti‐APC Microbeads (Biolegend). The obtained final cell fraction consisted of total mTECs (CD45−EpCAM+Ly51−I‐A/E+).
shRNA‐containing lentivirus production
pLKO.1 plasmids bearing the shRNA sequences (sh1: TRCN0000181363 and sh2: TRCN0000242035 for Raver2; sh1: TRCN0000238533 and sh2: TRCN0000238536 for Setd2; Ctr sh: TRCN0000072240 for LacZ; Sigma) were subcloned into the lentiviral pLKO.3G vector containing an eGFP cassette (Addgene #14748), by transferring the BamHI‐NdeI restriction fragments containing the shRNAs. Lentiviruses were produced into HEK293T cells by using the calcium phosphate co‐transfection method for each specific subcloned lentiviral pLKO.3G vector with the packaging plasmids gag/pol (Addgene #14887) and VSV‐G (Addgene #14888). HEK293T were grown in Dulbecco's modified Eagles medium (DMEM) with high glucose (4,500 mg/l), supplemented with 10% fetal bovine serum (FBS), pen/strep antibiotics. HEK293T cells were transfected at 70–80% confluence in two T175 flasks. The transfection solution was prepared with 500 µl of 1 M CaCl2 (Sigma), the lentiviral vector (32 µg), the packaging plasmids (VSV‐G: 16 µg, gag/pol: 24 µg), and DNAse‐free water (Invitrogen) for a final volume of 2 ml. Then, 2 ml of HEPES‐buffered saline pH 7.0 (2× for transfection; VWR) was added dropwise to the previous mixture, under constant agitation. The obtained solution was kept at room temperature for 15 min and subsequently equally transferred to the HEK293T flasks. The next day, the medium was changed. 48 h after transfection, the culture medium containing the lentiviral particles was collected and ultracentrifuged at 113,000 g for 90 min at 4°C using a Beckman SW28 ultracentrifuge rotor (Beckman Coulter) for the obtention of concentrated virus, that was resuspended in cold PBS 1X. Viral titration was performed in HEK293 cells. For individual evaluation of shRNAs, mTECs were transduced at a multiplicity of infection of 10.
3D organotypic culture of primary mTECs
Preparation of the fibrin gel
The 3D organotypic co‐culture system described in (Pinto et al, 2013) was set up using a ~ 1.2‐mm‐thick viscose‐coated nonwoven fibrous fabric: Jettex 2005/45 (Orsa) that was placed as a scaffold into 12‐well filter inserts (polyester capillary membrane with 3 μm pores; Dutscher). A semi‐solid fibrin gel inoculated in the insert that serves as support for the culture system was prepared seeding human dermal fibroblasts (200,000) re‐suspended in a fibrin gel made of fibrinogen (Merck‐Millipore), thrombin (Merck‐Millipore), and aprotinin (Euromedex) which prevents precocious fibrinolysis by serine proteases secreted by fibroblasts. Human dermal fibroblasts were generated from explant cultures of de‐epidermized dermis as described (Boehnke et al, 2007) and grown up to passage 4 in DMEM/F‐12 (Thermo Fisher) complemented with 10% FBS and pen/strep antibiotics. The fibrin gel polymerized at 37°C for 1 h or 2, forming a smooth upper surface for the culture of primary mTECs. A pre‐culture of 5 days is required for the fibroblast activation and is set up by the submersion of the polymerized gel in medium 1 containing DMEM/F‐12, 10% FBS, pen/strep antibiotics, l‐ascorbic acid (50 μg/ml; Sigma), and TGF‐β (1 ng/ml; R&D Systems). The medium 1 was changed every day. On the day of mTEC seeding, medium 1 was replaced by medium 2 containing DMEM high glucose + DMEM/F12 1:1 (v/v) with 10% FBS, pen/strep antibiotics, cholera toxin (10−10 M; Sigma), hydrocortisone (0.4 μg/μl; Sigma), l‐ascorbic acid (50 μg/ml), aprotinin (500 U/ml), and RankL (80 ng/ml; R&D Systems).
mTEC seeding and lentiviral infection
Freshly isolated mTECs were seeded at the top of the fibroblast‐containing fibrin gel with the addition of 10 µl of concentrated shRNA‐bearing lentiviruses that we produced against Raver2, Setd2, and the Ctr (LacZ). The next day, medium 2 was replaced by medium 3 that is similar to medium 2 but containing reduced amount of aprotinin (250 U/ml) and increased amount of RankL (100 ng/ml). The RankL supplemented in mediums 2 and 3 helped maintain mTEChi maturity.
Isolation of knockdown mTECs
Three days after infection, a first part of the 3D organotypic co‐culture was terminated by cutting out half of the fibrin gel to evaluate the Raver2 and Setd2 knockdown efficiencies by RT–qPCR. Two days later, the other half of the fibrin gel was processed to evaluate the impact of Raver2 and Setd2 knockdown on ASE inclusion of Aire‐sensitive and neutral genes by RNA‐seq. Using small dissecting scissors, the fibrin gel portions were cut into small pieces to be properly digested in an enzymatic solution of Collagenase/Dispase (0.5 mg/ml; Roche) and DNase I (0.5 mg/ml) at 37°C for 15 min for the obtention a single‐cell fraction. After three rounds of centrifugation and clean‐up in PBS containing 1% FBS and 5 mM of EDTA, the single‐cell fraction was stained for 20 min at 4°C with the I‐A/E‐APC (MHCII) antibody (1:1,200; eBioscience) and for 5 min at 4°C with the DAPI viability dye solution (1 µg/ml; Sigma) to identify dead cells. We then sorted by flow cytometry DAPI−I‐A/E+GFP+ cells corresponding to viable lentivirus‐infected mTECs.
shRNA knockdown efficiency
RNA extraction from knockdown mTECs cultured in the 3D organotypic system was performed using the Single Cell RNA Purification Kit (Norgen Biotek). First‐strand cDNA was synthesized using SuperScript IV VILO Master Mix (Thermo Fisher). cDNA was used for subsequent PCR amplification using the viia7 Real‐time PCR system (Thermo Fisher) and the Fast SYBR Green Master Mix (Thermo Fisher). Knockdown efficiency was assessed by comparing the level of expression of Raver2 (forward primer: AACCAGAAGACACCGCAGAG; reverse: TCTCCCAAGAGTGAAGTCTGATT) and Setd2 (forward primer: CAGCATGCAGATGTAGAAGTCA; reverse: TCCAGGACAAAGGTGTTCG) between the knockdown and control samples using Gapdh (forward primer: GGCAAATTCAACGGCACAGT; reverse: AGATGGTGATGGGCTTCCC) for normalization.
RNA‐seq of medullary epithelial cells
Total RNA of knockdown mTECs cultured in the 3D organotypic system was extracted using the Single Cell RNA Purification Kit (Norgen Biotek). Total RNA of mTEChi isolated from WT or Aire‐KO mice was extracted with TRIzol (Thermo Fisher) and used to generate the third replicate of two independent paired‐end RNA‐seq datasets that we previously generated and deposited in the GEO database (GSE140683). We used mTEClo paired‐end RNA‐seq data that we previously generated (GSE140815). Single‐cell full‐length and paired‐end (2 × 125 bp) RNA‐seq data of mTEChi were obtained from the GEO database (GSE114713). For mTECs in the 3D organotypic system, polyA‐selected transcriptome libraries were constructed using the SMART‐seq v4 Ultra Low Input RNA Kit (Takara) combined to the Nextera XT DNA Library Preparation Kit (Illumina) and sequenced on the Illumina NextSeq 500 machine as paired‐end data (2 × 150 bp). Sequences were deposited in the GEO database as GSE177063. For WT and Aire‐KO mTEChi, polyA‐selected transcriptome libraries were constructed using the TruSeq Stranded mRNA Library Prep (Illumina) and sequenced on the Illumina HiSeq 2000 machine as paired‐end data (2 × 100 bp). Sequences were deposited in the GEO database as GSE177062. All paired‐end reads of the different datasets were homogenized to 2 × 100 bp by read‐trimming and mapped to the mm10 genome using the TopHat 2 program (Trapnell et al, 2009) with default parameters. Levels of gene and transcript isoform expression were determined using the cufflinks 2 program (Trapnell et al, 2010) that assembles all reads on the RefSeq transcript annotations and estimates their abundances. We also used cuffdiff 2 (Trapnell et al, 2013) to compute differential gene expression between WT and Aire‐KO mTEChi and between mTEChi and mTEClo.
Human medullary epithelial cell isolation and RNA‐seq
The thymi from cardiac surgery pediatric patients (the ethical permission #170/T‐I from Ethics Review Committee on Human Research of the University of Tartu) were minced in 1× PBS to release the thymocytes and then treated four times with collagenase/dispase (1:200) and DNaseI (1:1,000) digestion, and the cell fractions were further dissociated with gentleMACS treatment. All fractions were combined and filtered through the 100 μm Falcon cell strainer. The cells were collected by centrifugation, washed, and transferred to Optiprep density gradient. The upper cell layer was combined into one tube, washed, and counted resulting in 0.4–6.5 × 108 thymic cells. The cell fraction was then depleted with human CD45 beads (100 µl beads per 2 × 108 cells; Miltenyi Biotec) using autoMACS. After the CD45 depletion, the cells were sorted with BD FACSAria for mTEC MHChi (CD45− CDR2− EpCAM+ HLA‐DRhi) directly into TRIzol (Thermo Fisher) solution, RNA was isolated with miRNAeasy Mini Kit (Qiagen) with DNase treatment. The RNA quality was assessed with TapeStation (Agilent D1000) before the processing with SMART‐Seq v4 Ultra Low Input RNA Kit for Sequencing, and the library preparation with Illumina Nextera XT DNA Library Preparation Kit. The paired‐end sequencing (2 × 150 bp) was performed on the Illumina NextSeq500. Sequences were deposited in the GEO database as GSE176445. The reads were homogenized to 2 × 100bp by read‐trimming and mapped to the hg38 genome with TopHat 2. Levels of gene and transcript isoform expression were determined using cufflinks 2 and expression data as well as GTF files were used for ASE analysis.
H3K36me3 ChIP‐seq. experiment and analysis
Nano‐ChIP‐seq on H3K36me3 was performed as previously described (Adli & Bernstein, 2011) on 50,000 isolated mTEClo. ChIP‐seq libraries were prepared with the TruSeq ChIP Sample Preparation Kit (Illumina) and 2 × 75 bp paired‐end reads were sequenced on an Illumina HiSeq 2000 machine. Sequences were deposited in the GEO database as GSE176371. Alignment to the mm10 genome was done using Bowtie 2 (Langmead et al, 2009). Duplicate alignments were removed using Samtools (Li et al, 2009) and the command line: “samtools view ‐S ‐hf 0x2 alignments.sam | grep ‐v "XS:i:" | foo.py > filtered.alignments.sam” the foo.py script been available online: “https://www.biostars.org/p/95929/”. Peak calling for a sample (in comparison to the inputs) was done using MACS2 callpeak (Zhang et al, 2008) and the options ‐f BAMPE, ‐‐SPMR and –broad. Enrichment to the inputs was calculated using MACS2 bdgcmp and the option ‐m FE. For multi‐tissue comparison, H3K36me3 ChIP‐seq data generated by the Bing Ren’s lab (http://renlab.sdsc.edu/), as part of the ENCODE project (ENCODE Project Consortium et al, 2007), were obtained from the GEO database: GSE31039 for heart, kidney, liver, small intestine, and testis of adult (8‐week‐old) mice. These data were available as signal (bigWig) and peak (bed) files processed on the mm9 genome following ENCODE standards. Finally, we used the CEAS distribution (Shin et al, 2009) on mTEClo and tissue data to calculate the average H3K36me3 enrichment within gene bodies and their upstream and downstream regions.
Splicing entropy calculation
The splicing entropy of a specific gene is a measure of diversity of the transcript isoforms that result from alternative splicing for this gene (Ritchie et al, 2008). The higher the number of transcript isoforms, the higher the splicing entropy. For a particular gene, the expression values of its transcript isoforms, computed by cufflinks, are used to calculate the splicing entropy:
n is the number of isoforms and Pi is the proportion that each isoform (i) contributes to the overall expression of the gene. We calculated the splicing entropy of each gene in our samples of interest and used the median splicing entropy for sample‐to‐sample comparison.
ASE inclusion analysis
We used the rMATS program (Multivariate Analysis of Transcript Splicing) (Shen et al, 2014) to identify all ASEs from the mm10 or hg19 RefSeq mRNA annotations database, including skipped exon (SE), alternative 5′ splicing site (5SS), alternative 3′ splicing site (3SS) or intron retention (IR). For a particular ASE, we computed the percent splicing inclusion (PSI), that is, the relative expression of transcript isoforms spliced in versus spliced in or out, using the R‐script that we developed (https://github.com/TeamGiraud/TEC_splicing_2021):
n spliced‐in is the number of isoforms with the ASE spliced in. m spliced‐in/‐out is the number of isoforms with the ASE spliced in or spliced out. To compare the PSI values between different set of genes and/or samples, we determined the splicing inclusion imbalance by dividing the number of ASEs showing some level of active inclusion (PSI > 0.1) by the number of excluded ASEs, that is, showing no or background inclusion (PSI < 0.1).
Multi‐tissue comparison analysis
Whole tissue level
We considered 14 homogeneously sequenced RNA‐seq datasets (paired‐end) of mouse peripheral tissues (Brain, Liver, Kidney, Adrenal Gland, Heart, Ovary, Testis, Stomach, Forestomach, Small intestine, Large Intestine, Muscle, Uterus, Vesicular Gland) generated by (Li et al, 2017) and obtained from the NCBI BioProject database (PRJNA375882). The reads were mapped to the mouse reference genome (mm10) with TopHat 2, using default parameters. Expression levels of transcript isoforms were calculated using Cufflinks. We determined the tissue specificity (one tissue of restricted expression) or selectivity (two‐to‐three tissues of restricted expression) of Aire‐sensitive genes, by using the specificity measurement (SPM) and the contribution measurement (CTM) methods (Pan et al, 2013), as described in Guyon et al (2020). For each considered gene, the SPM and CTM values were evaluated based on the level of gene expression in each tissue. A gene was considered tissue‐specific for a particular tissue if its SPM value in the tissue was > 0.9. A gene was considered tissue‐selective (2 or 3 tissue), if its SPM values were > 0.3 in those tissues and its CTM value for the corresponding tissue > 0.9. If the previous conditions were not met, the gene was left unassigned.
Epithelial cell level
To compare gene expression between TECs and epithelial cells from different mouse peripheral tissues, we collected public RNA‐seq datasets of epithelial cells isolated from skin (St‐Pierre et al, 2013) (GEO database: GSE44945); brain, caecum, large and small intestine, heart, kidney, lever, lung, lymph node, spleen (Krausgruber et al, 2020) (GSE134659); neural tube (ENCODE Project Consortium, 2012) (GSE78315); mammary glands (Pal et al, 2021) (GSE164307); olfactory system (Tan et al, 2020) (GSE146043); esophagus (Wiles et al, 2021) (GSE154129), and colon (Marincola Smith et al, 2021) (GSE100082). The reads were mapped to the mouse reference genome (mm10) with TopHat 2, using default parameters. Expression levels of transcript isoforms were calculated using Cufflinks.
Author contributions
Francine Padonou: Software; Formal analysis; Investigation; Visualization; Methodology; Writing ‐ original draft; Writing ‐ review & editing. Virginie Gonzalez: Investigation. Nathan Provin: Formal analysis; Investigation; Visualization. Sümeyye Yayilkan: Formal analysis; Investigation; Visualization. Nada Jmari: Investigation. Julia Maslovskaja: Investigation. Kai Kisand: Investigation. Pärt Peterson: Resources; Writing ‐ review & editing. Magali Irla: Conceptualization; Resources; Methodology; Writing ‐ review & editing. Matthieu Giraud: Conceptualization; Software; Formal analysis; Supervision; Funding acquisition; Visualization; Methodology; Writing ‐ original draft; Project administration; Writing ‐ review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
FP and MG designed the study and wrote the manuscript; FP and VG performed most of the experimental work; NP, SY, NJ, JM, and KK performed experiments. FP, NP, SY, and MG performed bioinformatics analyses; PP and MI provided key material, datasets and edited the manuscript.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
We thank Drs. D. Mathis and C. Benoist (Harvard Medical School) for Aire‐KO (B6) mice. This work was supported by the Agence Nationale de la Recherche (ANR) 2011‐CHEX‐001‐R12004KK to M.G., IHU‐Cesti funded by the «Investissements d’Avenir» ANR‐10‐IBHU‐005 as well as by Nantes Metropole and Region Pays de la Loire to M.G., and EJP‐Rare Disease JTC2019 program TARID project (EJPRD19‐208) funded by the ANR (ANR‐19‐RAR4‐0011‐5) to M.G., by Estonian Ministry of Social Affairs to P.P. The work was also supported by the University of Tartu Center of Translational Genomics (SP1GVARENG) and the Estonian Research Council grant PRG377 to P.P. and the Marie Curie Actions (Career Integration Grants, CIG_SIGnEPI4Tol_618541) to M.I. We would like to thank the members of the ‘Genomic’IC’ core facility head by Franck Letourneur at Cochin Institute, Paris, France, for RNA‐seq data production, as well as Ms. Maire Pihlap for her excellent technical assistance in preparing sequencing samples, Dr Mario Saare for his help in analyzing human mTEC data and Nicolas Richard for his help with the single‐cell analysis. F.P. was supported by the Labex IGO (project «Investissements d’Avenir» ANR‐ 11‐LABX‐0016‐01) and the RFI Bioregate grant (ThymIPS) from la Region Pays de la Loire to M.G. N.P. was supported by “la fondation d’entreprise ProGreffe”. S.Y. was supported by the ANR (ANR‐19‐RAR4‐0011‐5) to M.G. and la Region Pays de la Loire.
Disclosure statement and competing interests
The authors declare that they have no conflict of interest.
EMBO reports (2022) 23: e53576.
Data availability
RNA‐seq data for WT and Aire‐KO mTEChi, human mTEChi, and knockdown mouse mTECs can be accessed in GEO (https://www.ncbi.nlm.nih.gov/geo/) with accession codes GSE177062 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE177062), GSE176445 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE176445), and GSE177063 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE177063). H3K36me3 ChIP‐seq data are available with the accession code GSE176371 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE176371). We have provided the documented R code that we developed for ASE inclusion analysis as a Git repository available from GitHub (https://github.com/TeamGiraud/TEC_splicing_2021).
References
- Adli M, Bernstein BE (2011) Whole‐genome chromatin profiling from limited numbers of cells using nano‐ChIP‐seq. Nat Protoc 6: 1656–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SF, Carmo‐Fonseca M (2012) Design principles of interconnections between chromatin and pre‐mRNA splicing. Trends Biochem Sci 37: 248–253 [DOI] [PubMed] [Google Scholar]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C et al (2002) Projection of an immunological self shadow within the thymus by the Aire protein. Science 298: 1395–1401 [DOI] [PubMed] [Google Scholar]
- Aschenbrenner K, D'Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, Rolink A, Klein L (2007) Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol 8: 351–358 [DOI] [PubMed] [Google Scholar]
- Bartoletti‐Stella A, Gasparini L, Giacomini C, Corrado P, Terlizzi R, Giorgio E, Magini P, Seri M, Baruzzi A, Parchi P et al (2015) Messenger RNA processing is altered in autosomal dominant leukodystrophy. Hum Mol Genet 24: 2746–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehnke K, Mirancea N, Pavesio A, Fusenig NE, Boukamp P, Stark H‐J (2007) Effects of fibroblasts and microenvironment on epidermal regeneration and tissue function in long‐term skin equivalents. Eur J Cell Biol 86: 731–746 [DOI] [PubMed] [Google Scholar]
- Cowan JE, Parnell SM, Nakamura K, Caamano JH, Lane PJL, Jenkinson EJ, Jenkinson WE, Anderson G (2013) The thymic medulla is required for Foxp3+ regulatory but not conventional CD4+ thymocyte development. J Exp Med 210: 675–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danan‐Gotthold M, Guyon C, Giraud M, Levanon EY, Abramson J (2016) Extensive RNA editing and splicing increase immune self‐representation diversity in medullary thymic epithelial cells. Genome Biol 17: 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbinski J, Schulte A, Kyewski B, Klein L (2001) Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol 2: 1032–1039 [DOI] [PubMed] [Google Scholar]
- Dhalla F, Baran‐Gale J, Maio S, Chappell L, Holländer GA, Ponting CP (2020) Biologically indeterminate yet ordered promiscuous gene expression in single medullary thymic epithelial cells. EMBO J 39: e101828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium , Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET et al (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäbler J, Arnold J, Kyewski B (2007) Promiscuous gene expression and the developmental dynamics of medullary thymic epithelial cells. Eur J Immunol 37: 3363–3372 [DOI] [PubMed] [Google Scholar]
- Giraud M, Jmari N, Du L, Carallis F, Nieland TJF, Perez‐Campo FM, Bensaude O, Root DE, Hacohen N, Mathis D et al (2014) An RNAi screen for Aire cofactors reveals a role for Hnrnpl in polymerase release and Aire‐activated ectopic transcription. Proc Natl Acad Sci USA 111: 1491–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC, Sprent J, de St F, Groth B, Vinuesa CG (2005) Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 435: 590–597 [DOI] [PubMed] [Google Scholar]
- Guyon C, Jmari N, Padonou F, Li Y‐C, Ucar O, Fujikado N, Coulpier F, Blanchet C, Root DE, Giraud M (2020) Aire‐dependent genes undergo Clp1‐mediated 3'UTR shortening associated with higher transcript stability in the thymus. Elife 9: e52985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki Y, Fujita H, Kobayashi T, Choi Y, Scott HS, Matsumoto M, Minato N (2007) Medullary thymic epithelial cells expressing Aire represent a unique lineage derived from cells expressing claudin. Nat Immunol 8: 304–311 [DOI] [PubMed] [Google Scholar]
- Handel AE, Shikama‐Dorn N, Zhanybekova S, Maio S, Graedel AN, Zuklys S, Ponting CP, Holländer GA (2018) Comprehensively profiling the chromatin architecture of tissue restricted antigen expression in thymic epithelial cells over development. Front Immunol 9: 2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberg B, Swiniarski S, Becke S, Illenberger S (2010) A conserved peptide motif in Raver2 mediates its interaction with the polypyrimidine tract‐binding protein. Exp Cell Res 316: 966–979 [DOI] [PubMed] [Google Scholar]
- Hu Q, Greene CS, Heller EA (2020) Specific histone modifications associate with alternative exon selection during mammalian development. Nucleic Acids Res 48: 4709–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann D, Gozani O (2019) Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol 26: 880–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane P, Ceredig R, Seoighe C (2015) Promiscuous mRNA splicing under the control of AIRE in medullary thymic epithelial cells. Bioinformatics 31: 986–990 [DOI] [PubMed] [Google Scholar]
- Kekäläinen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pöntynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP (2007) A defect of regulatory T cells in patients with autoimmune polyendocrinopathy‐candidiasis‐ectodermal dystrophy. J Immunol 178: 1208–1215 [DOI] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, Hogquist KA (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol 14: 377–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinhenz B, Fabienke M, Swiniarski S, Wittenmayer N, Kirsch J, Jockusch BM, Arnold HH, Illenberger S (2005) Raver2, a new member of the hnRNP family. FEBS Lett 579: 4254–4258 [DOI] [PubMed] [Google Scholar]
- Kolasinska‐Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J (2009) Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 41: 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Muñoz MJ (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14: 153–165 [DOI] [PubMed] [Google Scholar]
- Krausgruber T, Fortelny N, Fife‐Gernedl V, Senekowitsch M, Schuster LC, Lercher A, Nemc A, Schmidl C, Rendeiro AF, Bergthaler A et al (2020) Structural cells are key regulators of organ‐specific immune responses. Nature 583: 296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Durbin R1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Qing T, Zhu J, Wen Z, Yu Y, Fukumura R, Zheng Y, Gondo Y, Shi L (2017) A comprehensive mouse transcriptomic BodyMap across 17 tissues by RNA‐seq. Sci Rep 7: 4200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira‐Smith OM, Misteli T (2010) Regulation of alternative splicing by histone modifications. Science 327: 996–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T (2011) Epigenetics in alternative pre‐mRNA splicing. Cell 144: 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP et al (2013) Aire‐dependent thymic development of tumor‐associated regulatory T cells. Science 339: 1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA (2016) Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 44: 1102–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marincola Smith P, Choksi YA, Markham NO, Hanna DN, Zi J, Weaver CJ, Hamaamen JA, Lewis KB, Yang J, Liu QI et al (2021) Colon epithelial cell TGFβ signaling modulates the expression of tight junction proteins and barrier function in mice. Am J Physiol Gastrointest Liver Physiol 320: G936–G957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE et al (1997) Positional cloning of the APECED gene. Nat Genet 17: 393–398 [DOI] [PubMed] [Google Scholar]
- Pal B, Chen Y, Milevskiy MJG, Vaillant F, Prokopuk L, Dawson CA, Capaldo BD, Song X, Jackling F, Timpson P et al (2021) Single cell transcriptome atlas of mouse mammary epithelial cells across development. Breast Cancer Res 23: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J‐B, Hu S‐C, Shi D, Cai M‐C, Li Y‐B, Zou Q, Ji Z‐L (2013) PaGenBase: a pattern gene database for the global and dynamic understanding of gene function. PLoS One 8: e80747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson P, Pitkänen J, Sillanpää N, Krohn K (2004) Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED): a model disease to study molecular aspects of endocrine autoimmunity. Clin Exp Immunol 135: 348–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Schmidt K, Egle S, Stark H‐J, Boukamp P, Kyewski B (2013) An organotypic coculture model supporting proliferation and differentiation of medullary thymic epithelial cells and promiscuous gene expression. J Immunol 190: 1085–1093 [DOI] [PubMed] [Google Scholar]
- Pomié C, Vicente R, Vuddamalay Y, Lundgren BA, van der Hoek M, Enault G, Kagan J, Fazilleau N, Scott HS, Romagnoli P et al (2011) Autoimmune regulator (AIRE)‐deficient CD8+CD28low regulatory T lymphocytes fail to control experimental colitis. Proc Natl Acad Sci USA 108: 12437–12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie W, Granjeaud S, Puthier D, Gautheret D (2008) Entropy measures quantify global splicing disorders in cancer. PLoS Comput Biol 4: e1000011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KR, Lawson CA, Lorenzi AR, Arkwright PD, Isaacs JD, Lilic D (2005) CD4+CD25+ T‐regulatory cells are decreased in patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J Allergy Clin Immunol 116: 1158–1159 [DOI] [PubMed] [Google Scholar]
- Sansom SN, Shikama‐Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, Hollander GA (2014) Population and single‐cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self‐antigen expression in thymic epithelia. Genome Res 24: 1918–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z‐X, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. Proc Natl Acad Sci USA 111: E5593–E5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, Liu XS (2009) CEAS: cis‐regulatory element annotation system. Bioinformatics 25: 2605–2606 [DOI] [PubMed] [Google Scholar]
- St‐Pierre C, Brochu S, Vanegas JR, Dumont‐Lagacé M, Lemieux S, Perreault C (2013) Transcriptome sequencing of neonatal thymic epithelial cells. Sci Rep 3: 1860–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St‐Pierre C, Trofimov A, Brochu S, Lemieux S, Perreault C (2015) Differential features of AIRE‐induced and AIRE‐independent promiscuous gene expression in thymic epithelial cells. J Immunol 195: 498–506 [DOI] [PubMed] [Google Scholar]
- Tan K, Jones SH, Lake BB, Dumdie JN, Shum EY, Zhang L, Chen S, Sohni A, Pandya S, Gallo RL et al (2020) The role of the NMD factor UPF3B in olfactory sensory neurons. Elife 9: e57525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nat Biotechnol 31: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojnic E, Simon B, Strahl BD, Sattler M, Cramer P (2006) Structure and carboxyl‐terminal domain (CTD) binding of the Set2 SRI domain that couples histone H3 Lys36 methylation to transcription. J Biol Chem 281: 13–15 [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Garcia‐Blanco MA (2002) RNAi‐mediated PTB depletion leads to enhanced exon definition. Mol Cell 10: 943–949 [DOI] [PubMed] [Google Scholar]
- Wiles KN, Alioto CM, Hodge NB, Clevenger MH, Tsikretsis LE, Lin FTJ, Tétreault M‐P (2021) IκB Kinase‐β regulates neutrophil recruitment through activation of STAT3 signaling in the esophagus. Cell Mol Gastroenterol Hepatol 12: 1743–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137–R139 [DOI] [PMC free article] [PubMed] [Google Scholar]