

Abstract
The integration of external control data, with patient-level information, in clinical trials has the potential to accelerate the development of new treatments in neuro-oncology, by contextualizing single arm studies and improving decision making (e.g. early stopping decisions) in randomized trials. Based upon a series of presentations at the 2020 Clinical Trials Think Tank hosted by the Society of Neuro-Oncology, we provide an overview on the use of external control data, representative of the standard of care, in the design and analysis of clinical trials. High quality patient-level records, rigorous methods and validation analyses are necessary to effectively leverage external data. We review study designs, risks and potential distortions in leveraging external data, data sources, evaluations of designs and methods based on data collections from completed trials and real world data, data sharing models, and ongoing work and applications in glioblastoma.
Introduction
Drug development is associated with inefficiency, high failure rate and long timelines with poor success rates in oncology where less than 10% of drug candidates are ultimately approved by the Food and Drug Administration (FDA).1,2 As new, unproven therapies emerge at an accelerated pace across oncology, there has been an increasing interest in novel approaches to clinical trial design that improve efficiency.3,4
Within neuro-oncology, the use of trial designs with potential for increased efficiency are of interest, particularly in the study of glioblastoma (GBM), a disease setting with a critical need for better therapies as it continues to be associated with a dismal prognosis.5 There are several distinctive challenges in drug development for GBM including the inability to completely resect tumors, the blood-brain barrier, tumor heterogeneity, challenges with imaging to monitor disease course, and the unique immune environment.6,7 With few treatment advances over the last two decades, the clinical trial landscape in GBM has been characterized by long development times, low patient participation, problematic surrogate outcomes, and poor go/no-go decision making.8,9 Poor early phase decision making has been repeatedly highlighted as a major problem in the development of therapeutics10 and continues to stimulate interest in novel clinical trial designs.
Randomized controlled trials (RCT) are the gold standard for clinical experimentation and evaluation of therapies. RCTs control for systematic bias from known and unknown confounders by randomizing patients to receive either an experimental therapy or standard of care, which allows for the evaluation of treatment effects. RCTs, however can be difficult to conduct in some neuro-oncology settings. A relatively small percentage of patients participate in clinical trials,11 and RCTs can suffer from slow accrual due to patient reluctance to enroll on studies with a control arm, which is a pronounced problem in settings with ineffective standard of care treatments (e.g. recurrent GBM).12–14 Precision medicine further complicates this picture by focusing trials on biomarker-defined subgroups of patients who may benefit from targeted therapies.15 These subgroups are often comprised of a small proportion of patients with a given tumor type, resulting in significant challenges to conducting RCTs with adequate sample sizes to detect treatment effects.15,16
Recently, the design and implementation of clinical trials that leverage external datasets, with patient level information on pre-treatment clinical profiles and outcomes to support testing of experimental therapies and study decision making, has attracted interest in neuro-oncology.17,18 A recent phase 2b recurrent GBM trial used a pre-specified eligibility-matched external control arm (ECA, a dataset which includes individual pre-treatment profiles and outcomes), developed with data from GBM patients from major neurosurgery centers, as a comparator arm to evaluate an experimental therapy (MDNA55). After implementation of this trial design, investigators reported evidence of improved survival in patients receiving MDNA55 relative to the matched ECA cohort.19 Several neuro-oncology trials under development are actively exploring similar approaches to leverage external data in the design and analysis of clinical studies.
The Society of Neuro-Oncology hosted the 2020 Clinical Trials Think Tank on November 6, 2020 with a virtual session dedicated to trial designs leveraging external data. Experts in the field of neuro-oncology were paired with experts in data science and biostatistics, and representatives from industry, patient advocacy, and the FDA. The interdisciplinary session focused on challenges in drug development, challenges to data sharing and access, regulatory considerations on novel trial designs, and emerging methodological approaches to leveraging external data. While there was broad participation, most participants were from the United States and provided a US-centric perspective on the topic. The discussion from the Think Tank serves as a framework for this review, which focuses on the use of external data to design, conduct, and analyze clinical trials, with an emphasis on possible applications in neuro-oncology. We review trial designs, methodologies, approaches for the evaluation of designs and external datasets, regulatory considerations, and current barriers to data sharing and access.
Search Strategy and Selection Criteria
We searched the literature using PubMed with the search terms “external control arms”, “synthetic control arms”, “neuro-oncology trial design”, “glioblastoma trial design”, from January 2000 until May 2021. Articles were also identified through searches of the authors’ own files. Only papers published in English were reviewed. The final reference list was generated based upon relevance to the scope of this review.
Early Phase Trial Designs
Early-phase trials are typically designed to obtain preliminary estimates of treatment efficacy and toxicity that will inform the decision to pursue a definitive phase 3 trial or stop drug development. Often in neuro-oncology, these early phase studies are single arm trials (SATs) that test the superiority of the experimental therapeutic compared to an established benchmark parameter for the current standard of care (e.g. median OS or other point estimates).20 Importantly, there can be significant differences between populations or standards to assess outcomes across trials,21 which can lead to inappropriate comparisons and inadequate evaluations of the experimental therapy. An additional major challenge with SATs is the choice of the primary efficacy endpoint. Response rate is difficult to interpret in GBM,22 and single arm studies are suboptimal for reliable inference on improvements of time-to-event endpoints such as survival. Based on these known limitations, single arm designs have been posited as a possible reason for poor go/no-go decision making and recent failed phase 3 trials in GBM.23,24
The risk of biased conclusions of SATs has been examined extensively and frameworks have been developed to help guide the choice between RCT vs. single-arm designs for GBM.10,25 Despite well documented limitations, SATs remain the most common trial design in early phase trials in GBM.25 Alternative trial designs have been proposed to overcome limitations of SATs and to improve the evaluation of therapeutic candidates in the early phase of a drug’s development, including the incorporation of randomization, seamless phase 2/3 study designs26 and Bayesian outcome-adaptive trials.27–29
Overview of Trial Designs that Leverage External Data
Trial designs that leverage external data can generate valuable inferences in settings where SATs are suboptimal and RCTs are infeasible.30 External data can play a role in supporting key decisions of the drug development process, including regulatory approvals and go/no-go decision making in early-phase trials. The use of external patient-level datasets has the potential to improve the accuracy of trial findings and inform decision making (e.g., determining the sample size of a subsequent confirmatory phase 3 trial, or selecting the phase 3 patient population). External data can also be incorporated into RCTs,31 for example within interim analyses,18 though these designs remain largely unexplored.
Externally augmented clinical trial (EACT) designs refer to the broad class of designs that leverage external data for decision making during a study or in the final analysis. EACTs rely on access to well-curated patient-level data for the standard of care treatment, from one or more relevant data sources, to allow for adjustments of differences of pre-treatment covariates between the enrolled patients and the external data, and to derive treatment effect estimates. Given the need for statistical adjustments, the external dataset ideally includes a comprehensive set of potential confounders.32 In considering such designs for gliomas, pre-treatment covariates have been thoroughly studied for adult primary brain tumors.33,34
An example of an EACT design consists of a single arm study combined with an ECA (i.e., an external dataset with patient-level outcomes and pre-treatment profiles), which is used as a comparator to evaluate the experimental treatment. This design, indicated as ECA-SAT (Figure 1A), is a type of EACT that infers the treatment effect by using adjustment methods, to account for differences in pre-treatment patient profiles between the external control group and the experimental arm.35 In this design the ECA is used to contextualize the outcome data from a single arm study. In contrast to the use of a benchmark estimates (e.g. median survival) of the standard of care efficacy in SATs, data analyses and treatment effect estimates are based on patient-level data from an external dataset.
Figure 1:
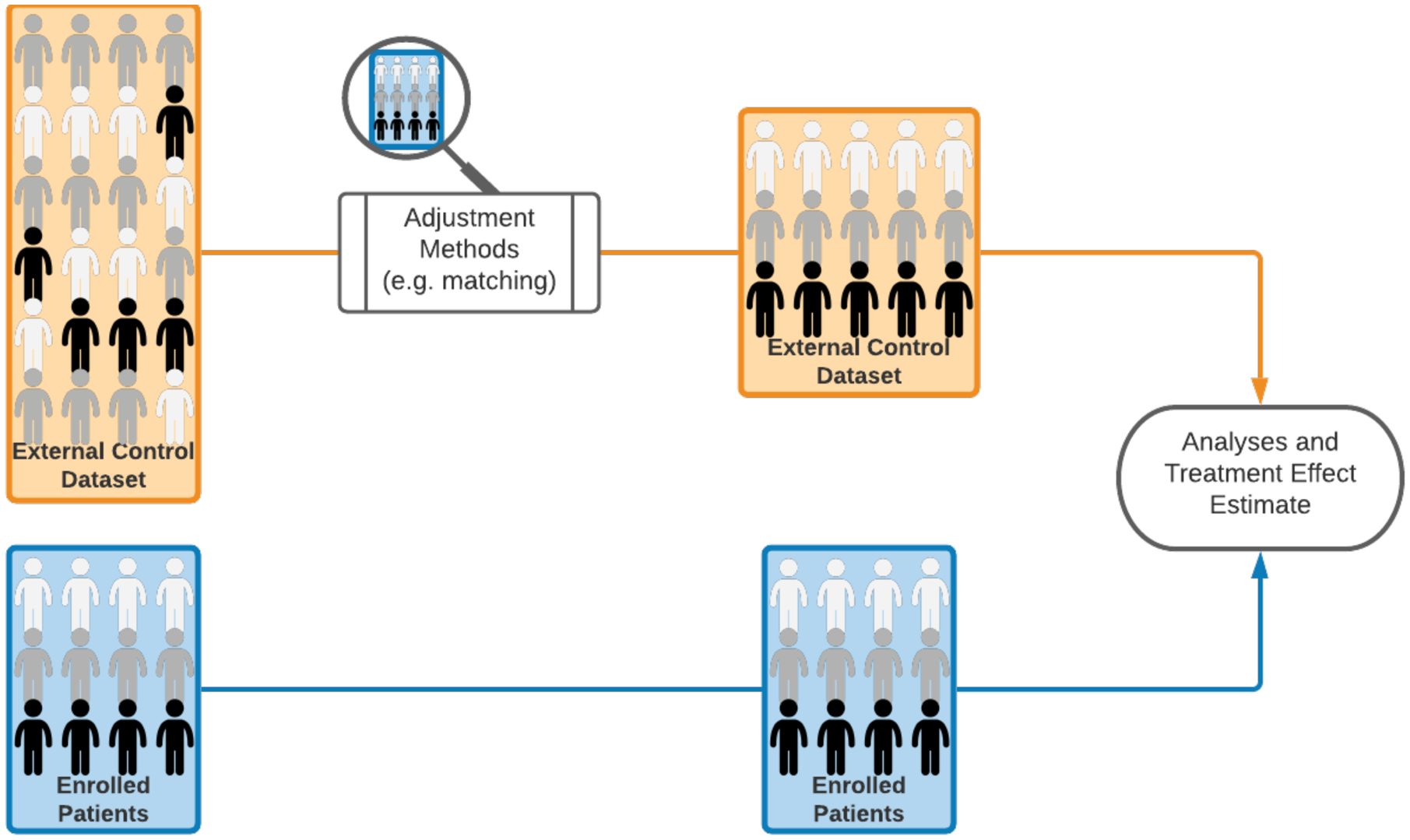
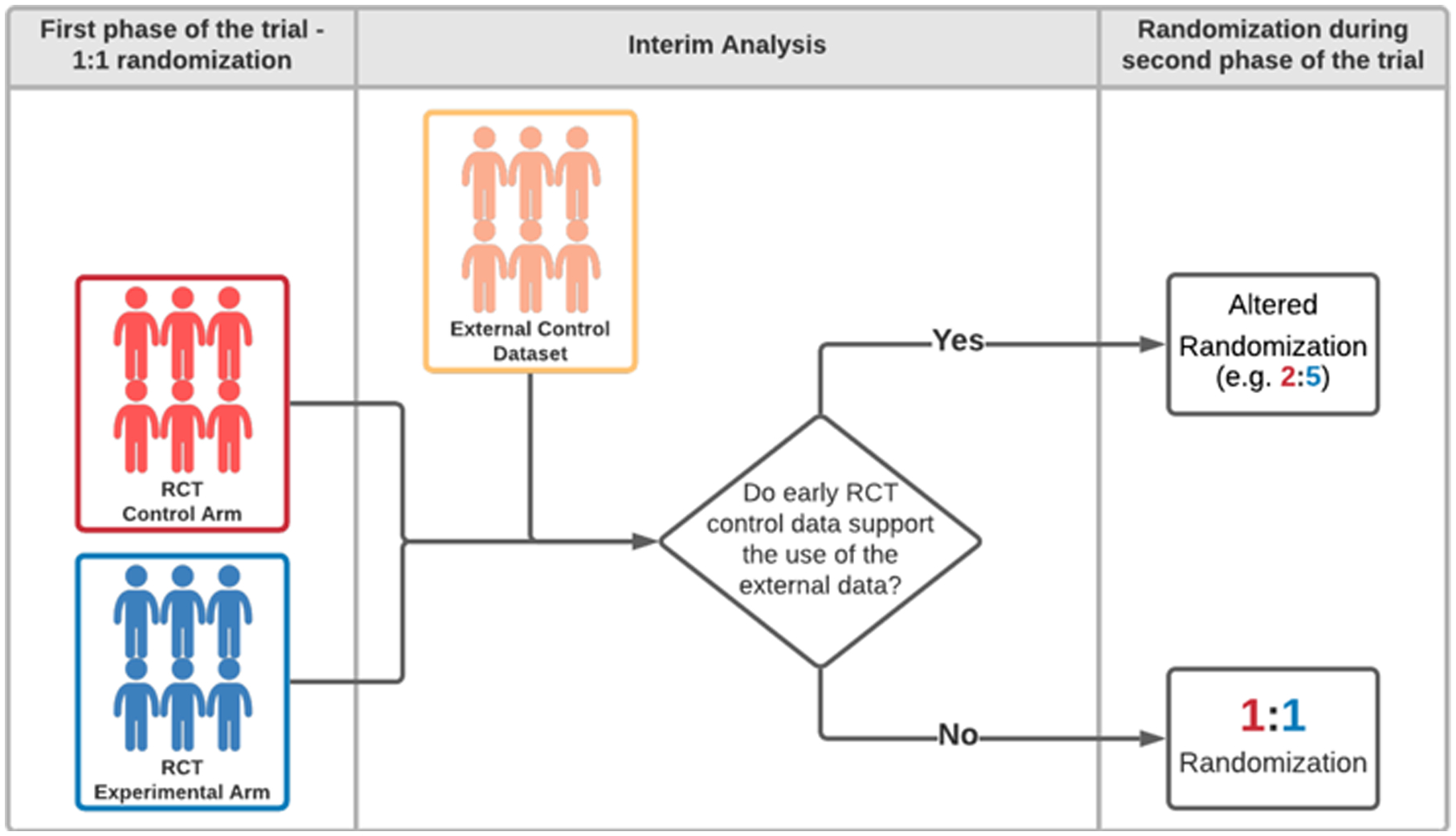
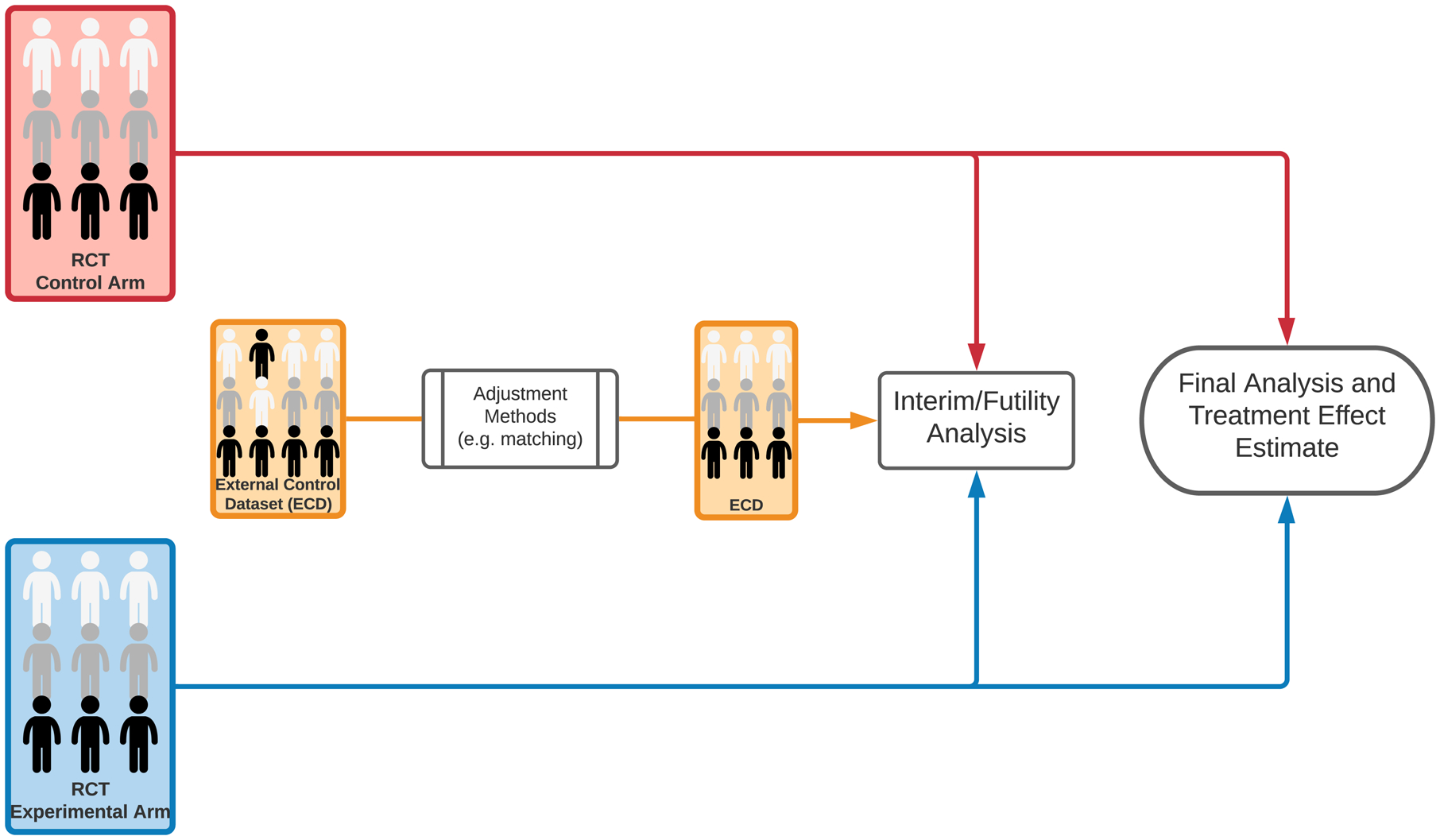
Schematic representation of clinical trial designs. (A) A clinical study with patients enrollment to a single experimental arm and an external control arm (ECA-SAT). Adjustment methods are used to compare the experimental arm and the external control arm. (B) An example of a two-staged hybrid randomized trial design. (C) An example of a randomized trial design that utilizes external data for interim futility analyses. The external dataset is used to support the decision to continue or discontinue the clinical study. If the trial is not discontinued for futility, the final analysis does not utilize external data.
Hybrid randomized trial designs constitute another type of EACT. These designs, with adequate external data and statistical plan36, have the potential of reducing the overall sample size while maintaining the benefits of randomization. We describe an example of a two-stage hybrid design (Figure 1B). The study has an initial 1:1 randomization to the experimental arm and the internal control arm. If the interim analysis does not identify differences between the adjusted primary outcome distributions in the internal (randomized) control group and the external control group, then different randomization ratios (e.g., 2:5) can be used in the second stage of the study. In contrast, if there is evidence of inconsistencies between the external and internal control groups (e.g., unmeasured confounders or different measurement standards of outcomes and prognostic variables), the trial can continue with 1:1 randomization.
In the outlined example, the potential increase of the randomization probability for the experimental arm can be attractive and may accelerate trial accrual. Indeed, brain tumor patients, with an inadequate standard of care, may be more likely to enroll onto a trial if the probability of receiving the experimental therapy is higher.25
EACT Designs
Along with high-quality and complete data, a statistically rigorous study design is the most important element of an EACT. As with any clinical trial, the design, including the sample size, a detailed plan for interim decisions, and statistical methods for data analyses, should be prespecified. Additionally, a plan for how missing data in the trial and external data sources will be handled is important. Potential distortion mechanisms that can bias the treatment effect estimates and undermine the scientific validity of EACT findings have been carefully examined and include unmeasured or misclassified confounders and data quality issues such as the use of different standards to capture or measure outcomes.37–39
The risks of introducing bias (Table 1) and of compromising the control of false positive and false negative results by leveraging external patient-level data can differ substantially across candidate EACT designs, which span from single-arm studies (ECA-SAT design, Figure 1A) to hybrid randomized studies (Figure 1B). Quantitative analyses of these and other risks (e.g., exposure of patients to inferior treatments) are necessary prior to trial initiation. The decision to leverage external data should account for several factors in addition to the study population and the available patient-level datasets, including:
the stage of the drug development process (e.g., early phase 2 vs. confirmatory trials);
the specific decisions (e.g., early stopping of a phase 2 study for futility40 or sample size re-estimation during the study41) that will be supported by external data;
resources (including maximum sample size); and
potential trial designs and statistical methodologies for data analyses.
Table 1:
Potential causes of bias in clinical trials with an external control group
| Description | Example | Methods to avoid or reduce the bias | |
|---|---|---|---|
| Measured confounders | The distributions of pre-treatment patient characteristics that correlates with the outcomes in the trial population and in the external control group are different. | The external control group has on average a higher Karnofsky performance status or age than the trial population. | Matching. Inverse probability weighting. Marginal structural models. |
| Unmeasured confounders | The distributions of unmeasured pre-treatment patient characteristics that correlates with the outcomes in the trial population and in the external control group are different. | Supportive care (not captured in the datasets) differs between patients in the clinical trial and in the external control group. | Validation analyses can indicate the risk of bias before the onset of the trial. |
| Differences in defining prognostic variables / outcomes | The definition of clinical measurements may vary between datasets leading to differences in the definitions of outcomes or prognostic variables between the clinical trial and the external control group. | Measurement of tumor response with different response criteria or at different intervals in external control arms | Data dictionaries and validation analyses can reveal these discrepancies before the onset of the trial. |
| Immortal time bias | In the external dataset the time-to-event outcome cannot occur during a time window, because of the study design or other causes. | In GBM, different real world datasets capture patient survival from diagnosis or from a different time points | Explicit and detailed definitions of the time-to-event outcomes for the trial and the external dataset can reveal the risk of bias. |
Candidate EACT designs and statistical methodologies for data analysis can present markedly different trade-offs between potential efficiencies (e.g., discontinuing early randomized studies of ineffective treatments by leveraging external data), and risks of poor operating characteristics (e.g., bias, poor control of false positive results). In other words, the value of integrating external data is context-specific, and it is strictly dependent on the specific EACT design and methodology selected for data analyses and decision making.
We describe three examples of EACTs with markedly different risks of poor operating characteristics. The purpose of these examples is to illustrate how external information can be leveraged for making different decisions during or at completion of a trial.
1). Single arm trial with an external control group (ECA-SAT).
We consider either binary primary outcomes (e.g., tumor response) or time to event outcomes with censoring (e.g., overall survival). The ECA-SAT design uses procedures developed for observational studies,42 such as matching, propensity score methods,43,44 or inverse probability weighting,45 which are applicable to the comparison of (i) data from a SAT (experimental treatment) and (ii) external patient-level data, representative of the standard of care therapy (external control, Figure 1A). These procedures have been developed to estimate treatment effects in non-randomized studies and have generated an extensive number of contributions in the statistical literature.46 They compare outcome data Y under the experimental and control treatment with adjustments that account for confounders X.
(A). Evaluation of the ECA-SAT design using a collection of datasets.
The literature on adjustment methods applicable to EACT designs (e.g., matching)43,44 is largely anchored to assumptions that are difficult or impossible to demonstrate,42 including the absence of unmeasured confounders.47 In the context of ECA-SAT designs, where these assumptions might be violated, the investigator can attempt to evaluate the risk of bias and other statistical properties of treatment effects estimates (e.g., the coverage of confidence intervals) computed using adjustment methods. Patient-level data from a library of recently completed RCTs in a specific clinical setting, e.g. newly diagnosed GBM patients, facilitate the comparison of ECA-SAT, RCT, and SAT designs. For example, a treatment effect estimate computed using only data from a previously completed RCT can be compared to a second treatment effect estimate, computed using only the experimental arm of the same RCT and external data (Figure 2).48 The comparison can be repeated considering different RCTs, adjustment methods, and external datasets. These comparisons allow one to describe the consistency between the RCT results and hypothetical results obtained from a smaller ECA-SAT (i.e., the experimental arm of the RCT, or part of it) leveraging external data; similar evaluation frameworks have been discussed recently.49,50
Figure 2:
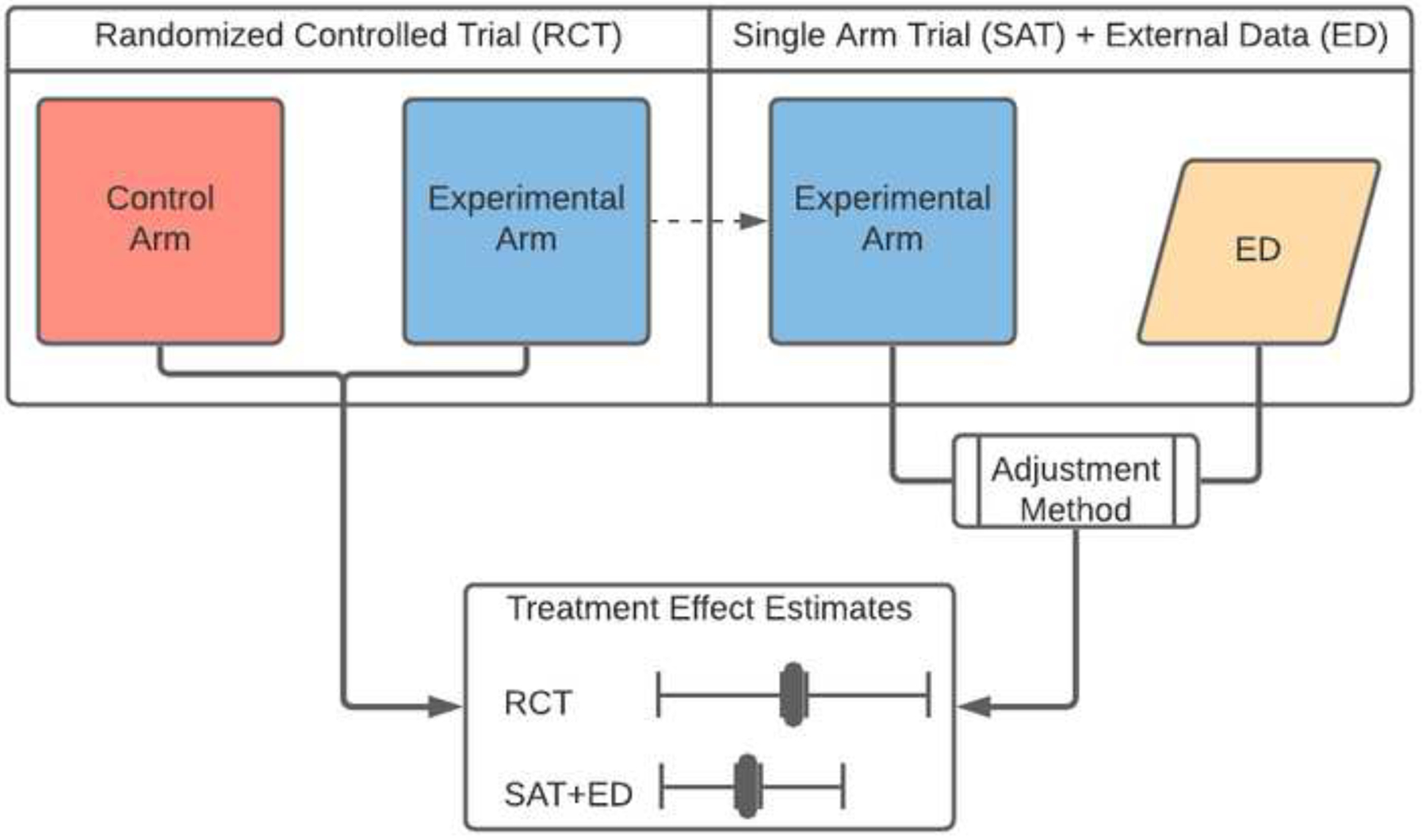
Schematic representation of a validation schema. A treatment effect estimate computed using only data from a previously completed RCT is compared to a second treatment effect estimate, computed using only the experimental arm of the same RCT and external control data.
(B). Leave-one-out algorithm.
An alternative evaluation approach that requires a collection of recently completed RCTs with the same control treatment has been proposed recently.17 The algorithm has been used to compare the application of candidate causal inference methods in ECA-SATs. This approach has been applied to a collection of newly diagnosed GBM studies, and it requires only pre-treatment profiles and outcomes from patients treated with the standard of care therapy, radiation and temozolomide (RT/TMZ). This is relevant because data access barriers can be substantially different for the control and the experimental arms. The algorithm17 iterates the following three operations for each RCT in the data collection:
Experimental Treatment
-
i
it randomly selects n (the sample size of a hypothetical ECA-SAT trial) patients (without replacement) from the RT/TMZ arm (control) of the trial, and uses patient pre-treatment profiles X and outcomes Y of these patients to define a fictitious single-arm study (i.e., the treatment group);
Control
-
ii
the data on patients treated with RT/TMZ in the remaining studies are used as external data (these datasets are combined into a single data matrix, the control group); and
Analysis
-
iii
a treatment effect estimate is computed by comparing the (fictitious) single-arm ECT study (step i) and the external data (step ii), using a candidate adjustment method, which is also used to test the null hypothesis H0 that the treatment does not improve the primary outcome.
For each study in the data collection, these steps (i-iii), which are similar to cross-validation, can be repeated to evaluate bias, variability of the treatment effect estimate, and the risk of false positive results. By construction, the treatment effect in this fictitious comparison (steps i-iii) is null, as patients receiving RT/TMZ are being compared to other patients receiving RT/TMZ from a different study (RT/TMZ vs. RT/TMZ). This facilitates interpretability and produces bias summaries for the ECA-SAT statistical plan. A recent analysis using this leave-one-out algorithm approach in newly diagnosed GBM illustrated high false positive error rates of standard SAT designs (above the alpha-level),10 which can be considerably reduced (up to 30% reduction) by using external control data from previously completed clinical trials in ECA-SAT designs.17
The first approach (A) attempts to replicate the results of a completed RCT, while the leave-one-out algorithm approach (B) is based on subsampling a control arm. Both approaches are valuable strategies that can detect potential distortion mechanisms (e.g., unmeasured confounders or inconsistent definitions of primary outcomes), which undermine the scientific validity of ECA-SAT designs. These approaches require patient-level data from several RCTs, with adequate sample sizes, to produce reliable analyses of the risk of bias and false positive results in future ECA-SATs. It is also important to not overinterpret positive findings from retrospective analyses using either approach, as relevant changes of the available treatments, technologies, or other factors can rapidly make the entire data collection obsolete and inadequate.51
2). Hybrid randomized trial designs with internal and external control groups.
Hybrid randomized trial designs combine external and randomized control data to estimate potential treatment effects.52 Figure 1B represents a two-stage hybrid design. In the first stage, n(E,1) and n(C,1) patients are randomized to the experimental arm and the (internal) control arm, respectively. The interim analysis is used for futility early stopping and to determine sample sizes n(E,2) and n(C,2), for the experimental and control arm in the second stage. These decisions are based on (i) a similarity measure comparing estimates of the conditional outcome distributions Pr(Y|X) of the external and internal control groups, and (ii) preliminary treatment effect estimates. The proportion of patients randomized to the internal control arm during the second phase can be reduced or increased based on the pre-specified interim analysis, which involve summaries in support or against the integration of external data to estimate the effects of the experimental treatment.
Based on recent results from a phase 2 study of an experimental therapeutic MDNA55,19 investigators are currently planning an open-label phase 3 registration study with implementation of a hybrid randomized design in recurrent IDH-wildtype GBM patients. The study team is considering a 3:1 randomization ratio for allocation to the experimental and control arms, with a final comparison of overall survival between patients receiving the experimental agent (MDNA55) and the control groups (external and internal control arms).53
3). Randomized controlled trials that incorporate external data to support futility analyses.
External data can be incorporated into RCTs for other purposes31 such as leveraging external data for interim decisions.18 In the design illustrated in Figure 1C, interim analyses utilize predictions based on early data from the RCT combined with external data. These predictions express the probability that the trial will generate significant evidence of positive treatment effects. The trial is discontinued by design if the predictive probability becomes smaller than a fixed threshold. The final analysis, after completing the enrollment and follow-up phases, does not utilize external data. Indeed, the primary result of the trial is positive, indicating evidence of improved outcomes with the experimental treatment, if a standard p-value, computed using only the RCT data (excluding external information) has a value below the targeted control of false positive results α.
In ideal settings, without unmeasured confounders and other distortion mechanisms, leveraging external data for interim futility analyses can (i) reduce the expected sample size of the RCT when the experimental treatment is ineffective, and (ii) reduce the early stopping probability when the experimental therapy is superior, thus increasing the power.18 Additionally, the outlined design maintains a rigorous control of the RCT type I error probability, even in presence of unmeasured confounders, because the external data are excluded from the final data analyses. The efficiency gains and risks associated with the described integration of external data into interim decisions have been quantified for newly diagnosed GBM trials, with evaluation analyses that built upon a collection of datasets from completed RCTs, the leave-one-out algorithm outlined above and other similar procedures.18
As EACTs require a number of context-specific considerations, from relevant aspects of the external datasets to the feasibility of alternative designs, a discussion with regulatory agencies in early stages of trial planning is strongly recommended.
External Data Sources
The use of external controls to evaluate new treatments is dependent on the availability of high-quality external data. Selecting appropriate datasets is critical and checklists have been developed to provide guidance on data quality.54 Data considerations for external controls include appropriate capture of patient-level data,55 consistent definition of covariates and endpoints, and adequate temporality of the data, as small temporal lags can significantly affect the trial analysis.49 Investigators should consider potential biases that occur if the endpoints definitions are inconsistent across studies. For example, survival can be measured from the date of diagnosis, the date of randomization, or the initial date of adjuvant treatment. In other words, the definition of the “time zero” should be explicit and consistent, during the trial and in the external datasets.56 Missing data is another important consideration in analyses with external data.57,58 While there are methods to address missing data (e.g., multiple imputation and likelihood-based methods), their use within EACT designs has not been well studied.
Statistical methods should be employed to adjust for differences, but in general, the population of the external control and the trial population should be similar to reduce the risk of bias. Potential unmeasured confounders, inconsistencies in definitions, and differential measurement standards of covariates and outcomes across datasets need to be scrutinized using data dictionaries and study protocols. Contemporaneous controls are ideal, but historical controls with patient-level data may be helpful in appropriate contexts. For example, disease settings without a recent change in the standard of care (e.g. GBM) or long-track record of time-stable outcomes may have more flexibility in the temporality of data, but this should be weighed against possibility of unmeasured aspects of care such as advances in imaging, radiation therapy, surgical techniques, and supportive care that may change over time.59 Additionally, in order to support a marketing application, the data should be traceable (i.e. an audit trail should be available to document the data management processes).54
The two most relevant sources of external data are previously completed clinical trials and non-trial real world data (RWD) derived from clinical practice, each of which have strengths and weaknesses. The use of data from previously completed clinical trials can be advantageous, given that the data are typically collected in a rigorous trial environment with vetting procedures. Clinical trials are often conducted in specialized institutions and enroll clearly defined segments of their patient populations. Patients previously enrolled on RCTs and treated with the standard of care may be more likely than RWD cohorts to contain pre-treatment profiles similar to patients that will be enrolled in future trials. The use of detailed data collection forms, intensive monitoring, and specialized personnel facilitates adherence to clear protocols that produce standardized data.15 In GBM in recent years, there have been several negative phase 3 randomized trials with hundreds of patients receiving current standard of care therapy (RT/TMZ) in studies conducted by cooperative groups and industry.60–63 While external data from previously completed clinical trials are more likely to be complete and accurate, data access can be challenging due to impediments to data sharing64 and contemporary trial data may not be made available by trial sponsors.
RWD represents a distinct data source derived from registries, claims and billing data, personal devices or applications, or electronic health records (EHR). As RWD is generally not collected for research purposes, there can be concerns about data organization, data quality, confounding, selection mechanisms, and ultimately bias.65–67 Advances in the quality of EHR data have created opportunities, however, with newer datasets that can be well curated and linked with molecular or radiologic data with high fidelity. Efforts to harmonize RWD from disparate data sources and novel methods to incorporate such data into clinical studies provide an avenue to inform trial designs68 and regulatory decision making,69 but further work is required to validate these approaches.
Although differences between RWD and data from clinical studies have been reported,70 methodological work on the use of joint models and analyses, to compensate for the scarcity of trial data and the potential distortions of RWD (e.g. measurement errors or unknown selection mechanisms), is currently in its early stages.
Existing methodological work has primarily focused on overall survival,17,48 which is more likely to be adequately captured in external datasets relative to other outcomes. Radiologic endpoints such as progression-free survival require caution because of the risk of inconsistent assessments across datasets. In RWD data sources, radiologic outcomes may not be determined by formal response assessment criteria, and central radiologic review would likely not be routinely implemented. Although recently completed brain tumor clinical trials often use consensus guidelines produced by the Response Assessment in Neuro-Oncology working group for response assessment,71,72 these criteria include subjective components73 and datasets of previously completed trials can still include misclassification errors. Quality of life, neurologic function, and neurocognitive outcomes are increasingly incorporated into clinical trials.72,74 These non-survival outcomes can provide meaningful measures of the clinical benefit of a therapy, and they can serve as valuable endpoints in neuro-oncology trials.75 Nonetheless, missing measurements of these outcomes are common across datasets from completed trials and RWD. Exploration of the use of external data, to analyze radiographic outcomes, patient centered outcomes, and safety outcomes in neuro-oncology trials remains limited.
Examples of External Control Groups Beyond Neuro-Oncology
Carrigan et al. leveraged a curated RWD dataset of 48,856 patients (EHR from the Flatiron database) to re-analyze 11 completed trials in advanced non-small cell lung cancer.48 In this study, the external control arms were defined with matching methods. The external control arms were able to recapitulate the treatment effect estimates (hazard ratios) for 10 of the 11 RCTs. The study suggests the potential utility of RWD as external controls. This is likely due, at least in part, to the large number of patients in the external data, with large subsets of patient records that met the RCTs inclusion and exclusion criteria. Of note, the EHR-derived external control arms did not re-capitulate the results for one of the 11 RCTs. On inspection, the authors felt this discordance was due to a biomarker-subgroup population not sufficiently represented in the EHR dataset. These findings underscore the need to account for biomarkers and well represented subpopulations in the external control groups.
Another recent example supports the utility of external data to contextualize SATs.76 In a FDA-led retrospective analysis, the outcomes (invasive disease-free survival) from a single arm study77 of adjuvant paclitaxel and trastuzumab in HER2-positive early breast cancer patients were analyzed using an external control group derived from clinical trials (control therapy: anthracycline/cyclophosphamide/taxane/trastuzumab or taxane/carboplatin/trastuzumab).78 The de-escalated regimen that combines adjuvant paclitaxel and trastuzumab had been adopted in clinical practice based upon initial SAT results.77 This retrospective analysis used propensity score matching to adjust for differences in pre-treatment patient profiles in the SAT and in the external control dataset. The analysis estimated comparable outcome distributions for the adjuvant paclitaxel and trastuzumab regimen and the control regimen, which supported the use of the de-escalated regimen, particularly in light of higher toxicity with the control regimen.
Considerations and Implications for Regulatory Decision Making
In the United States, the 21st Century Cures Act directed the FDA to develop guidance for the evaluation and use of RWD, and to consider potential roles for RWD in drug development and regulatory decision making. For example, RWD could be used to support approvals for new indications or be integrated into existing monitoring requirements after approval.79 Accordingly, the FDA launched a RWD program to lay the foundation for rigorous use of such data in regulatory decisions.80 Several ongoing initiatives are providing guidance on data quality, data standards, and study designs that incorporate RWD.81 Also, other regulatory institutions such as the European Medicines Agency and Health Canada have demonstrated an openness towards better understanding and potentially leveraging RWD for drug development.82,83
The use of external control data, within a regulatory scope, can support expedited approval, extend the label for a therapy to a new indication or subgroup, and more generally support regulatory decision making.84 For example, in a rare disease setting, the FDA approved blinatumomab for adult relapsed/refractory acute lymphoblastic leukemia in a study that used data from a previously conducted clinical trial as a comparator.85
In a regulatory context, there is an understandably high burden of proof for investigators to demonstrate the scientific rigor of study designs and analyses that leverage external control data, with an appropriate risk level. Comparative analyses with standard RCT designs are fundamental to evaluate robustness and efficiencies of EACT designs. The external data, study design, and analytics should be tailored to each specific clinical context and intended regulatory use. Each of these elements should carefully considered and scrutinized in evaluating the risks of biased treatment effect estimates and inadequate control of false positive findings.
Data Sharing Models
Despite the appeal of patient-level data from prior clinical trials, data access is a barrier to studying and implementing EACT designs, in both early-phase and late-phase trials. Data sharing efforts from industry-funded RCTs are increasing, but there remains substantial room for improvement.86,87 Beyond implications for EACTs, clinical trial data sharing allows investigators to carry out analyses that can generate new knowledge, analyses which have been deemed to be “essential for expedited translation of research results into knowledge, products and procedures to improve human health” by the National Institutes of Health.88 Significant challenges and appropriate concerns about data sharing remain,89 including the need to ensure patient privacy and academic credit; the use of adequate standards for combining data from different sources; and the allotment of resources required to deidentify patient records and to provide infrastructures for data sharing. The patient perspective serves as an important counterpoint; assuming that privacy is protected, studies indicate that patients are in favor of having data shared for purposes that can help advance clinical outcomes.90
Advances towards simpler data access could transform the ability to perform secondary analyses91 and to leverage external data in future clinical studies. For many data-sharing platforms, a gatekeeper model is utilized, often with long approval processes, restrictive criteria for data access, and limitations on data use. These requirements act as a mechanism of passive resistance and delay access to data from completed trials. An increasing number of data sharing platforms such as Vivli,92 YODA,93 and Project Data Sphere,94 are aligned with more open-sharing models for clinical trial datasets.95 Nonetheless, data from previously completed neuro-oncology trials remain largely difficult to access.
New policies may be necessary for data sharing and to accelerate the study of new therapeutics. An important consideration is the modification of incentives for data sharing.96 A systematic effort from cooperative groups, industry, academics, and other stakeholders could help achieve this goal. Regulatory requirements that ensure timely data sharing and patient advocacy groups could play key roles in hastening this process. In addition, initiatives and agreements to prospectively share patient-level data from the control arms of multiple cooperative RCTs could be beneficial to the participating studies and create opportunities to extend data sharing.
Future directions and conclusion
At the conclusion of our think tank session, there was a strong interest and desire to continue to collaborate and to critically investigate, validate and implement EACT designs in GBM and on a broader level, in neuro-oncology. Efforts to form industry-cooperative group partnerships, and selection of datasets and statistical methods were set as goals to continue towards an advancement of our understanding of the role of EACTs for drug development in neuro-oncology.
The use of external data to design and analyze clinical studies has the potential to accelerate drug development and can contribute to rigorous evaluation of new treatments. RCTs will remain the indisputable gold standard for the evaluation of treatments, but external datasets can supplement information gleaned from RCTs and single arm studies. Further methodological work can help identify the appropriate clinical contexts, data, and statistical designs for EACTs that generate inference on treatment effects of experimental therapies, with well controlled risks on their accuracy and scientific validity.
There is a continuum of approaches for leveraging external data, and the use of EACTs should be tailored to the disease context. An emphasis on high quality patient-level data, rigorous methods and biostatistical expertise are critical in the successful implementation of EACTs. Data access to previously completed clinical trials and RWD is improving, but new policies and initiatives for data sharing could further unlock the value of external data. Continued collaborations between stakeholders including industry, academics, biostatisticians, clinicians, regulatory agencies, and patient advocates are crucial to understand the appropriate use of EACT designs in neuro-oncology.
Acknowledgements:
The authors thank Amy Barone, Pallavi Mishra-Kalyani and the Food and Drug Administration for contributing and helping with preparation of this review.
The authors thank the Society for Neuro-Oncology and their staff for arrangements and coordination for the 2020 Clinical Trials Think Tank meeting. The authors thank Johnathan Rine for help with preparation of the figures. RR has been supported by the Joint Center for Radiation Therapy Foundation Grant. LT and SV have been supported by the National Institutes of Health (NIH Grant 1R01LM013352-01A1).
Declaration of interests:
RR has received research support from Project Data Sphere, Inc, outside of submitted work. IR reports employment and owning stocks of Roche, owning stocks of Genentech, outside of submitted work. FM reports employment at Medicenna Therapeutics, Corp. LEA reports employment and owning stocks of Novartis. JEA reports employment and owning of stocks from Chimerix, Inc, outside of submitted work. LA and EA report employment at Candel Therapuetics. SB reports grants and personal fees from Novocure, grants from Incyte, grants from GSK, grants from Eli Lilly, personal fees from Bayer, personal fees from Sumitomo Dainippon, outside the submitted work. MK reports personal fees from Ipsen, Pfizer Roche, and Jackson Laboratory for Genomic Medicine and research funding paid to his institution from Specialized Therapeutics, all outside the submitted work. TC reports personal fees from Roche, Trizel, Medscape, Bayer, Amgen, Odonate Therapeutics, Pascal Biosciences, Del Mar, Tocagen, Karyopharm, GW Pharma, Kiyatec, AbbVie, Boehinger Ingelheim, VBI, Dicephera, VBL, Agios, Merck, Genocea, Puma, Lilly, BMS, Cortice, Wellcome Trust, other from Notable Labs; outside the submitted work. TC has a patent 62/819,322 with royalties paid to Katmai and Member of the board for the 501c3 Global Coalition for Adaptive Research, outside the submitted work. PW reports personal fees from Abbvie, Agios, Astra Zeneca, Blue Earth Diagnostics, Eli Lilly, Genentech/Roche, Immunomic Therapeutics, Kadmon, Kiyatec, Merck, Puma, Vascular Biogenics, Taiho, Tocagen, Deciphera, VBI Vaccines and research support from Agios, Astra Zeneca, Beigene, Eli Lily, Genentech/Roche, Karyopharm, Kazia, MediciNova, Merck, Novartis, Oncoceutics, Sanofi-Aventis, VBI Vaccines, outside the submitted work. BA reports employment at Foundation Medicine. BA reports personal fees from AbbVie, Bristol-Myers Squibb, Precision Health Economics, and Schlesinger Associates, outside of submitted work. He reports research support from Puma, Eli Lilly, Celgene, outside of submitted work. SV, JM, BL, MP, DA, KT, LT have nothing to disclose.
Contributor Information
Rifaquat Rahman, Department of Radiation Oncology, Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, MA, USA..
Steffen Ventz, Department of Data Sciences, Dana-Farber Cancer Institute, Harvard T.H. Chan School of Public Health, Boston, MA, USA..
Jon McDunn, Project Data Sphere, Morrisville, NC, USA..
Bill Louv, Project Data Sphere, Morrisville, NC, USA..
Irmarie Reyes-Rivera, F. Hoffmann-La Roche Ltd, Basel, Switzerland..
Mei-Yin Chen Polley, Department of Public Health Sciences, University of Chicago, Chicago, IL, USA..
Fahar Merchant, Medicenna Therapeutics Corp, Toronto, Canada..
Lauren E. Abrey, Novartis AG, Basel Switzerland.
Joshua Allen, Chimerix, Inc, Durham, NC, USA..
Laura K. Aguilar, Candel Therapeutics, Needham, MA, USA.
Estuardo Aguilar-Cordova, Candel Therapeutics, Needham, MA, USA..
David Arons, National Brain Tumor Society, Newton, MA, USA..
Kirk Tanner, National Brain Tumor Society, Newton, MA, USA..
Stephen Bagley, Division of Hematology/Oncology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA..
Mustafa Khasraw, Preston Robert Tisch Brain Tumor Center at Duke, Departments of Neurosurgery, Duke University Medical Center, Durham, NC, USA..
Timothy Cloughesy, Neuro-Oncology Program and Department of Neurology, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA..
Patrick Y. Wen, Center for Neuro-Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA.
Brian M. Alexander, Foundation Medicine, Inc., Cambridge, MA, USA; Department of Radiation Oncology, Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, MA, USA..
Lorenzo Trippa, Department of Data Sciences, Dana-Farber Cancer Institute, Harvard T.H. Chan School of Public Health, Boston, MA, USA..
References
- 1.Hwang TJ, Carpenter D, Lauffenburger JC, Wang B, Franklin JM, Kesselheim AS. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern Med 2016; 176: 1826–33. [DOI] [PubMed] [Google Scholar]
- 2.Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics 2019; 20: 273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu M, Yang M, Liu Y. Statistical adaptation to oncology drug development evolution. Contemp Clin Trials 2020; 99: 106180. [DOI] [PubMed] [Google Scholar]
- 4.Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J Clin Oncol 2009; 27: 4027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander BM, Cloughesy TF. Adult Glioblastoma. J Clin Oncol 2017; 35: 2402–9. [DOI] [PubMed] [Google Scholar]
- 6.Wen PY, Weller M, Lee EQ, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020; 22: 1073–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbaro M, Fine HA, Magge RS. Scientific and Clinical Challenges within Neuro-Oncology. World Neurosurg 2021; published online Feb 18. DOI: 10.1016/j.wneu.2021.01.151. [DOI] [PubMed] [Google Scholar]
- 8.Vanderbeek AM, Rahman R, Fell G, et al. The clinical trials landscape for glioblastoma: is it adequate to develop new treatments? Neuro-oncology 2018; published online March 6. DOI: 10.1093/neuonc/noy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EQ, Chukwueke UN, Hervey-Jumper SL, et al. Barriers to Accrual and Enrollment in Brain Tumor Trials. Neuro-oncology 2019; published online June 7. DOI: 10.1093/neuonc/noz104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanderbeek AM, Ventz S, Rahman R, et al. To randomize, or not to randomize, that is the question: using data from prior clinical trials to guide future designs. Neuro-oncology 2019; 21: 1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol 2020; 22: iv1–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eborall HC, Stewart MCW, Cunningham-Burley S, Price JF, Fowkes FGR. Accrual and drop out in a primary prevention randomised controlled trial: qualitative study. Trials 2011; 12: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Featherstone K, Donovan JL. ‘Why don’t they just tell me straight, why allocate it?’ The struggle to make sense of participating in a randomised controlled trial. Soc Sci Med 2002; 55: 709–19. [DOI] [PubMed] [Google Scholar]
- 14.Feinberg BA, Gajra A, Zettler ME, Phillips TD, Phillips EG, Kish JK. Use of Real-World Evidence to Support FDA Approval of Oncology Drugs. Value Health 2020; 23: 1358–65. [DOI] [PubMed] [Google Scholar]
- 15.Eichler H-G, Pignatti F, Schwarzer-Daum B, et al. Randomized Controlled Trials Versus Real World Evidence: Neither Magic Nor Myth. Clin Pharmacol Ther 2021; 109: 1212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry DA. The Brave New World of clinical cancer research: Adaptive biomarker-driven trials integrating clinical practice with clinical research. Mol Oncol 2015; 9: 951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ventz S, Lai A, Cloughesy TF, Wen PY, Trippa L, Alexander BM. Design and Evaluation of an External Control Arm Using Prior Clinical Trials and Real-World Data. Clin Cancer Res 2019; 25: 4993–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ventz S, Comment L, Louv B, et al. The Use of External Control Data for Predictions and Futility Interim Analyses in Clinical Trials. Neuro Oncol 2021; published online June 9. DOI: 10.1093/neuonc/noab141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sampson JH, Achrol A, Aghi MK, et al. MDNA55 survival in recurrent glioblastoma (rGBM) patients expressing the interleukin-4 receptor (IL4R) as compared to a matched synthetic control. JCO 2020; 38: 2513–2513. [Google Scholar]
- 20.Thall PF, Simon R. Incorporating historical control data in planning phase II clinical trials. Stat Med 1990; 9: 215–28. [DOI] [PubMed] [Google Scholar]
- 21.Neuenschwander B, Capkun-Niggli G, Branson M, Spiegelhalter DJ. Summarizing historical information on controls in clinical trials. Clin Trials 2010; 7: 5–18. [DOI] [PubMed] [Google Scholar]
- 22.Reardon DA, Galanis E, DeGroot JF, et al. Clinical trial end points for high-grade glioma: the evolving landscape. Neuro-oncology 2011; 13: 353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma MR, Karrison TG, Jin Y, et al. Resampling phase III data to assess phase II trial designs and endpoints. Clin Cancer Res 2012; 18: 2309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang H, Foster NR, Grothey A, Ansell SM, Goldberg RM, Sargent DJ. Comparison of error rates in single-arm versus randomized phase II cancer clinical trials. J Clin Oncol 2010; 28: 1936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grossman SA, Schreck KC, Ballman K, Alexander B. Point/counterpoint: randomized versus single-arm phase II clinical trials for patients with newly diagnosed glioblastoma. Neuro Oncol 2017; 19: 469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stallard N, Todd S. Seamless phase II/III designs. Stat Methods Med Res 2011; 20: 623–34. [DOI] [PubMed] [Google Scholar]
- 27.Alexander BM, Trippa L, Gaffey S, et al. Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): A Bayesian Adaptive Platform Trial to Develop Precision Medicines for Patients With Glioblastoma. JCO Precision Oncology 2019; : 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander BM, Ba S, Berger MS, et al. Adaptive Global Innovative Learning Environment for Glioblastoma: GBM AGILE. Clin Cancer Res 2018; 24: 737–43. [DOI] [PubMed] [Google Scholar]
- 29.Buxton MB, Alexander BM, Berry DA, et al. GBM AGILE: A global, phase II/III adaptive platform trial to evaluate multiple regimens in newly diagnosed and recurrent glioblastoma. JCO 2020; 38: TPS2579–TPS2579. [Google Scholar]
- 30.Thorlund K, Dron L, Park JJH, Mills EJ. Synthetic and External Controls in Clinical Trials – A Primer for Researchers. Clin Epidemiol 2020; 12: 457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viele K, Berry S, Neuenschwander B, et al. Use of historical control data for assessing treatment effects in clinical trials. Pharm Stat 2014; 13: 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.VanderWeele TJ, Shpitser I. On the definition of a confounder. Ann Stat 2013; 41: 196–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pignatti F, van den Bent M, Curran D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol 2002; 20: 2076–84. [DOI] [PubMed] [Google Scholar]
- 34.Gittleman H, Lim D, Kattan MW, et al. An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG Oncology RTOG 0525 and 0825. Neuro Oncol 2017; 19: 669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davi R, Mahendraratnam N, Chatterjee A, Dawson CJ, Sherman R. Informing single-arm clinical trials with external controls. Nat Rev Drug Discov 2020; published online Aug 18. DOI: 10.1038/d41573-020-00146-5. [DOI] [PubMed] [Google Scholar]
- 36.Normington J, Zhu J, Mattiello F, Sarkar S, Carlin B. An efficient Bayesian platform trial design for borrowing adaptively from historical control data in lymphoma. Contemp Clin Trials 2020; 89: 105890. [DOI] [PubMed] [Google Scholar]
- 37.Webster-Clark M, Jonsson Funk M, Stürmer T. Single-arm Trials With External Comparators and Confounder Misclassification: How Adjustment Can Fail. Med Care 2020; 58: 1116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson D Replication of Randomized, Controlled Trials Using Real-World Data: What Could Go Wrong? Value Health 2021; 24: 112–5. [DOI] [PubMed] [Google Scholar]
- 39.Seeger JD, Davis KJ, Iannacone MR, et al. Methods for external control groups for single arm trials or long-term uncontrolled extensions to randomized clinical trials. Pharmacoepidemiol Drug Saf 2020; 29: 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snapinn S, Chen M-G, Jiang Q, Koutsoukos T. Assessment of futility in clinical trials. Pharm Stat 2006; 5: 273–81. [DOI] [PubMed] [Google Scholar]
- 41.Gould AL. Sample size re-estimation: recent developments and practical considerations. Stat Med 2001; 20: 2625–43. [DOI] [PubMed] [Google Scholar]
- 42.Imbens GW, Rubin DB. Causal Inference for Statistics, Social, and Biomedical Sciences: An Introduction, 1st Edition. New York: Cambridge University Press, 2015. [Google Scholar]
- 43.Rubin DB. The Use of Matched Sampling and Regression Adjustment to Remove Bias in Observational Studies. Biometrics 1973; 29: 185–203. [Google Scholar]
- 44.ROSENBAUM PR, RUBIN DB. The central role of the propensity score in observational studies for causal effects. Biometrika 1983; 70: 41–55. [Google Scholar]
- 45.Li L, Greene T. A weighting analogue to pair matching in propensity score analysis. Int J Biostat 2013; 9: 215–34. [DOI] [PubMed] [Google Scholar]
- 46.Pearl J An Introduction to Causal Inference. Int J Biostat 2010; 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin W-J, Chen JJ. Biomarker classifiers for identifying susceptible subpopulations for treatment decisions. Pharmacogenomics 2012; 13: 147–57. [DOI] [PubMed] [Google Scholar]
- 48.Carrigan G, Whipple S, Capra WB, et al. Using Electronic Health Records to Derive Control Arms for Early Phase Single-Arm Lung Cancer Trials: Proof-of-Concept in Randomized Controlled Trials. Clin Pharmacol Ther 2020; 107: 369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abrahami D, Pradhan R, Yin H, Honig P, Baumfeld Andre E, Azoulay L. Use of Real-World Data to Emulate a Clinical Trial and Support Regulatory Decision Making: Assessing the Impact of Temporality, Comparator Choice, and Method of Adjustment. Clin Pharmacol Ther 2021; 109: 452–61. [DOI] [PubMed] [Google Scholar]
- 50.Franklin JM, Patorno E, Desai RJ, et al. Emulating Randomized Clinical Trials With Nonrandomized Real-World Evidence Studies: First Results From the RCT DUPLICATE Initiative. Circulation 2021; 143: 1002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beaulieu-Jones BK, Finlayson SG, Yuan W, et al. Examining the Use of Real-World Evidence in the Regulatory Process. Clin Pharmacol Ther 2020; 107: 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hobbs BP, Carlin BP, Sargent DJ. Adaptive adjustment of the randomization ratio using historical control data. Clinical Trials 2013; 10: 430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medicenna Provides MDNA55 rGBM Clinical Program Update Following Positive End of Phase 2 Meeting with the U.S. Food and Drug Administration (FDA) - Medicenna Therapeutics. https://ir.medicenna.com/news-releases/news-release-details/medicenna-provides-mdna55-rgbm-clinical-program-update-following/ (accessed June 4, 2021).
- 54.Miksad RA, Abernethy AP. Harnessing the Power of Real-World Evidence (RWE): A Checklist to Ensure Regulatory-Grade Data Quality. Clin Pharmacol Ther 2018; 103: 202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghadessi M, Tang R, Zhou J, et al. A roadmap to using historical controls in clinical trials - by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG). Orphanet J Rare Dis 2020; 15: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Backenroth D How to choose a time zero for patients in external control arms. Pharm Stat 2021; 20: 783–92. [DOI] [PubMed] [Google Scholar]
- 57.Kilburn LS, Aresu M, Banerji J, Barrett-Lee P, Ellis P, Bliss JM. Can routine data be used to support cancer clinical trials? A historical baseline on which to build: retrospective linkage of data from the TACT (CRUK 01/001) breast cancer trial and the National Cancer Data Repository. Trials 2017; 18: 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Little RJ, D’Agostino R, Cohen ML, et al. The prevention and treatment of missing data in clinical trials. N Engl J Med 2012; 367: 1355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basch E, Deal AM, Dueck AC, et al. Overall Survival Results of a Trial Assessing Patient-Reported Outcomes for Symptom Monitoring During Routine Cancer Treatment. JAMA 2017; 318: 197–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gilbert MR, Dignam J, Won M, et al. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin Oncol 2013; 31: 1.23129739 [Google Scholar]
- 61.Gilbert MR, Dignam JJ, Armstrong TS, et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N Engl J Med 2014; 370: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014; 370: 709–22. [DOI] [PubMed] [Google Scholar]
- 63.Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017; 18: 1373–85. [DOI] [PubMed] [Google Scholar]
- 64.Mbuagbaw L, Foster G, Cheng J, Thabane L. Challenges to complete and useful data sharing. Trials 2017; 18: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearl J Causality: models, reasoning and inference, 2nd ed. New York: Cambridge University Press, 2009. [Google Scholar]
- 66.Collins R, Bowman L, Landray M, Peto R. The Magic of Randomization versus the Myth of Real-World Evidence. N Engl J Med 2020; 382: 674–8. [DOI] [PubMed] [Google Scholar]
- 67.Larrouquere L, Giai J, Cracowski J-L, Bailly S, Roustit M. Externally Controlled Trials: Are We There Yet? Clin Pharmacol Ther 2020; 108: 918–9. [DOI] [PubMed] [Google Scholar]
- 68.Liu R, Rizzo S, Whipple S, et al. Evaluating eligibility criteria of oncology trials using real-world data and AI. Nature 2021; 592: 629–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Corrigan-Curay J, Sacks L, Woodcock J. Real-World Evidence and Real-World Data for Evaluating Drug Safety and Effectiveness. JAMA 2018; 320: 867–8. [DOI] [PubMed] [Google Scholar]
- 70.Unger JM, Barlow WE, Martin DP, et al. Comparison of survival outcomes among cancer patients treated in and out of clinical trials. J Natl Cancer Inst 2014; 106: dju002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chukwueke UN, Wen PY. Use of the Response Assessment in Neuro-Oncology (RANO) criteria in clinical trials and clinical practice. CNS Oncol 2019; 8: CNS28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen PY, Chang SM, Van den Bent MJ, Vogelbaum MA, Macdonald DR, Lee EQ. Response Assessment in Neuro-Oncology Clinical Trials. J Clin Oncol 2017; 35: 2439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang RY, Neagu MR, Reardon DA, Wen PY. Pitfalls in the neuroimaging of glioblastoma in the era of antiangiogenic and immuno/targeted therapy - detecting illusive disease, defining response. Front Neurol 2015; 6: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilbert MR, Rubinstein L, Lesser G. Creating clinical trial designs that incorporate clinical outcome assessments. Neuro Oncol 2016; 18 Suppl 2: ii21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blakeley JO, Coons SJ, Corboy JR, Kline Leidy N, Mendoza TR, Wefel JS. Clinical outcome assessment in malignant glioma trials: measuring signs, symptoms, and functional limitations. Neuro Oncol 2016; 18 Suppl 2: ii13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ray EM, Carey LA, Reeder-Hayes KE. Leveraging existing data to contextualize phase II clinical trial findings in oncology. Annals of Oncology 2020; 0. DOI: 10.1016/j.annonc.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 77.Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015; 372: 134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amiri-Kordestani L, Xie D, Tolaney SM, et al. A Food and Drug Administration analysis of survival outcomes comparing the Adjuvant Paclitaxel and Trastuzumab trial with an external control from historical clinical trials. Ann Oncol 2020; published online Aug 28. DOI: 10.1016/j.annonc.2020.08.2106. [DOI] [PubMed] [Google Scholar]
- 79.Franklin JM, Glynn RJ, Martin D, Schneeweiss S. Evaluating the Use of Nonrandomized Real-World Data Analyses for Regulatory Decision Making. Clin Pharmacol Ther 2019; 105: 867–77. [DOI] [PubMed] [Google Scholar]
- 80.Framework for FDA’s Real-World Evidence Program. 2018; published online Dec. https://www.fda.gov/media/120060/download (accessed June 2, 2021).
- 81.Complex Innovative Trial Design Pilot Meeting Program. 2021; published online May 26. https://www.fda.gov/drugs/development-resources/complex-innovative-trial-design-pilot-meeting-program. [DOI] [PubMed]
- 82.European Medicines Agency. EMA Regulatory Science to 2025. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf (accessed June 3, 2021).
- 83.Health Canada. Strengthening the use of real world evidence for drugs. https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/strengthening-use-real-world-evidence-drugs.html (accessed June 3, 2021).
- 84.Burcu M, Dreyer NA, Franklin JM, et al. Real-world evidence to support regulatory decision-making for medicines: Considerations for external control arms. Pharmacoepidemiol Drug Saf 2020; 29: 1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gökbuget N, Kelsh M, Chia V, et al. Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J 2016; 6: e473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boutron I, Dechartres A, Baron G, Li J, Ravaud P. Sharing of Data From Industry-Funded Registered Clinical Trials. JAMA 2016; 315: 2729–30. [DOI] [PubMed] [Google Scholar]
- 87.Miller J, Ross JS, Wilenzick M, Mello MM. Sharing of clinical trial data and results reporting practices among large pharmaceutical companies: cross sectional descriptive study and pilot of a tool to improve company practices. BMJ 2019; 366: l4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.National Institute of Health (NIH). FINAL NIH STATEMENT ON SHARING RESEARCH DATA. 2003; published online Feb 26. https://grants.nih.gov/grants/guide/notice-files/not-od-03-032.html#:~:text=We%20believe%20that%20data%20sharing,and%20other%20important%20scientific%20goals.
- 89.Longo DL, Drazen JM. Data Sharing. N Engl J Med 2016; 374: 276–7. [DOI] [PubMed] [Google Scholar]
- 90.Mello MM, Lieou V, Goodman SN. Clinical Trial Participants’ Views of the Risks and Benefits of Data Sharing. N Engl J Med 2018; 378: 2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arfè A, Ventz S, Trippa L. Shared and Usable Data From Phase 1 Oncology Trials-An Unmet Need. JAMA Oncol 2020; 6: 980–1. [DOI] [PubMed] [Google Scholar]
- 92.Bierer BE, Li R, Barnes M, Sim I. A Global, Neutral Platform for Sharing Trial Data. N Engl J Med 2016; 374: 2411–3. [DOI] [PubMed] [Google Scholar]
- 93.Krumholz HM, Waldstreicher J. The Yale Open Data Access (YODA) Project--A Mechanism for Data Sharing. N Engl J Med 2016; 375: 403–5. [DOI] [PubMed] [Google Scholar]
- 94.Bertagnolli MM, Sartor O, Chabner BA, et al. Advantages of a Truly Open-Access Data-Sharing Model. N Engl J Med 2017; 376: 1178–81. [DOI] [PubMed] [Google Scholar]
- 95.Pisani E, Aaby P, Breugelmans JG, et al. Beyond open data: realising the health benefits of sharing data. BMJ 2016; 355: i5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lo B, DeMets DL. Incentives for Clinical Trialists to Share Data. N Engl J Med 2016; 375: 1112–5. [DOI] [PubMed] [Google Scholar]