

Abstract
Motivation
Recent technological developments have facilitated an expansion of microbiome–metabolome studies, in which samples are assayed using both genomic and metabolomic technologies to characterize the abundances of microbial taxa and metabolites. A common goal of these studies is to identify microbial species or genes that contribute to differences in metabolite levels across samples. Previous work indicated that integrating these datasets with reference knowledge on microbial metabolic capacities may enable more precise and confident inference of microbe–metabolite links.
Results
We present MIMOSA2, an R package and web application for model-based integrative analysis of microbiome–metabolome datasets. MIMOSA2 uses genomic and metabolic reference databases to construct a community metabolic model based on microbiome data and uses this model to predict differences in metabolite levels across samples. These predictions are compared with metabolomics data to identify putative microbiome-governed metabolites and taxonomic contributors to metabolite variation. MIMOSA2 supports various input data types and customization with user-defined metabolic pathways. We establish MIMOSA2’s ability to identify ground truth microbial mechanisms in simulation datasets, compare its results with experimentally inferred mechanisms in honeybee microbiota, and demonstrate its application in two human studies of inflammatory bowel disease. Overall, MIMOSA2 combines reference databases, a validated statistical framework, and a user-friendly interface to facilitate modeling and evaluating relationships between members of the microbiota and their metabolic products.
Availability and implementation
MIMOSA2 is implemented in R under the GNU General Public License v3.0 and is freely available as a web server at http://elbo-spice.cs.tau.ac.il/shiny/MIMOSA2shiny/ and as an R package from http://www.borensteinlab.com/software_MIMOSA2.html.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Microbial community metabolism contributes to global nutrient cycling (McGuire and Treseder, 2010), metabolic dysregulation in human disease (Kasubuchi et al., 2015) and detoxification of pollutants (Hazen et al., 2010), among other crucial processes. A growing number of studies investigate these processes by profiling the composition and metabolism of host-associated and environmental microbial communities using genomic and metabolomic technologies (Shaffer et al., 2017). Such studies commonly seek to answer two important questions: whether metabolic differences between environments (e.g. between healthy and diseased settings) can be attributed to differences in microbial composition and ecology, and if so, which specific microbial community members might be the key players generating such differences.
Broad surveys of microbial taxa and metabolites, which we refer to here as ‘microbiome–metabolome studies’, have great potential utility to answer these two questions by uncovering associations between taxa and metabolite abundances across samples. However, we have recently shown that calculating univariate correlations between taxa and metabolites may not identify mechanistic links between them with very high accuracy, depending on properties of the communities under study (Noecker et al., 2019). Comparisons of taxon–metabolite associations with metabolite production in monoculture have also suggested a high false positive rate for this approach (Hoyles et al., 2018). Some recently introduced tools address this challenge by using machine learning to infer complex models predicting metabolite abundances from microbial taxa (Mallick et al., 2019; Morton et al., 2019; Reiman et al., 2021). However, these can be difficult to interpret in terms of possible mechanisms, and in general do not account for or incorporate prior knowledge of microbial metabolism.
An alternative or complementary approach is to use metabolic reference databases, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000), as well as collections of genome-scale metabolic reconstructions, such as AGORA (Magnúsdóttir et al., 2017) and embl_gems (Machado et al., 2018), to generate and evaluate more precise hypotheses on the relationships between microbes and metabolites. We previously demonstrated that this conceptual approach can lead to new insights and released a preliminary but widely used R package (Noecker et al., 2016), Model-based Integration of Metabolite Observations and Species Abundances (MIMOSA), for performing these analyses (Adamovsky et al., 2020; Casero et al., 2017; Ilhan et al., 2019; Sharon et al., 2019; Snijders et al., 2016; Stewart et al., 2017). Other methods to integrate microbiome and metabolome data with metabolic reference databases have also recently been introduced (Garza et al., 2018; McHardy et al., 2013; Pedersen et al., 2018; Shaffer et al., 2019). To date, however, these tools all have some limitations in usability and/or compatibility with common data types and reference databases. Furthermore, none have been systematically evaluated for their ability to accurately infer direct mechanistic links between microbes and metabolites.
Here, we introduce MIMOSA2, an R package and web application (http://borensteinlab.com/software_MIMOSA2.html) for model-based integration of microbiome and metabolome data. MIMOSA2 uses metabolic reference databases to analyze paired microbiome and metabolite profiles, identifying metabolite differences that can be explained by specific microbiome features. MIMOSA2 expands and improves on the original proof-of-concept version of MIMOSA in several ways (described in detail in Supplementary Table S1): first, it applies a new statistical algorithm for quantifying links between taxa and metabolites, which we have validated here using simulated microbiome–metabolome data and experimental comparisons. It also implements several distinct strategies for constructing community metabolic models from different types of microbiome data and reference databases. Finally, it can be run either locally or via a web server with greatly improved ease of use, flexibility and documentation.
2 Implementation
MIMOSA2 integrates paired microbiome–metabolome datasets with reference reaction databases to generate specific mechanistic hypotheses. It constructs community metabolic models using microbiome compositional data and a reaction database, assesses whether measured metabolite concentrations are consistent with estimated community metabolic potential (CMP) across a set of samples, and identifies specific taxa and reactions that can explain observed metabolite variation (Fig. 1). Below, we describe the analysis workflow steps and how to run an analysis using the web application.
Fig. 1.

Summary of the MIMOSA2 analysis pipeline. In a MIMOSA2 analysis, microbiome data features are first linked to pre-processed reference databases to construct a community metabolic model describing the predicted metabolic reaction capabilities of each community member taxon (Step 1). Next, this network model is combined with microbiome feature abundances to calculate CMP scores for each metabolite, taxon and sample, representing the approximate relative capacity to synthesize or utilize that metabolite (Step 2). Total CMP scores for each metabolite are linked to metabolomics measurements using a regression model (Step 3). For metabolites with a significant relationship between concentration and potential, the specific taxonomic contributors to each metabolite are then analyzed (Step 4)
2.1 Data input
The basic input requirement for MIMOSA2 is a pair of datasets from the same set of samples: one of microbiome measurements and the other of metabolites. Each of these datasets may take a variety of forms (Fig. 1), depending on the study’s design, experimental assays and processing techniques.
Users can provide microbiome data generated from 16S rRNA or shotgun metagenomic sequencing studies. 16S rRNA data can be provided as a feature table of amplicon sequence variants, or of closed-reference operational taxonomic units (OTUs) using either the GreenGenes or SILVA databases (McDonald et al., 2012; Quast et al., 2013). Data from a shotgun metagenomic study can be provided in the form of a table of KEGG Ortholog (Kanehisa and Goto, 2000) functional abundances. Metagenomic data can be either unstratified (total KEGG Ortholog abundances in each sample) or stratified by microbial taxa [in the formats produced by the latest versions of either HUMAnN or PICRUSt2 software (Beghini et al., 2021; Douglas et al., 2020)].
Metabolite data can be produced from any metabolomics platform (Fig. 1), but it must contain putative metabolite identifications in the form of metabolite names, Human Metabolome Database (HMDB) IDs or KEGG compound IDs. If metabolite names or HMDB IDs are provided, they are mapped to KEGG IDs for the main analysis using the Chemical Translation Service database (Wohlgemuth et al., 2010), accessed via the R package webchem (Szöcs et al., 2020).
2.2 Reference data and metabolic model construction
MIMOSA2 uses reference data to estimate how CMP varies across a set of microbiome samples. It takes advantage of genome-scale metabolic model data from multiple alternative sources to use the most appropriate reference data for a given dataset. Specifically, MIMOSA2 can currently generate a metabolic model based on one of three sources. First, it can use a curated set of reactions from the KEGG database. If OTU data are provided, KEGG reactions are inferred for each OTU using PICRUSt (Langille et al., 2013) pre-computed outputs. Alternatively, MIMOSA2 can link 16S rRNA amplicon data to one of two large collections of genome-scale metabolic reconstructions: the AGORA collection of genome-scale metabolic models of gut microbial species (v1.0.2) (Magnúsdóttir et al., 2017), or the embl_gems library of genome-scale metabolic models for all 5587 reference and representative bacterial genomes in RefSeq (Machado et al., 2018).
The method used to map microbiome taxa abundances to metabolic reactions depends on the input data type (described fully in Supplementary Text and Fig. S1). 16S rRNA sequence variants are mapped using vsearch to either RNA genes for the AGORA genome collection (see Supplementary Text), or to Greengenes 99% OTU representative sequences. Greengenes OTUs are mapped to KEGG using the pre-computed genome inferences from PICRUSt 1.1.3 (Langille et al., 2013), and are linked to AGORA models using a pre-calculated alignment between the two databases. KEGG Ortholog abundances provided from a shotgun metagenomic dataset or another method [such as from PICRUSt2 (Douglas et al., 2020)] can be directly linked to KEGG reactions.
Additionally, users can specify custom additions, subtractions or modifications to any of the MIMOSA2 model templates. These can be provided at the gene, reaction or taxon level. Example modification files are provided in the MIMOSA2 documentation. Adding or removing a particular reaction can be useful to assess the impact of suspected incomplete or incorrect annotations in the reference database, as we have observed in analyses using MIMOSA version 1 (Sharon et al., 2019). Full details on the implementation of each of the metabolic model construction options are provided in the Supplementary Text.
2.3 Core algorithm and identification of species–metabolite contributors
Using the constructed metabolic models, MIMOSA2 next calculates CMP scores for each taxon, sample and metabolite. These scores represent the predicted capacity of each taxon in each sample to synthesize or utilize the metabolite in question, facilitating downstream analysis of the links between individual taxa and metabolites. These metabolic potential scores are then aggregated at the community level and compared with the relevant metabolite measurements. An example analysis for a toy metabolite and dataset is shown in Figure 2.
Fig. 2.
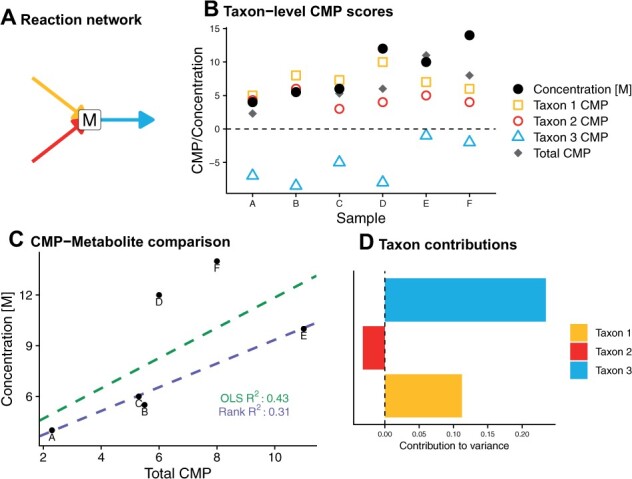
An example contribution analysis of hypothetical metabolite M. (A) In this toy example, metabolite M is produced by two taxa and utilized by a third. (B) Overview of CMP scores and metabolite measurements for metabolite M across a dataset of six samples. Taxon-level CMP scores are based on the estimated ability of each species to synthesize/utilize the metabolite and on the abundance of the taxon in the sample. (C) Comparison of CMP scores and measurements of M across this dataset. The solution found by OLS regression is affected more strongly by the outlier samples D and F than that found by rank-based regression. (D) Final summary contribution plot for metabolite M using the rank-based regression option. The bars represent the contributions to metabolite variance explained by each taxon. Most of the variance is attributed to differences in the amount of utilization of M by Taxon 3 across samples, reflecting the larger variability in Taxon 3’s CMP scores shown in panel (B)
Specifically, metabolic potential scores are calculated as a linear combination of the abundances of genes predicted to contribute to synthesis or utilization of a metabolite multiplied by their expected stoichiometric effects (Fig. 2A and B). This method is similar to the approach used by MIMOSA version 1 (Noecker et al., 2016), but importantly, these scores are now calculated at the level of individual community members, rather than for the community as a whole. The score smij for metabolite m, taxon i and sample j can be expressed as the sum of the estimated effects of all synthesis reactions (s) minus the effects of all utilization reactions (u). The estimated effect of a reaction is approximated as the product of the estimated abundance of linked gene families (A) and a normalized stoichiometric coefficient for the metabolite in question (:
The total CMP score for metabolite m in sample j is then the sum of the taxon-level scores:
MIMOSA2 next evaluates the relationship between total CMP scores and metabolites by fitting a linear regression model of metabolite levels across samples (Fig. 2C). By default, the regression model is fit using rank-based estimation (Kloke and McKean, 2012) (referred to here as MIMOSA2-rank), although an option for ordinary least-squares (OLS) estimation is also provided (MIMOSA2-OLS). Notably, rank-based estimation achieves a more robust fit for noisy data (Kloke and McKean, 2012), as the error function incorporates both the rank and the magnitude of the model residuals. OLS estimation, in contrast, provides greater computational efficiency in both the model fitting and the subsequent calculation of the contributions of specific taxa.
A metabolite is identified as putatively microbiome-governed if the overall regression model fit meets a significance threshold (F test or drop-in-deviance test P < 0.1 by default). MIMOSA2 then decomposes the share of metabolite variation explained by the model into linear contributions from each taxon (Fig. 2D), and microbial taxa with large contributions to model variation are identified as potential contributors. The calculation of this decomposition is analogous to our previous approach for calculating taxonomic contributors based on simulated metabolic fluxes (Noecker et al., 2019). This metric prioritizes taxa whose estimated metabolic potential is relatively abundant, variable and associated with the measured metabolite concentrations (Fig. 2B and D). For analyses using OLS regression, contributions are calculated as the covariance of the metabolic potential scores from each taxon with metabolite concentrations. For analyses using rank-based regression, this metric is calculated using a permutation-based analysis of the importance of each taxon’s scores to the model fit. A full explanation of these contribution calculations can be found in the Supplementary Text. Notably, this approach is different from the taxonomic contributor metric used in MIMOSA version 1, which prioritized taxa based on the correlation coefficient of their metabolic potential with metabolites across samples. It also differs from standard approaches for assessing covariate importance in a regression model in that it accounts for both the association and relative magnitude of contributions from each taxon, without any scaling.
Importantly, each of the steps in the MIMOSA2 workflow is modular. For instance, CMP is currently calculated from the community metabolic model using the gene abundance-based scoring approach described above, but these values could in the future be replaced with estimates of metabolic fluxes using Flux Balance Analysis or other simulation methods (Varma and Palsson, 1994).
2.4 Interface, results and visualization
The MIMOSA2 workflow can be run via either a web application or an R package. In both options, the input file names and analysis parameters are encoded in a configuration file, which can be used to reproduce the analysis.
MIMOSA2 produces several detailed results for each analyzed metabolite: the model fit between CMP and metabolite measurements, the primary taxa contributors to metabolite variation and the specific reactions that formed the basis of the metabolic potential calculation. When an analysis is run using the web application, these results are summarized in an interactive table with a row for each metabolite (Fig. 3). Processed result tables are made available for download, along with the analysis configuration file. The web application also includes an option to view results of an example analysis of cecal samples from mice treated with the antibiotic cefoperazone (Theriot et al., 2014).
Fig. 3.

Interactive results interface for the MIMOSA2 web application. In addition to making all processed results available for download, the application displays an interactive table in which each row summarizes the results for a single metabolite. The best-predicted metabolites are shown first, along with associated information including model statistics, plots of the data and top contributors and lists of the top contributing taxa and reactions predicted via both synthesis and utilization
3 Results
3.1 Application of MIMOSA2 to simulated datasets with known microbe–metabolite interactions
We validated the MIMOSA2 method by applying it first to two simulated microbiome–metabolome datasets [previously described in Noecker et al. (2019)]. We used these simulation datasets to evaluate the ability of MIMOSA2 to achieve two main objectives: (i) to classify whether metabolite levels are likely determined by microbiome metabolism, and (ii) to identify key contributors for those metabolites and the relevant genes and reactions. In our previous study, we defined ‘key contributors’ as the set of microbial taxa (or exogenous environmental influences) that are responsible for a substantial share of the variation in the levels of a metabolite across samples, based on their quantitative metabolite fluxes. Notably, these two objectives differ from the objectives of other recently published microbiome–metabolome analysis methods (Mallick et al., 2019; Morton et al., 2019), which are designed primarily to predict metabolite levels from microbiome data with high accuracy without incorporating additional information or generating mechanistic hypotheses. In this study, we compared the performance of MIMOSA2 using both rank-based and OLS regression estimation with five alternative approaches: a species-metabolite pairwise Spearman correlation analysis, a modified correlation analysis in which significant taxa-metabolite correlations are only retained if the taxon is known to possess reactions linked to the metabolite (see Supplementary Text for additional details), a cross-validated random forest regression model following Muller et al. (2021), MelonnPan (Mallick et al., 2019) and the original implementation of MIMOSA.
The two simulation datasets describe divergent sets of hypothetical microbiome samples (Fig. 4A) and have been previously used to evaluate microbiome–metabolome analysis (Noecker et al., 2019). They were generated using a multi-species dynamic Flux Balance Analysis framework to simulate gut bacterial metabolism in a fixed nutrient environment, using metabolic network reconstructions from the AGORA collection (Magnúsdóttir et al., 2017; Noecker et al., 2019). In this framework, simulated microbial uptake and secretion dynamically impact concentrations of metabolites in the shared environment. Both datasets consisted of species and metabolite concentrations at the final time point of several 144-h dynamic multi-species simulations. Both datasets included small random variations in the simulated environmental metabolite concentrations available to the simulated species across samples (see Supplementary Text), but microbial fluxes are the primary drivers of final concentrations for a large share of compounds. Using this method, Dataset 1 consists of communities with varying compositions of 10 representative gut species and 3% variation in environmental metabolite concentrations. Dataset 2 is more complex, consisting of 57 samples whose initial compositions were designed to emulate Human Microbiome Project (HMP) gut samples (Huttenhower et al., 2012), along with 1% variation in nutrient inflow. These species compositions were determined by aligning HMP 16S rRNA sequencing variants against genomes linked to AGORA reconstructions, resulting in a total of 131 species unevenly distributed across the dataset. To construct community metabolic network models for MIMOSA2, we used the unconstrained community metabolic reconstructions, which formed the basis for each set of simulations.
Fig. 4.

Identification of key microbe–metabolite contributions by MIMOSA2 from simulated datasets. (A) Descriptive summary statistics for the two simulated datasets analyzed, including number of samples, species and metabolites included. (B) Precision, specificity and sensitivity of MIMOSA2 analysis for recovering true key microbial contributors to metabolite variation from metabolites identified as potentially microbiome-governed by MIMOSA2 in two simulated datasets, compared with correlation-based approaches, MIMOSA version 1, MelonnPan L1 linear models (Mallick et al., 2019) and feature importance from a cross-validated random forest regression (RF) (Muller et al., 2021). Standard thresholds were used to designate contributors for each method (correlation: 0.01 q-value, MIMOSA2: 5% contribution, MIMOSA1: correlation >0.25 and q-value <0.1, MelonnPan: non-zero model weight, RF: Altman P-value <0.1). (C) Precision-recall curves for identifying key contributors for the same set of metabolites in simulated Datasets 1 and 2 as in panel (B), by MIMOSA2 and the same alternative correlation-based methods. Colors for each method are the same as labeled in panel (B). (D) Precision, specificity and sensitivity of MIMOSA2 analysis for recovering true key microbial contributors to metabolite variation [as in panel (B)], but for all metabolites. All thresholds are the same as above. (E) Precision-recall curves for identifying key taxon–metabolite contributors in simulated Datasets 1 and 2, as in panel (C), but for all metabolites
We first evaluated the ability of MIMOSA2 to identify true microbiome-governed metabolites in these datasets. We found that MIMOSA2 identifies these metabolites with somewhat low sensitivity but very high precision, especially in the higher-complexity dataset. Specifically, in Dataset 1, 50 of 85 simulated metabolites were true microbiome-governed metabolites (i.e. at least 10% of variation in their concentrations is determined by the microbiome as opposed to external fluctuations). Using a significance cutoff of P < 0.1, MIMOSA2-OLS recovered 19 of these metabolites with only 3 false positives (86% precision), and MIMOSA2-rank recovered 17 with only 2 false positives (89% precision). A large share of the metabolites missed by either MIMOSA2 method (n = 19) were not able to be analyzed at all by MIMOSA2 due to a lack of linked non-reversible reactions. MIMOSA version 1 had a higher false positive rate, as it identified 32 metabolites as consistent with metabolic potential of which 21 were true microbiome-governed metabolites (65.6% precision). Using significant taxon-metabolite correlations (q-value <0.01) as a basis for inferring microbiome-governed metabolites resulted in similar performance, correctly identifying 30 of the 50 metabolites as microbiome-governed, with 2 false positives; as did MelonnPan (37 correct with 2 false positives) and the random forest method (36 correct with 2 false positives). In Dataset 2, nearly all simulated metabolites were classified as true microbiome-governed compounds: 193 of 221. MIMOSA2 again identified these with high precision: 73 metabolites were detected by MIMOSA2-rank, with 0 false positives (MIMOSA2-OLS, 68 with 0 false positives), out of 156 analyzed metabolites, while MIMOSA 1 identified 93 microbiome-governed compounds and 6 false positives (93.9% precision), MelonnPan identified 99 with 4 false positives (96.1%) and the random forest method identified 158 with 0 false positives. We investigated factors that may influence MIMOSA2’s ability to detect metabolites as microbiome-governed, and found that larger numbers of taxa capable of producing or utilizing a metabolite were negatively associated with predictability (Supplementary Fig. S3). However, a subset of metabolites modified by large numbers of taxa are still correctly identified as microbiome-governed in both datasets, and the compounds linked to many taxa and not detected are often abundant and universal metabolites, such as phosphate, which are accordingly less likely to have substantial interindividual variation attributable to microbiome composition. The larger share of metabolites identified as microbiome-governed by MIMOSA2-rank compared to MIMOSA2-OLS suggests that it may be able to detect microbial contributions with higher sensitivity in complex datasets. Overall, MIMOSA2 displayed high precision and lower sensitivity than other methods in identifying microbiome-governed metabolites. This result is somewhat expected, since MIMOSA2’s sensitivity is constrained by the quality of reference reaction information used to calculate metabolic potential.
A central advantage of MIMOSA2 is its ability to link metabolites with microbial taxa and mechanisms that appear to be responsible for the observed variation. Notably, MIMOSA2 only aims to identify contributors for metabolites that are predicted well by its model, and this prioritization of metabolites that are meaningfully associated with microbial profiles is an important advantage of the framework. Therefore, we focused our evaluation on each method’s performance in identifying key microbial contributors to metabolites for only the set of metabolites that were detected as microbiome-governed by either MIMOSA2-rank and/or MIMOSA2-OLS (22 from Dataset 1, 78 from Dataset 2), although results of the same evaluation across all metabolites are shown in Figure 4D and E. Key microbial contributors were identified with comparable sensitivity and higher specificity and precision overall by MIMOSA2 than any alternative approach at standard significance cutoffs (Fig. 4B and D). Specifically, both MIMOSA2 methods outperform all other methods across a range of significance thresholds, particularly at low recall thresholds and in the noisier and more diverse Dataset 2 (Fig. 4C and E), highlighting that methods optimized to predict metabolite abundances from microbiome data (such as MelonnPan) may not be optimal for precise identification of the key microbial players. We further confirmed that the MIMOSA2-OLS and MIMOSA2-rank models are not overfitted by performing the same evaluations on datasets with permuted sample labels (Supplementary Text and Fig. S2). Overall, these results, including MIMOSA2’s higher precision and lower sensitivity, can be understood in light of its reliance on an approximate metabolic model that acts as a filter for spurious associations, but cannot detect mechanisms that are not described by the model.
3.2 MIMOSA2 results compare favorably with experimentally inferred microbe–metabolite links in insect microbiomes
We next applied MIMOSA2 to re-analyze a publicly available dataset of taxonomic and metabolomics measurements from the gut microbiota of the honeybee Apis mellifera (Kešnerová et al., 2017). This dataset consists of samples from medium-complexity natural communities as well as monocolonization experiments, which allowed us to compare MIMOSA2’s inferences of taxon–metabolite links with independent experimental data. We applied MIMOSA2 to a set of 18 samples from gnotobiotic bees, each colonized with a synthetic community of resident gut microbial strains. We then compared MIMOSA2’s inference with metabolomics data from microbiota-depleted (i.e. nearly germ-free) and monocolonized samples (Fig. 5A). Specifically, we used the metabolomics data from microbiota-depleted and monocolonized samples to define experimentally inferred microbiome-governed metabolites (EIMM) and key contributing strains. We defined EIMM as those that differed in colonized bee samples compared with microbiota-depleted control bee samples. Similarly, experimentally inferred key contributor strains (EIKC) were defined by comparing metabolite levels in bees monocolonized with each individual gut strain with levels from community-colonized bees, following the approach used in the original study (see Supplementary Text). We compared these experimentally inferred metabolites and contributions with the results of MIMOSA2-rank applied to just the set of 18 community samples (Fig. 5A).
Fig. 5.
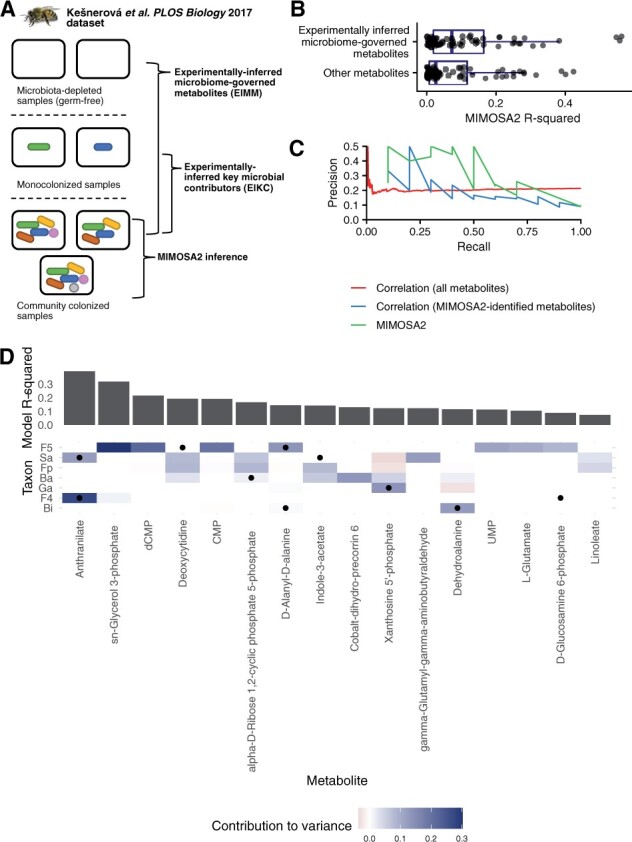
Partial overlap between MIMOSA2 results and experimental inferences in honeybee gut microbiota. (A) Summary of experiments from Kešnerová et al. (2017) reanalyzed here. MIMOSA2 was applied to a dataset of 18 samples from community-colonized bees, and its results were compared with inferences from metabolomics of microbiota-depleted (germ-free) bees and bees monocolonized with individual bacterial strains. (B) Experimentally inferred microbial metabolites in 11-strain communities are significantly better predicted by MIMOSA2 than other metabolites. (C) MIMOSA2 identifies experimentally inferred microbial contributors with higher precision and recall than microbe–metabolite correlation analysis. (D) Metabolite-level comparison of experimentally inferred and MIMOSA2-inferred microbial key contributors. Cell color indicates a microbe’s contribution to variance in a metabolite as inferred by MIMOSA2; black dots indicate experimentally inferred contributors based on metabolomics of monocolonized samples. Metabolites shown are microbiome-governed as determined by the MIMOSA2 model. Ba, Bartonella apis; Bi, Bifidobacterium asteroides; F4, Lactobacillus Firm-4; F5, Lactobacillus Firm-5; Fp, Frischella perrara; Ga, Gilliamella apicola; Sa, Snodgrassella alvi
MIMOSA2 results were largely consistent with experimental findings. The abundances of EIMM were predicted with higher accuracy by MIMOSA2-rank than other metabolites (Fig. 5B) (P = 0.04, Wilcoxon rank-sum test), suggesting that MIMOSA2 correctly identified metabolites affected by microbial composition. Strikingly, MIMOSA2-inferred key contributors were 6.02 times more likely than expected by chance to also be EIKC (Fisher exact test, P = 0.0099). MIMOSA2-identified contributors were also more predictive of EIKC than correlation-based contributors: MIMOSA2 predicted EIKC with 23% precision and an area under the ROC curve (AUC) of 0.76 among microbiome-governed metabolites, compared with 20.6% precision and AUC of 0.68 for correlation analysis in the same set of metabolites. Across all EIMM (i.e. including those not identified as microbiome-governed by MIMOSA2), microbe-metabolite correlations were strikingly not predictive of EIKC, with a precision of 18.8% and AUC of 0.52 for correlation magnitude across all EIMM (Fig. 5C), suggesting that metabolite shifts in this dataset are not well described by univariate associations. Filtering correlations based on genomic potential of the associated strain only slightly improved these values (precision 21.7%, AUC 0.53).
The contributors identified by both methods are shown for microbiome-governed metabolites in Figure 5D. These span a variety of metabolic categories including vitamins, amino acid metabolites, fatty acids and nucleoside metabolites, which were noted as a particularly enriched category in the original study (Kešnerová et al., 2017). Notably, although some of these metabolites could theoretically be produced or modified by both the host and the microbiota, the MIMOSA2 analysis provides evidence that variation in their levels is best explained by shifts in microbial metabolism. For example, the metabolite best explained by MIMOSA2, anthranilate, is an aromatic compound in the tryptophan biosynthesis pathway, and variation in its levels is attributed to the microbes Lactobacillus Firm-5 and Snodgrassella alvi, coinciding exactly with the contributors identified in the monocolonization data. It should also be noted that discrepancies observed between experimental inferences and MIMOSA2 findings for other metabolites are not necessarily due to inaccurate inference by MIMOSA2, but could alternatively indicate context-dependent metabolism, in which the metabolic effects of a microbe differ between a community setting and monocolonized samples. For example, the strong contribution to sn-glycerol-3-phosphate by Lactobacillus Firm-5 inferred by MIMOSA2 could indicate a true effect, but one that occurs at lower levels in isolation than in a complex community. Overall, these results show that insights on the metabolic contributions of specific members of the honeybee gut microbiota, which were obtained in the original study through extensive microbial depletion and monocolonization experiments, can be partially recapitulated by MIMOSA2 using metabolic models and interindividual variation across a small number of community samples.
3.3 MIMOSA2 analysis of human gut microbiota identifies potential microbial modulators of disease-associated metabolites
To demonstrate the use of MIMOSA2 in a more complex setting, we applied it to two large datasets describing the fecal microbiome and metabolome of patients with inflammatory bowel disease (IBD) and healthy controls (Franzosa et al., 2018; IBDMDB Investigators et al., 2019) (Supplementary Text). Notably, although these datasets have hundreds of samples, species and metabolites, MIMOSA2 runs quickly (Supplementary Fig. S4), as the community metabolic network model only needs to be constructed once and the workflow fits a single parameter for each metabolite.
A relatively small number of metabolites were identified as microbiome-governed in these datasets (eight for Franzosa and six for IBDMDB) (Fig. 6A and B), likely due to the heterogeneous and dynamic gut environment. However, the identified compounds, including lactate, fumarate and bile acids, are generally consistent with prior knowledge, and some were reported as associated with specific taxa in the original studies. Interestingly, the compounds pyrocatechol and 4-methylcatechol are decreased in subjects with IBD in both datasets, and both were identified as microbiome-governed in at least one dataset. Catechol compounds, which may be derived from the diet or from microbial sources and can have a range of biological functions (Maini Rekdal et al., 2020), were predicted by MIMOSA2 on the basis of shifts in abundance of catechol 2,3-dioxygenase ring-opening enzymes in Streptococcus and Enterococcus. Because the reactions catalyzed by these enzymes are oxygen-dependent, this result suggests catechol depletion could be a possible metabolic consequence of increased barrier permeability and oxidative stress in IBD (Bourgonje et al., 2020).
Fig. 6.
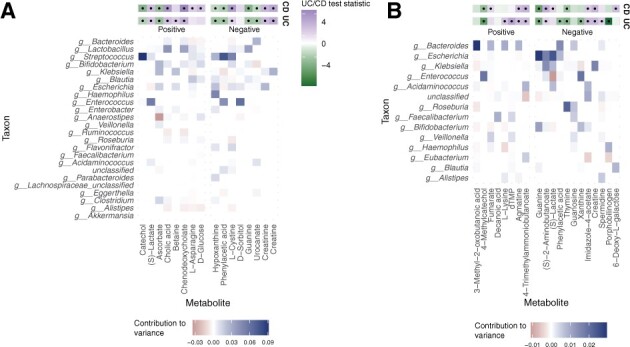
Taxonomic contributors to microbiome-governed metabolites in two studies of IBD. (A) Heatmap of genus-level microbial contributors to fecal metabolites identified by MIMOSA2-OLS in Franzosa et al. (2018). The upper two bars show the association of each metabolite with Crohn’s disease (CD) and/or ulcerative colitis (UC), with a black dot indicating a significant association (FDR-adjusted P<0.1). Metabolites with a false discovery rate-adjusted P-value <0.25 are shown. (B) Heatmap of genus-level microbial contributors to fecal metabolites in IBDMDB Investigators et al. (2019). As in panel (A), the upper two bars show association with CD and UC (same color scale), and metabolites with an FDR-adjusted P-value <0.25 are shown
Several compounds were also negatively predicted by metabolic potential (Fig. 6A and B), including phenylacetate in both datasets. While negative associations can occur for multiple reasons [as we previously showed with MIMOSA version 1 (Snijders et al., 2016)], in this setting a strong association may suggest that the metabolite is microbiome-governed but the utilization and synthesis reactions and their distribution across taxa may not be represented well by the metabolic network model. Indeed, the KEGG model used for this analysis included two phenylacetate production reactions and two utilization reactions, distributed broadly across 44 different taxa, suggesting that custom curation of the linked genes and reaction annotations may improve the model.
4 Conclusions
Here, we have described MIMOSA2, a comprehensive software framework for generating and evaluating mechanistic metabolic hypotheses from microbiome–metabolome datasets. MIMOSA2 evaluates whether metabolite levels across a set of microbial communities are consistent with the communities’ estimated metabolic capacities. The software has multiple interfaces, is compatible with several microbiome data formats, and can provide evidence of metabolic mechanisms from varied microbiomes measured in their natural context. The MIMOSA2 framework can assess possible microbial causes of differing metabolite phenotypes between health and disease (Sharon et al., 2019), and can evaluate the metabolic consequences of microbiome shifts across lifestyle or environmental factors (Snijders et al., 2016).
MIMOSA2 supports a variety of analysis options with distinct benefits and caveats. For example, 16S rRNA amplicon sequencing datasets are cost-accessible and easy to link to reference databases, but sequence divergence in this gene is imperfectly linked to metabolic and phenotypic divergence (Bauer et al., 2015). Nevertheless, a large share of functional information can be accurately predicted from 16S rRNA amplicon datasets (Langille et al., 2013; Mallick et al., 2019). Additionally, the use of metatranscriptomic data may provide better information on metabolic activity compared to metagenomics or amplicon sequencing, but it also presents difficulties in data acquisition and processing that may outweigh these benefits in some settings (Yin et al., 2020). The best database options may be similarly context dependent—e.g. the AGORA v1.0.3 database (Magnúsdóttir et al., 2017) contains high-quality metabolic network reconstructions, but only for 818 microbial species commonly found in the human gut.
Although our validation analyses have demonstrated that MIMOSA2 infers true microbe–metabolite links with high precision, it does have several limitations. First, a strong statistical relationship between CMP and metabolite concentrations can arise for reasons other than direct microbial production, including selection, regulation and differences in host metabolism or the surrounding environment. This possibility is more likely if the MIMOSA2 metabolic model is a poor representation of the core metabolic activities of the community (i.e. in environments not well represented in reference databases). Moreover, MIMOSA2 currently uses a relatively simple approach to calculate CMP. Therefore, its ability to detect microbiome effects involving large numbers of taxa may be lower (Supplementary Fig. S3), and it is not optimized to handle dynamic changes in metabolic activity or non-monotonic relationships between microbes and metabolites. Finally, we have not fully evaluated how MIMOSA2 performance may be affected by other properties of microbiome–metabolome studies, including sample size and number of metabolites, although we expect these to have relatively small effects on performance given the structure of the model and workflow.
Notably, a diverse set of tools for microbiome–metabolome analysis are now available, with different approaches, aims and priorities. These tools include machine-learning methods to predict the metabolome from the microbiome (Mallick et al., 2019; Morton et al., 2019), methods to construct detailed constraint-based community metabolic models (Baldini et al., 2018), resources to discover links between biosynthetic gene clusters and specialized metabolites (Schorn et al., 2021) and methods to annotate metabolites based on microbial genes (Shaffer et al., 2019). MIMOSA2 fills a unique niche, performing a generalizable and user-friendly analysis that generates hypotheses about metabolic mechanisms by quantitatively synthesizing microbiome and metabolome data with reference knowledge.
Availability of data and materials
The simulated microbiome–metabolome datasets analyzed in this study and the code used to generate them are available from http://www.borensteinlab.com/pub/mmcorr/SimulationData.zip. The honeybee microbiota data are available from the Supplementary Data S1 and S2 of Kešnerová et al. (2017). Metadata and processed metabolomics data from Franzosa et al. (2018) were obtained from the publication’s supplementary data, and metagenomic data were obtained from NCBI SRA PRJNA400072. The data described in IBDMDB Investigators et al. (2019) was obtained from the IBDMDB data portal (https://ibdmdb.org/tunnel/public/summary.html). The processed versions of the AGORA and embl_gems databases generated for use in the MIMOSA2 software are available at http://elbo-spice.cs.tau.ac.il/shiny/MIMOSA2shiny/refData/. The processed version of KEGG data can be provided upon request with confirmation of a KEGG subscription. The code for processing raw KEGG files is available at www.github.com/borenstein-lab/MIMOSA2shiny.
Supplementary Material
Acknowledgements
We thank members of the Borenstein lab for suggestions and for software testing and support.
Funding
This work was supported in part by National Institutes of Health [Grant 1R01GM124312 to E.B.]; National Institutes of Health [Grants R01DK095869, U19AG057377]; and Israel Science Foundation [Grant 2435/19 to E.B.]. E.B. is a Faculty Fellow of the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. E.M. is supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University.
Conflict of Interest: none declared.
Contributor Information
Cecilia Noecker, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.
Alexander Eng, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.
Efrat Muller, Blavatnik School of Computer Science, Tel Aviv University, Tel Aviv 6997801, Israel.
Elhanan Borenstein, Blavatnik School of Computer Science, Tel Aviv University, Tel Aviv 6997801, Israel; Department of Clinical Microbiology and Immunology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv 6997801, Israel; Santa Fe Institute, Santa Fe, NM 87501, USA.
References
- Adamovsky O. et al. (2020) Evaluation of microbiome-host relationships in the zebrafish gastrointestinal system reveals adaptive immunity is a target of bis(2-ethylhexyl) phthalate (DEHP) exposure. Environ. Sci. Technol., 54, 5719–5728. [DOI] [PubMed] [Google Scholar]
- Baldini F. et al. (2018) The Microbiome Modeling Toolbox: from microbial interactions to personalized microbial communities. Bioinformatics, 35, 2332–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer E. et al. (2015) Phenotypic differentiation of gastrointestinal microbes is reflected in their encoded metabolic repertoires. Microbiome, 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghini F. et al. (2021) Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife, 10, e65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgonje A.R. et al. (2020) Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends Mol. Med., 26, 1034–1046. [DOI] [PubMed] [Google Scholar]
- Casero D. et al. (2017) Space-type radiation induces multimodal responses in the mouse gut microbiome and metabolome. Microbiome, 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas G.M. et al. (2020) PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol., 38, 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa E.A. et al. (2018) Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol., 4, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza D.R. et al. (2018) Towards predicting the environmental metabolome from metagenomics with a mechanistic model. Nat. Microbiol., 3, 456–460. [DOI] [PubMed] [Google Scholar]
- Hazen T.C. et al. (2010) Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science, 330, 204–208. [DOI] [PubMed] [Google Scholar]
- Hoyles L. et al. (2018) Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome, 6, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C. et al. (2012) Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBDMDB Investigators. et al. (2019) Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature, 569, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilhan Z.E. et al. (2019) Deciphering the complex interplay between microbiota, HPV, inflammation and cancer through cervicovaginal metabolic profiling. EBioMedicine, 44, 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasubuchi M. et al. (2015) Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients, 7, 2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kešnerová L. et al. (2017) Disentangling metabolic functions of bacteria in the honey bee gut. PLoS Biol., 15, e2003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloke J.D., McKean J.W. (2012) Rfit: rank-based estimation for linear models. R J., 4, 57–64. [Google Scholar]
- Langille M.G.I. et al. (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol., 31, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado D. et al. (2018) Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res., 46, 7542–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S. et al. (2017) Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol., 35, 81–89. [DOI] [PubMed] [Google Scholar]
- Maini Rekdal V. et al. (2020) A widely distributed metalloenzyme class enables gut microbial metabolism of host- and diet-derived catechols. Elife, 9, e50845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick H. et al. (2019) Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun., 10, 3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D. et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J., 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire K.L., Treseder K.K. (2010) Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol. Biochem., 42, 529–535. [Google Scholar]
- McHardy I.H. et al. (2013) Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome, 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton J.T. et al. (2019) Learning representations of microbe–metabolite interactions. Nat. Methods, 16, 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller E. et al. (2021) A meta-analysis study of the robustness and universality of gut microbiome-metabolome associations. Microbiome, 9, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noecker C. et al. (2016) Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems, 1, e00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noecker C. et al. (2019) Defining and evaluating microbial contributions to metabolite variation in microbiome-metabolome association studies. mSystems, 4, e00579-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen H.K. et al. (2018) A computational framework to integrate high-throughput ‘-omics’ datasets for the identification of potential mechanistic links. Nat. Protoc., 13, 2781–2800. [DOI] [PubMed] [Google Scholar]
- Quast C. et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res., 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman D. et al. (2021) MiMeNet: exploring microbiome-metabolome relationships using neural networks. PLoS Comput. Biol., 17, e1009021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorn M.A. et al. (2021) A community resource for paired genomic and metabolomic data mining. Nat. Chem. Biol., 17, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer M. et al. (2017) Microbiome and metabolome data integration provides insight into health and disease. Transl. Res., 189, 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer M. et al. (2019) AMON: annotation of metabolite origins via networks to integrate microbiome and metabolome data. BMC Bioinformatics, 20, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon G. et al. (2019) Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell, 177, 1600–1618.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders A.M. et al. (2016) Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat. Microbiol., 2, 16221. [DOI] [PubMed] [Google Scholar]
- Stewart C.J. et al. (2017) Associations of nasopharyngeal metabolome and microbiome with severity among infants with bronchiolitis. A multiomic analysis. Am. J. Respir. Crit. Care Med., 196, 882–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöcs E. et al. (2020) webchem: an R package to retrieve chemical information from the web. J. Stat. Softw., 93, 1–17. [Google Scholar]
- Theriot C.M. et al. (2014) Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun., 5, 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A., Palsson B.O. (1994) Metabolic flux balancing: basic concepts, scientific and practical use. Bio/Technology, 12, 994–998. [Google Scholar]
- Wohlgemuth G. et al. (2010) The chemical translation service–a web-based tool to improve standardization of metabolomic reports. Bioinformatics, 26, 2647–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X. et al. (2020) A comparative evaluation of tools to predict metabolite profiles from microbiome sequencing data. Front. Microbiol., 11, 595910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The simulated microbiome–metabolome datasets analyzed in this study and the code used to generate them are available from http://www.borensteinlab.com/pub/mmcorr/SimulationData.zip. The honeybee microbiota data are available from the Supplementary Data S1 and S2 of Kešnerová et al. (2017). Metadata and processed metabolomics data from Franzosa et al. (2018) were obtained from the publication’s supplementary data, and metagenomic data were obtained from NCBI SRA PRJNA400072. The data described in IBDMDB Investigators et al. (2019) was obtained from the IBDMDB data portal (https://ibdmdb.org/tunnel/public/summary.html). The processed versions of the AGORA and embl_gems databases generated for use in the MIMOSA2 software are available at http://elbo-spice.cs.tau.ac.il/shiny/MIMOSA2shiny/refData/. The processed version of KEGG data can be provided upon request with confirmation of a KEGG subscription. The code for processing raw KEGG files is available at www.github.com/borenstein-lab/MIMOSA2shiny.