

Summary
Genetic constraint metrics such as the gnomAD probability of being loss-of-function (LoF) intolerant (pLI) are used to prioritize candidate genes but the mode of inheritance of highly constrained genes has never specifically been studied. We compared 605 genes with a pLI of 1 (pLI1 group) with a random sample of 635 genes from gnomAD (the random group) in terms of genetic constraint metrics, associations with Mendelian disease, modes of inheritance, and two intragenic constraint scores: the percentage of constraint coding regions (CCR) in the 99th percentile and the gene variation intolerance rank (GeVIR). The proportion of genes associated with a Mendelian disease was 35.9% (217/605) in the pLI1 group and 19.5% (124/635) in the random group (p < 0.0001). The modes of inheritance in the random group were autosomal dominant for 35 genes (28.2%), autosomal recessive for 69 (55.6%), mixed for 14 (11.3%) and X-linked for 6 genes (4.8%). The corresponding distribution in the pLI1 group was 150 (69.1%), 26 (12.0%), 14 (6.5%) and 27 (12.4%) (p < 0.0001). The percentage of CCRs in the 99th percentile was 0.3 in the random group versus 1.12 in the pLI1 group (p < 0.0001). The GeVIR score was 50.9 for the random group versus 15.1 for the pLI1 group (p < 0.0001). High genetic constraint does not seem to be associated with a particular mode of inheritance but does seem to be associated with the intragenic constraint scores considered here. Some highly constrained genes are associated with two different modes of inheritance.
Keywords: Mendelian inheritance, pLI, gnomAD, ExAC
1. Introduction
The Exome Aggregation Consortium (ExAC) database, created in October 2014, contains exome sequence data from 60 706 individuals and has rapidly become an essential tool in the study of Mendelian diseases (1). The ExAC database has allowed levels of genetic constraint to be estimated (2) and a popular metric is the probability of loss-of-function (LoF) intolerance (pLI). The pLI ranges from 0 to 1 and genes with a pLI ≥ 0.9 are very likely to be intolerant to loss-of-function variations and are often associated with haploinsufficiency and dominant genetic diseases. Despite some limitations, the pLI has been widely used to prioritize candidate genes (3). The successor of the ExAC database, the genome aggregation database (gnomAD) (4), contains more than 100,000 human exome and genome sequences along with annotations including the pLI and missense and synonymous Z-scores. Just as for the pLI, higher (more positive) Z-scores indicate greater intolerance to the corresponding type of variation. Other measures of genetic constraint derived from gnomAD data have been proposed to identify candidate genes, including the gene variation intolerance rank (GeVIR) (5) and the mapping of constraint coding regions (CCRs) in genes (6). While modes of inheritance clearly affect genetic constraints (4,7), the Mendelian mode of inheritance of highly constrained genes has never been specifically studied. The aim of this study was therefore to analyze the modes of inheritance of the most constrained genes (with a pLI of 1) in comparison with those of a random selection.
2. Material and Methods
The gnomAD constraint metric by gene table (4) containing 19,704 genes was downloaded from the gnomAD website (https://gnomad.broadinstitute.org/downloads, file "pLoF Metrics by Gene TSV") on 15 October 2019. Gene constraint metrics (pLI, missense and synonymous Z-scores) and chromosome location were extracted for the 605 genes with a pLI = 1 (the pLI1 gene group) and a random sample of 650 genes (the random gene (RG) group). Manual searches were performed for each gene on the Online Mendelian Inheritance in Man (OMIM website, https://omim.org/) between 15 October 2019 and 20 May 2020. The data retrieved were the existence of an associated Mendelian disease (non-diseases and multifactorial disorders were not included), and for each disease, the mode of inheritance (autosomal dominant, autosomal recessive, or X-linked). For genes associated with multiple phenotypes, the number of associated Mendelian diseases was also recorded and the mode of inheritance was recorded as mixed if it varied between phenotypes. The number of CCRs in the 99th percentile for each gene was obtained from Abramov et al. (5) and GeVIRs were obtained from Havrilla et al. (6).
Continuous variables were expressed as mean (standard deviation). Comparisons were made with t-tests when comparing highly constrained and randomly selected genes. Kruskal-Wallis tests were used when comparing the 4 groups according to the mode of inheritance. Chi-square test was used for comparison of categorical variables. The alpha level was set at 0.05 for all two-tailed tests. The analyses were conducted using IBM SPSS Statistics 27.0 (IBM Inc., New York, USA). Differences in gene ontology terms for biological processes, molecular function and cellular components were analyzed with Panther (http://pantherdb.org/) (8).
No ethics approval was required under French law as the study only involved data analysis. Database data were used in accordance with the corresponding data use agreements. Tables of raw data (genetic constraint, GeVIR score, Number of CCRs in 99th percentile, Mendelian mode of inheritance) are available upon request.
3. Results and Discussion
One thousand two hundred and forty genes were analyzed, 605 in the pLI1 group and 635 in the RG group (15 of the 650 randomly selected genes were removed because they had a pLI of 1 and were therefore part of the pLI1 group). Their characteristics are compared in Table 1. One hundred and fifty-nine genes were not present in the OMIM database (131 in the RG group and 18 in pLI1 group, p < 0.0001) and 342 genes were associated with at least one Mendelian disease (124 in the RG group and 217 in the pLI1 group, p < 0.0001). The groups differed significantly in terms of the distribution of modes of inheritance (AD, AR, XL or mixed; p < 0.0001), the number of CCRs in the 99th percentile (higher in the pLI1 group, p < 0.0001), the GEVIR score (lower for pLI1 genes; p < 0.0001) and borderline significantly in terms of the mean number of OMIM phenotypes per disease-associated gene (higher in the pLI1 group, p = 0.071; Table 1). The genes in both groups were first associated with a Mendelian disease in 2008 on average (Table 1).
Table 1. Gene characteristics.
| Characteristics | Highly constrained genesa | Randomly selected genes | p |
|---|---|---|---|
| Genes | 605 | 635 | |
| Present in OMIM database | 577 (95.4%) | 504 (79.4%) | < 0.0001 |
| Associated with Mendelian disease in OMIM database | 217 (37.6%) | 124 (24.6%) | < 0.0001 |
| Autosomal dominant inheritance | 150 (69.1%) | 35 (28.2%) | |
| Autosomal recessive inheritance | 26 (12%) | 69 (55.6%) | < 0.0001 |
| Mixed inheritance | 14 (6.5%) | 14 (11.3%) | |
| X linked inheritance | 27 (12.4%) | 6 (4.8%) | |
| OMIM phenotypes per disease-associated gene | 1.5 (1.3) | 1.3 (0.7) | 0.071 |
| Missense Z-Score | 3.1 (1.8) | 0.7 (1.2) | < 0.0001 |
| Synonymous Z-Score | -0.5 (2.0) | -0.3 (1.4) | 0.014 |
| Number of CCRs in 99th percentile | 1.1 (2.2) | 0.03 (0.29) | < 0.0001 |
| GeVIR score | 15.1 (14.4) | 50.9 (28.5) | < 0.0001 |
| Year of first molecular association with a Mendelian disease | 2008.8 (8.6) | 2008.0 (7.8) | 0.42 |
| Year of first molecular association with Mendelian disease for all phenotypes | 2008.0 (8.7) | 2008.1 (7.6) | 0.92 |
Data are reported as frequency (%) or mean (standard deviation). aWith a probability of loss-of-function intolerance of 1. OMIM, Online Inheritance in Man; GeVIR, gene variation intolerance rank; CCR, constraint coding region.
Considering genes with different modes of inheritance separately (Supplemental Table S1, http:// www.irdrjournal.com/action/getSupplementalData.php?ID=90), the mean missense Z-score and the number of CCRs in the 99th percentile were in each case significantly higher in the pLI1 group than in the RG group, and the mean GEVIR score was significantly lower. The first association with a Mendelian disease occurred significantly later in the pLI group for autosomal recessive diseases.
Within the pLI1 group, the variables significantly associated with the mode of inheritance were the mean GEVIR score and number of CCRs in the 99th percentile (Figure 1 and Table 2; p < 0.001 and p = 0.001 respectively), while in the RG group, the variables significantly associated with the mode of inheritance were the GEVIR score and the missense Z-score (Table 3; p < 0.001 in both cases).
Figure 1.
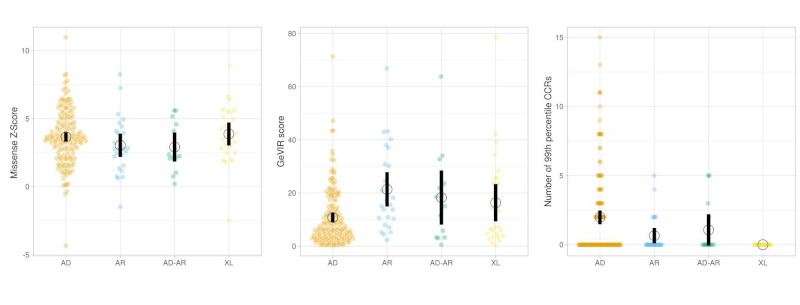
Dot plot distributions of missense Z-scores, GeVIR, number of CCRs in 99th percentile for highly constrained genes (those with a probability of loss-of-function intolerance of 1) for different Mendelian modes of inheritance. Figure prepared with https://huygens.science.uva.nl/PlotsOfData/.
Table 2. Comparison of constraint metrics for highly constrained (pLI = 1) genes in terms of their mode of inheritance.
| Characteristics | Autosomal dominant inheritance | Autosomal recessive inheritance | Mixed inheritance | X linked inheritance | p |
|---|---|---|---|---|---|
| Genes | 150 | 26 | 14 | 27 | |
| Missense Z-Score | 3.7 (2.0) | 3.0 (2.1) | 2.9 (1.8) | 3.9 (2.1) | 0.15 |
| Synonymous Z-Score | -0.9 (2.4) | -0.2 (1.4) | -0.8 (1.2) | -0.4 (1.3) | 0.46 |
| Number of CCRs in 99th percentile | 2.0 (3.1) | 0.7 (1.4) | 1.1 (1.9) | 0 | < 0.001 |
| GeVIR score | 10.8 (10.9) | 21.4 (15.8) | 18.3 (17.1) | 16.3 (17.5) | 0.001 |
Data are reported as mean (standard deviation). pLI, probability of loss-of-function intolerance; GeVIR, gene variation intolerance rank; CCR, constraint coding region.
Table 3. Comparison of constraint metrics for randomly selected genes in terms of their mode of inheritance.
| Characteristics | Autosomal dominant inheritance | Autosomal recessive inheritance | Mixed inheritance | X linked inheritance | p |
|---|---|---|---|---|---|
| Genes | 35 | 69 | 14 | 6 | |
| Missense Z-Score | 1.3 (1.5) | 0.3 (1.0) | 2.9 (1.8) | 1 (0.9) | 0.0004 |
| Synonymous Z-Score | -0.6 (1.9) | -0.6 (1.4) | -0.8 (1.2) | -0.7 (1.8) | 0.86 |
| Number of CCRs in 99th percentile | 0.06 (0.34) | 0 | 1.1 (1.9) | 0 | 0.24 |
| GeVIR score | 28.6 (25.1) | 52.5 (20.1) | 18.3 (17.1) | 41.9 (19.6) | < 0.0001 |
Data are reported as mean (standard deviation). GeVIR, gene variation intolerance rank; CCR, constraint coding region.
Among highly constrained genes (pLI1 group), those associated with a Mendelian disease did not differ significantly from those not associated with a Mendelian disease in terms of gene ontologies. Among pLI1 genes associated with a Mendelian disease, genes with autosomal dominant inheritance were significantly more likely than those with autosomal recessive inheritance to be associated with DNA binding (fold enrichment, FE = 11.2, p = 0.001) and significantly less likely to be associated with guanyl-nucleotide exchange (FE = 0.06, p = 0.004). None of the other associations between pLI1 gene ontology and mode of inheritance were statistically significant.
Considering genes with different modes of inheritance separately, there were no significant differences in terms of gene ontologies between the pLI1 and RG groups for genes with autosomal dominant or mixed inheritance. Among genes with X linked inheritance, GO:005634 (cellular component, nucleus) was significantly overrepresented (FE = 2.9; p = 0.048). Among genes with autosomal recessive inheritance, 11 gene ontologies were significantly overrepresented in the pLI1 group compared with the RG group: five biological process gene ontologies (GO:0001932: regulation of protein phosphorylation, FE = 10.9, p = 0.022; GO:0031175: neuron projection development, FE = 8.2, p = 0.018; GO:0007010: cytoskeleton organization, FE = 8.2, p = 0.018; GO:0035556: intracellular signal transduction, FE = 6.14, p = 0.048; GO:0034613: cellular protein localization FE = 6.14, p = 0.048), four molecular function gene ontologies (GO:0005096: GTPase activator activity, FE = 19.1, p = 0.014; GO:0008092: cytoskeletal protein binding, FE = 16.4, p = 0.045; GO:0005198: structural molecule activity, FE = 9.6, p = 0.042; GO:0140096: catalytic activity, acting on a protein activity, FE = 7.3, p = 0.0333), and two cellular component gene ontologies (GO:0070161: anchoring junction activity, FE = 21.9, p = 0.004; and GO:0005856: cytoskeleton, FE = 4.4, p = 0.007).
Although it has been clear from the first articles on ExAC and gnomAD that constrained genes are overrepresented in haploinsufficiency diseases, the Mendelian inheritance of the most constrained genes has never been analyzed in detail. The results of the present study confirm that highly constrained genes are mostly (69.1%) autosomal dominant, whereas randomly selected genes are mostly (55.6%) autosomal recessive. Nevertheless, around one in five highly constrained genes (18.5%) was found to be autosomal recessive, and this mode of inheritance should therefore not be ruled out even for the most constrained genes. Interestingly furthermore, a small fraction of genes were associated with two different modes of inheritance and with several OMIM phenotypes, indicating that even if a gene is associated with a phenotype and a mode of inheritance, the existence of another phenotype with a different mode of inheritance cannot be excluded either.
Compared with a random group of genes, highly constrained genes were significantly more likely to be associated with a Mendelian disease. Although it cannot be ruled out that this difference simply reflects the fact that constrained gene are more readily suspected and investigated, the data show that on average the constrained genes were not associated with diseases earlier than those in the randomly selected group, suggesting on the contrary that this result is not due to selection bias.
Genes with autosomal dominant inheritance were found to have more CCRs in the 99th percentile and lower mean GEVIR scores than autosomal recessive genes did, with the scores of mixed inheritance genes roughly half way between those of dominant and recessive genes. This suggests that exon specific metrics may be better indicators of the mode of Mendelian inheritance. However, the ranges of the scores considered here overlapped between the three modes of Mendelian inheritance. The only significant difference between autosomal dominant and autosomal recessive genes identified by the analysis of gene ontology terms was that autosomal dominant genes were more likely to be associated with DNA binding.
A possible limitation of this study is the use of pLI instead of the more recently proposed loss-of-function observed/expected upper bound fraction (LOEUF). However, since all genes with a pLI = 1 also have a LOEUF < 0.24,which is less than the proposed value for constrained gene (< 0.35) (9), these results probably hold for genes with low LOEUF scores.
The emergence of genes associated with two different modes of inheritance is intriguing. Whether continued sequencing efforts will lead to all genes being associated with two modes of inheritance or whether this will remain a property of a small subset is unclear.
In conclusion, this study shows that even the most highly constrained genes are not necessarily autosomal dominant. Gene-specific constraint scores are useful indicators of the mode of inheritance, whose precision will likely improve as genomic databases continue to expand.
Acknowledgements
Pr. Alexandre Belot for useful discussion which lead to this work. We thank Paul Guerry (GreenGrow Scientific) for editing the article.
Funding:
None.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Bennett CA, Petrovski S, Oliver KL, Berkovic SF. ExACtly zero or once: A clinically helpful guide to assessing genetic variants in mild epilepsies. Neurol Genet. 2017; 3:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ziegler A, Colin E, Goudenège D, Bonneau D. A snapshot of some pLI score pitfalls. Hum Mutat. 2019; 40:839-841. [DOI] [PubMed] [Google Scholar]
- 4. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abramovs N, Brass A, Tassabehji M. GeVIR is a continuous gene-level metric that uses variant distribution patterns to prioritize disease candidate genes. Nat Genet. 2020; 52:35-39. [DOI] [PubMed] [Google Scholar]
- 6. Havrilla JM, Pedersen BS, Layer RM, Quinlan AR. A map of constrained coding regions in the human genome. Nat Genet. 2019; 51:88-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cassa CA, Weghorn D, Balick DJ, Jordan DM, Nusinow D, Samocha KE, O'Donnell-Luria A, MacArthur DG, Daly MJ, Beier DR, Sunyaev SR. Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat Genet. 2017; 49:806-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mi H, Ebert D, Muruganujan A, Mills C, Albou LP, Mushayamaha T, Thomas PD. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021; 49:D394-D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Francioli L. gnomAD v2. https://macarthurlab.org/2018/10/17/gnomad-v2-1/ (accessed February 25, 2022).