
Figure 3: Cardiomyocyte Molecular Signalling Pathways Triggered by Ischaemia.
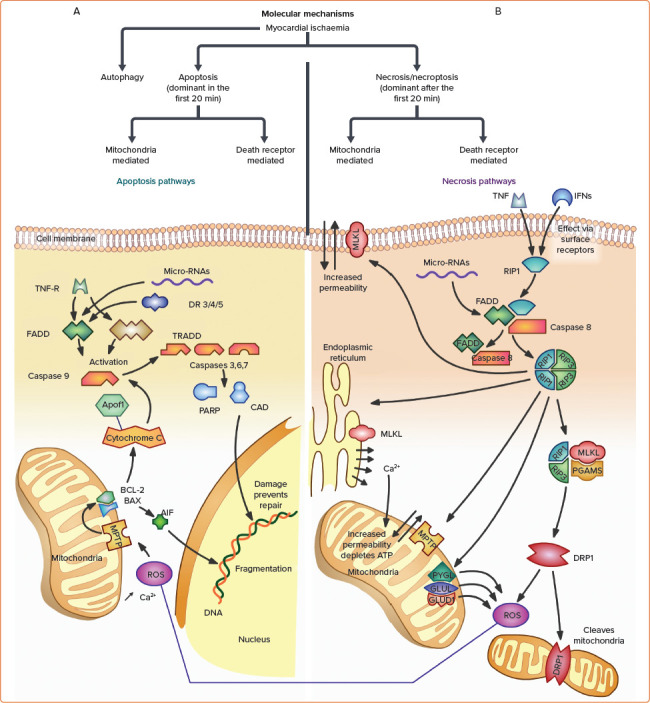
A: Cardiomyocyte apoptosis. Cellular apoptosis, like necrosis, occurs predominantly via the mitochondrial pathway mediated by ROS and internal organelle-associated calcium, and, to a lesser extent, via the death receptor pathway. In the mitochondrial apoptotic pathway, ROS and intracellular calcium trigger MPTP opening, allowing increased mitochondrial permeability. Through mediators, this activates mitochondrial membrane proteins Bax and Bcl-2, which then initiate an efflux of the two primary instigators of cell damage in apoptosis: cytochrome C and AIF.[105] AIF causes fragmentation of DNA, while cytochrome C activates Apaf-1 and forms the apoptosome, which then activates multiple caspases downstream. Proteolytic caspases degrade kinases, cytoskeletal proteins, and transcriptional regulators resulting in cell destruction. Degradation activates CAD and PARP, which both feed back to further degrade DNA along with AIF. In death-receptor-mediated apoptosis, locally secreted external proteins bind with death receptors, such as TNF-R, which leads to interactions with the proteins FADD and TRADD. This triggers a cascade involving procaspases 3, 7, and 8, ultimately converging on protein degradation with the mitochondrial pathway. Compared with necrosis, apoptosis is much more tightly regulated and involves more DNA fragmentation and cell shrinkage without significant surface cell membrane leakiness.[105] B: Cardiomyocyte necrosis. Necrosis also occurs via death receptor- and mitochondria-mediated pathways. In death receptor-mediated necrosis, TNF-α and local IFNs act at surface receptors (e.g. TNF-R) to initiate necrosome regulatory machine formation, made up of receptor-interacting protein kinases RIP1 and RIP3.[54] The necrosome complex activates RIP3 itself, leading to downstream effectors of cell death. These effectors include MLKL phosphorylation, which disturbs membrane permeability; toxic ROS production by PYGL, GLUL, and GLUD1; DRP1, which cleaves mitochondria; and opening of MPTP, which leads to mitochondria-mediated necrosis via ionic disturbances and depleted ATP.[54] MPTP can be induced by ionised calcium released through MLKL disruption above, or by ROS produced by the necrosome complex or through apoptotic pathways. During ischaemia, MPTP opening is the major cause of cell death in the first few minutes, and contributes up to 50% of infarct size.[54] There is considerable communication between the death receptor and mitochondrial necrotic pathways, and indeed between necrotic and apoptotic pathways. Common mediators are often part of positive feedback mechanisms that can quickly result in large-scale cellular death. AIF = apoptosis-inducing factor; CAD = caspase activated deoxyribonuclease; DR 3/4/5 = death receptor 3/4/5; DRP1 = dynaminrelated protein 1; FADD = Fas-associated protein with death domain; GLUD1 = glutamate dehydrogenase 1; GLUL = glutamate-ammonia ligase; IFN = interferon; MLKL = mixed-lineage kinase domain-like protein; MPTP = mitochondrial permeability transition pore; PARP = poly adenosine diphosphate-ribose polymerase; PGAMS = mitochondrial protein phosphatase; ROS = reactive oxygen species; TNF = tumour necrosis factor; TNF-R = tumour necrosis factor receptor; TRADD = tumour necrosis factor receptor type 1 associated death domain protein.