
Figure 4.
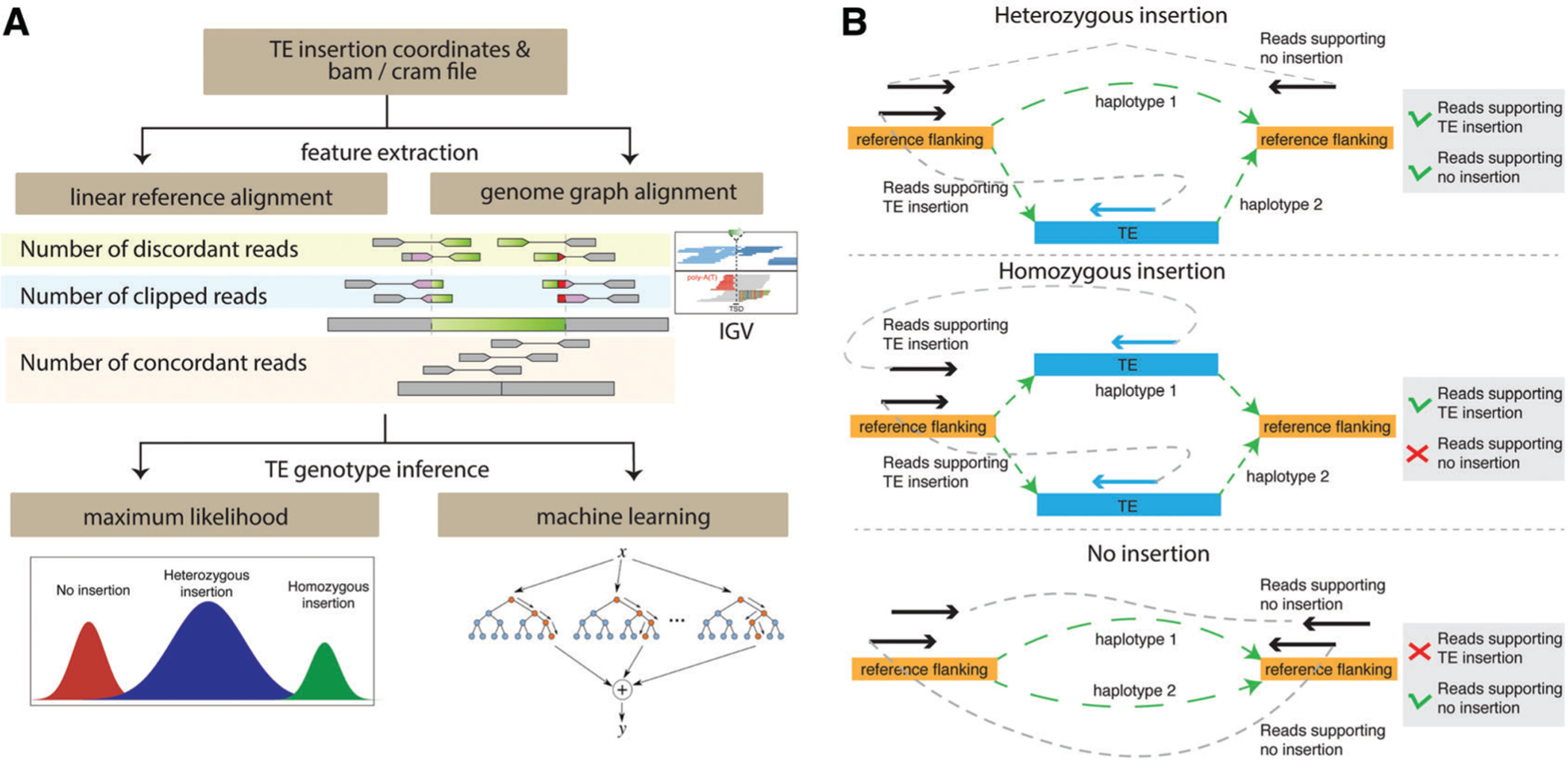
Overview of TE insertion genotyping. (A) In order to genotype TE insertions, informative features are extracted from read alignment to the linear reference genome or genome graphs. The IGV screenshot shows the local alignment patterns of a non-reference insertion with discordant and soft-clipped reads. Based on the extracted features, machine-learning or maximum-likelihood approaches are taken to estimate the genotype of each insertion. (B) Genome graph−based feature extraction is illustrated. Genome graphs represent two haplotypes of a heterozygous insertion (top), a homozygous insertion (middle), and no insertion (bottom). Using each genome graph, read pairs with one read aligned to the flanking region and the mate-pair read aligned to the TE insertion are counted as pairs supporting the presence of a TE insertion, while read pairs with both reads aligned to the reference flanking regions are counted as pairs supporting no TE insertion.