
FIGURE 2.
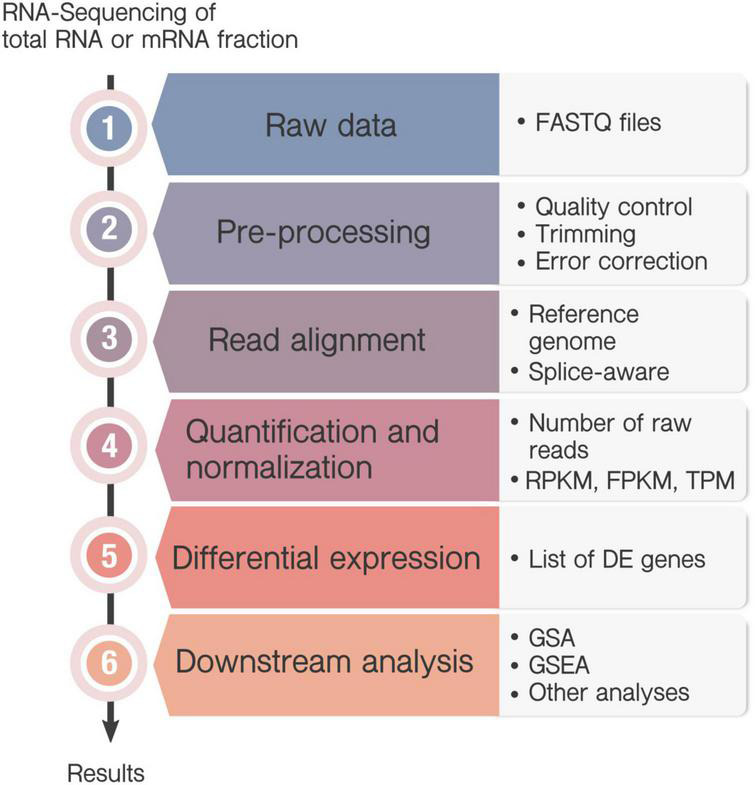
Workflow in RNA-Seq analysis. Transcriptome can be sequenced either from messenger RNA (mRNA) fraction, or total RNA, which includes also ribosomal RNA and transfer RNA. (1) RNA-sequencing generate a large amount of data from the millions of sequenced fragments (reads), and converts the information into a FASTQ file. (2) Pre-processing steps are commonly performed including quality check, trimming, filtering or error correction. (3) If an annotated genome is available, the sequenced reads are mapped onto the reference genome to identify each transcript and the correspondent gene. In this case, it is recommended to use splice-aware aligners, that align reads across splice junctions. However, if a reference genome is not available, then the reads will be assembled de novo by their overlapping regions to form contigs. (4) Next, quantification determines the number of raw reads that map to each transcript or gene and commonly normalized them to be compared between samples. The most commonly used normalizations are the “Reads Per Kilobase Million” (RPKM) or its alternative “Fragments Per Kilobase Million” (FPKM) and the “Transcripts per Kilobase” (TPM). (5) Then, differential expression (DE) analysis allows the identification of those genes whose expression change under particular circumstances indicates the gene expression profile associated to a certain condition through different statistical methods. (6) The result of a differential expression analysis is a list of DE genes that can sometimes contain hundreds or even thousands of genes. A downstream analysis is usually needed to interpret the results, as Gene Set Analysis (GSA) or Gene Set Enrichment Analysis (GSEA). Besides, there are many other options for the analysis of RNA-seq data, as the identification of Single Nucleotide Polymorphisms, or nucleotide insertions and deletions.