

Abstract
Insulin resistance is one of the earliest defects in the pathogenesis of type 2 diabetes. Over the past 50 years, elucidation of the insulin signalling network has provided important mechanistic insights into the abnormalities of glucose, lipid and protein metabolism that underlie insulin resistance. In classical target tissues (liver, muscle and adipose tissue), insulin binding to its receptor initiates a broad signalling cascade mediated by changes in phosphorylation, gene expression and vesicular trafficking that result in increased nutrient utilisation and storage, and suppression of catabolic processes. Insulin receptors are also expressed in non-classical targets, such as the brain and endothelial cells, where it helps regulate appetite, energy expenditure, reproductive hormones, mood/behaviour and vascular function. Recent progress in cell biology and unbiased molecular profiling by mass spectrometry and DNA/RNA-sequencing has provided a unique opportunity to dissect the determinants of insulin resistance in type 2 diabetes and the metabolic syndrome; best studied are extrinsic factors, such as circulating lipids, amino acids and other metabolites and exosomal microRNAs. More challenging has been defining the cell-intrinsic factors programmed by genetics and epigenetics that underlie insulin resistance. In this regard, studies using human induced pluripotent stem cells and tissues point to cell-autonomous alterations in signalling super-networks, involving changes in phosphorylation and gene expression both inside and outside the canonical insulin signalling pathway. Understanding how these multi-layered molecular networks modulate insulin action and metabolism in different tissues will open new avenues for therapy and prevention of type 2 diabetes and its associated pathologies.
Keywords: Cell-autonomous, Insulin action, Insulin resistance, iPS cells, Phosphorylation, Review, The metabolic syndrome, Tissue crosstalk, Type 2 diabetes
Introduction
The ground-breaking discovery of insulin 100 years ago [1] turned diabetes from a death sentence into a manageable condition. However, it soon became clear that most individuals with diabetes are not insulin deficient, but rather have increased insulin levels and are resistant to exogenous insulin [2]. Major breakthroughs in understanding insulin action and insulin resistance came in the early 1970s, with the demonstration of the existence of insulin receptors on the membrane of cells [3] and the subsequent recognition of their intrinsic tyrosine kinase activity [4]. Here, we review current and evolving concepts of the mechanisms of insulin signalling and how these are modified by extrinsic and intrinsic factors that underlie insulin resistance in type 2 diabetes.
Defining the insulin signalling network
Although key components involved in insulin signal transduction are present in virtually every cell, the biological outcomes following activation or disruption of this pathway are highly dependent on the cell type and physiological context (Fig. 1). In skeletal muscle, insulin promotes glucose transport and utilisation, stimulates glycogen synthesis and inhibits protein catabolism (Fig. 1a). In adipose tissue, insulin promotes glucose transport and lipogenesis and inhibits lipolysis (Fig. 1b). In liver, insulin action inhibits glucose production and fatty acid oxidation and stimulates glycogen synthesis and lipogenesis (Fig. 1c). In addition to these direct cellular effects, insulin can also regulate metabolism indirectly. For example, insulin suppression of lipolysis in fat and inhibition of protein catabolism in muscle reduces substrate supply for gluconeogenesis in the liver [5, 6]. In states of insulin resistance, all or only some of these pathways may be altered, with the exact combination leading to differing clinical presentations.
Fig. 1.
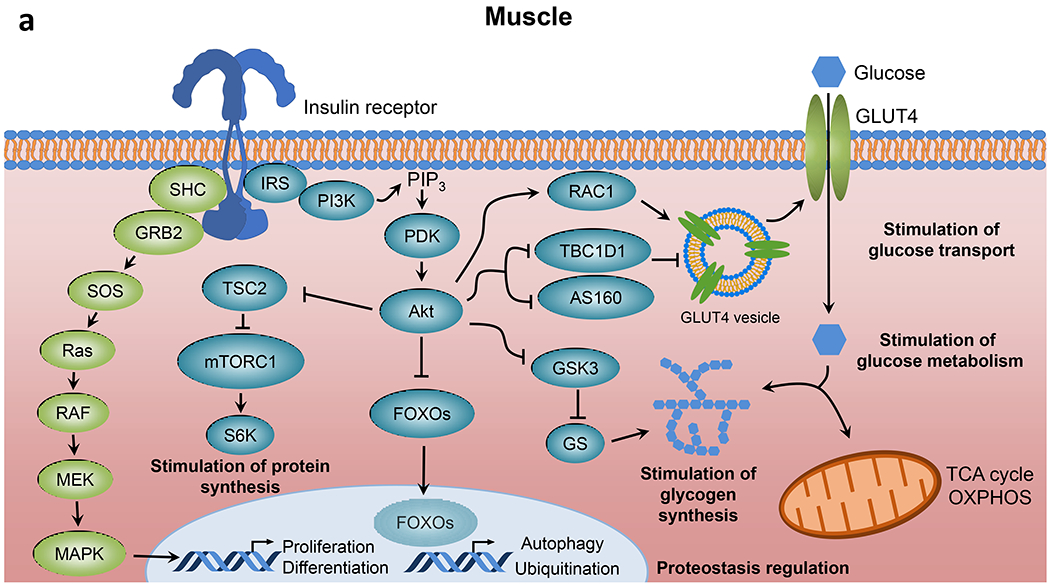
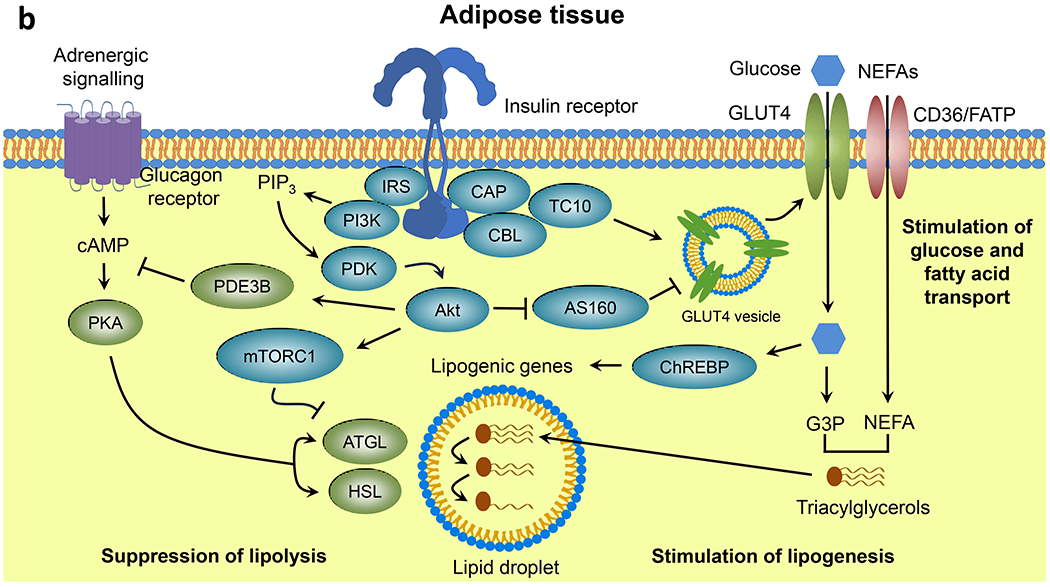
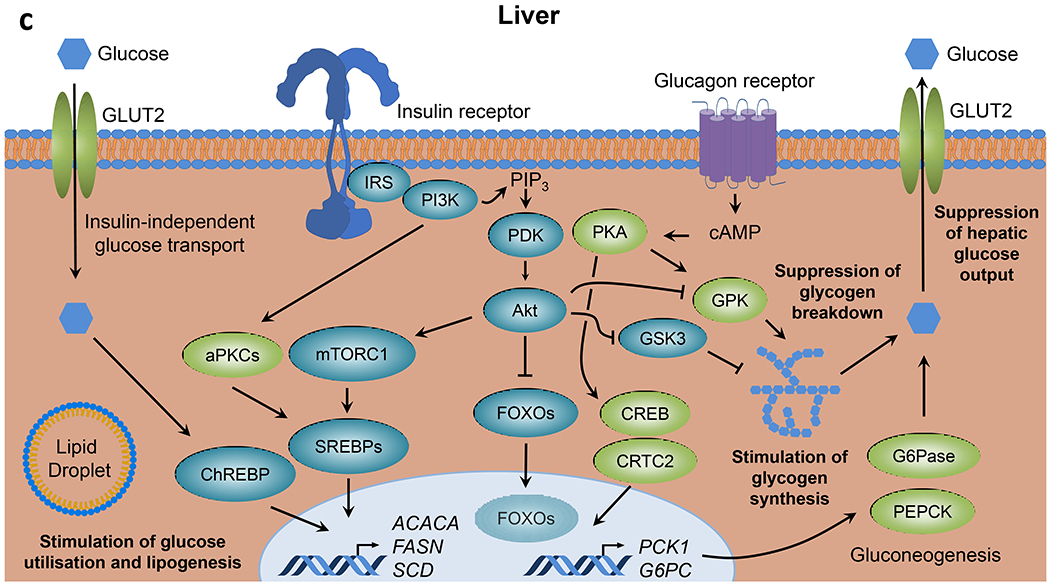
Insulin signalling in classical tissues. Insulin binding to the insulin receptor leads to activation of intrinsic tyrosine kinase activity and multisite insulin receptor and IRS phosphorylation. Tyrosine-phosphorylated IRS serves as docking sites for PI3K leading to PIP3 formation and PDK-dependent Akt activation, which in turn promotes nutrient utilisation, storage and other anabolic processes, and concomitantly suppresses catabolic pathways in (a) skeletal muscle, (b) adipose tissue and (c) liver. aPKC, atypical PKC; ATGL, adipose triglyceride lipase; CAP, Cbl-associated protein; CBL, Cbl proto-oncogene; ChREBP, carbohydrate-responsive element binding protein; CREB, cAMP responsive element binding protein; CRTC2, CREB-regulated transcription coactivator 2; FATP, long-chain fatty acid transport protein; G3P, glyceraldehyde 3-phosphate; G6Pase, glucose-6-phosphatase; GRB2, growth factor receptor bound protein 2; GS, glycogen synthase; HSL, hormone-sensitive lipase; MAPK, mitogen-activated protein kinases; MEK, MAPK kinase; OXPHOS, oxidative phosphorylation; PDE3B, phosphodiesterase 3B; PHK, phosphorylase kinase; PKA, protein kinase A; RAC1, RAC family small GTPase 1; RAF, RAF proto-oncogene serine/threonine kinase; Ras, Ras GTPase; SHC, SH2 domain containing transforming protein; S6K, p70 ribosomal S6 kinase; SOS, son of sevenless homolog; TBC1D1, TBC1 domain family member 1; TC10, Rho-related GTP binding protein RhoQ; TCA, tricarboxylic acid; TSC2, tuberous sclerosis 2. This figure is available as part of a downloadable slideset.
Insulin and IGF-1 receptors
Insulin and IGF-1 regulate growth and metabolism through binding to their cognate receptors on the cell surface. The insulin receptor and IGF-1 receptor (IGF1R) are highly homologous heterodimers composed of two α and two β subunits stabilised by disulfide bonds. These subunits are derived from single-chain proreceptors (encoded by the INSR and IGF1R genes), which are processed to the mature α2β2 receptor tetramer. The α subunits are completely extracellular and create the insulin binding sites through their three-dimensional inverted-V structure, while the transmembrane β subunits contain an intracellular tyrosine kinase domain that is required for catalytic activity and signal transduction [4, 7]. Alternative splicing of exon 11 in the INSR mRNA results in a shorter insulin receptor isoform (insulin receptor isoform A [IR-A]), which is predominantly expressed in neurons and less-differentiated cellular progenitors, and a longer isoform (insulin receptor isoform B [IR-B]), which is predominant in mature cells and tissues with prominent roles in glucose, lipid and protein metabolism. Both IR-A and IR-B display similar affinity for insulin, while IR-A has higher affinity for IGF-1 and IGF-2 than IR-B [8].
Functionally, the insulin receptor and IGF1R are members of the family of receptor tyrosine kinases. Despite their high degree of homology and many shared downstream signalling pathways, activation of each receptor results in different physiological outcomes, with the insulin receptor primarily regulating metabolic functions and IGF1R being more involved in mitogenesis and growth. Some of these functional differences are explained by distinct cellular distribution but, even in the same cell type, these receptors exert differential effects. Studies have demonstrated that these receptor-specific effects depend on differences in both the extracellular and intracellular domains of these receptors and their relative affinity for different intracellular substrates, with the insulin receptor favouring phosphorylation of IRS proteins and IGF1R favouring phosphorylation of src homology 2 (SH2) domain containing transforming protein (SHC) [9, 10].
Studies using x-ray crystallography and cryo-electron microscopy have shown that insulin/IGF-1 binding to the extracellular domains of the insulin receptor/IGF1R involves both the N- and C-terminal regions of the α-subunit, leading to conformational changes that bring together the intracellular β-subunit within each receptor [11, 12]. This leads to activation of the intrinsic tyrosine kinase, resulting in transphosphorylation of the β-subunits and phosphorylation of multiple tyrosine residues within the receptors themselves and their immediate substrates.
Insulin receptor substrates
For metabolic action, the two most important substrates are IRS1 and IRS2. Structurally, IRS proteins are defined by phosphotyrosine binding (PTB) and pleckstrin-homology domains located in the N-terminal region, which are required for their interaction with phosphorylated insulin receptor and targeting to the plasma membrane, and by multiple tyrosine residues in the mid- and C-terminal regions, which are phosphorylated by activated insulin receptor and serve as docking sites for proteins containing SH2 domains [13]. Genetic ablation in mice has shown a predominant role of IRS1 in insulin signalling in skeletal muscle and adipose tissue [14, 15] and IRS2 in liver, pancreatic beta cells and neurons [16]. Consistent with their complementary roles, MS-based phosphoproteomic studies of IRS1 and IRS2 knockout in pre-adipocytes have revealed regulation of distinct signalling pathways downstream of each substrate and suggest that IRS1/2 do not fully compensate for each other [17].
In addition to signalling through canonical substrates and downstream elements, the insulin receptor and its signalling are also regulated, both positively and negatively, by interaction with membrane and intracellular proteins. These include glycosylphosphatidylinositol (GPI)-linked proteins, such as glypican-4 [18], membrane pyrophosphatases and phosphodiesterases, such as ectonucleotide pyrophosphatase/phosphodiesterase 1 (PC-1) [19], α-arrestin adaptors [20] and, even, transcription factors [21] and cell-cycle regulators [22].
Downstream insulin signalling
The critical step linking insulin receptor activation to downstream metabolic functions of insulin is the binding of class IA phosphoinositide 3-kinase (PI3K) to tyrosine-phosphorylated IRS proteins, leading to the formation of phosphatidylinositol (3,4,5)-triphosphate (PIP3). Downstream effects of PIP3 lead to activation of 3-phosphoinositide dependent protein kinase (PDK)1 and subsequent activation of a variety of kinases, of which Akt1–3 are the best studied, but which also include p70 ribosomal S6 kinase (S6K), serum- and glucocorticoid-induced protein kinase (SGK) and protein kinase C (PKC) isoforms [23].
PI3K is a lipid kinase consisting of a catalytic subunit (either p110α, p110β or p110δ, encoded by the PIK3CA, PIK3CB and PIK3CD genes, respectively) and a regulatory subunit (either p85α [and its splice variants p55α and p50α], p85β or p55γ, encoded by PIK3R1, PIK3R2 and PIK3R3 genes, respectively) [24]. The binding of SH2 domains in the regulatory subunits to phosphotyrosines on IRS proteins reduces the constitutive inhibitory effects exerted on the catalytic subunits, leading to increased kinase activity towards phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane, resulting in PIP3 formation. PIP3 then serves as a docking site for proteins containing pleckstrin-homology domains, including Akt, PDK1 and the mechanistic target of rapamycin complex (mTORC) 2 component mitogen-activated protein kinase associated protein 1 (SIN1), which represent critical steps in downstream signalling. Thus, Akt undergoes PDK1-dependent phosphorylation at T308 within the kinase domain and mTORC2-dependent phosphorylation at S473 in a C-terminal hydrophobic motif, resulting in full kinase activation [25]. In addition to mTORC2, the Akt S473 residue is phosphorylated by DNA-dependent protein kinase (DNA-PK) [26].
A wide range of insulin’s metabolic actions are linked to Akt-dependent phosphorylation: Akt-induced phosphorylation inactivates glycogen synthase kinase-3 (GSK3)α/β, allowing dephosphorylation and activation of glycogen synthase, which increases glycogen synthesis [27]; phosphorylation of forkhead box (FOX)O transcription factors results in their nuclear exclusion, thus, inhibiting their effect on the expression of gluconeogenic genes in the liver [28, 29] and autophagy genes in muscle [30]; phosphorylation of tuberous sclerosis 2 (TSC2) and the 40 kDa proline-rich Akt substrate (PRAS40) leads to activation of mTORC1, resulting in stimulation of protein synthesis and suppression of autophagy [31, 32]; phosphorylation of TBC1 domain family member 1/Akt substrate of 160 kDa (TBC1D4/AS160) regulates trafficking of intracellular GLUT4 vesicles to the plasma membrane and increases glucose uptake [33, 34]. In addition, PI3K/Akt signalling plays a role in cell survival, proliferation and cytoskeleton organisation. While some of these actions occur through phosphorylation of targets, such as GSK3, FOXO1 and mTORC1, Akt also directly phosphorylates proteins in the apoptotic pathway (B cell lymphoma 2 [BCL2]-associated agonist of cell death [BAD], X-linked inhibitor of apoptosis [XIAP] and BCL2-interacting mediator of cell death [BIM]) and regulates cell division through phosphorylation of cyclin-dependent kinase 2 (CDK2) and the cell cycle arrest protein cyclin-dependent kinase inhibitor 1B (p27) [25].
Regulation of gene expression
The pleiotropic effects of insulin action on cell growth and metabolism result from a complex interaction between rapid phosphorylation-dependent signalling [35, 36] and slower changes in gene expression [37]. For example, the effect of insulin on glucose transport in skeletal muscle and adipocytes is dependent on the movement of pre-existing vesicles containing GLUT4 glucose transporters to the plasma membrane [38] and is dependent on AS160 phosphorylation by Akt [39], while glycogen synthesis and glycolytic and oxidative glucose metabolism are supported by increased mRNA expression of glycogen synthase 1 [40], hexokinase 2 [41] and many components of the mitochondrial electron transport chain [42]. Insulin also regulates several key mechanisms involved in gene expression, with the regulation of mRNA transcription being the best studied [43]. This important aspect of insulin action is accomplished by insulin-induced changes in phosphorylation, expression, processing and translocation of a variety of transcription factors, leading to stimulation or inhibition of gene transcription.
FOX proteins represent a large family of transcription factors, of which FOXOs (FOXO1, FOXO3, FOXO4 and FOXO6) are the most well-characterised regulators of downstream insulin signalling. Here, the effect of insulin is one of negative regulation (Fig. 2a). Upon insulin stimulation, FOXOs undergo multisite phosphorylation by Akt and/or SGK kinase. This creates interaction sites for FOXOs with phosphoserine-binding 14-3-3 proteins, resulting in their retention in the cytoplasm and decreased transcriptional activity in the nucleus [44, 45]. Thus, insulin-induced phosphorylation of FOXOs results in reduced hepatic gluconeogenesis [46], inhibition of muscle autophagy and protein degradation [30, 47] and regulation of adipocyte differentiation [48]. Because of the negative nature of insulin action on FOXO-regulated gene expression, FOXO deletion can reverse many of the metabolic abnormalities caused by insulin resistance resulting from deletion of the insulin receptor/IGF1R [30, 49] or deletion of Akt1/Akt2 [50] or those caused by streptozotocin-induced diabetes [51].
Fig. 2.
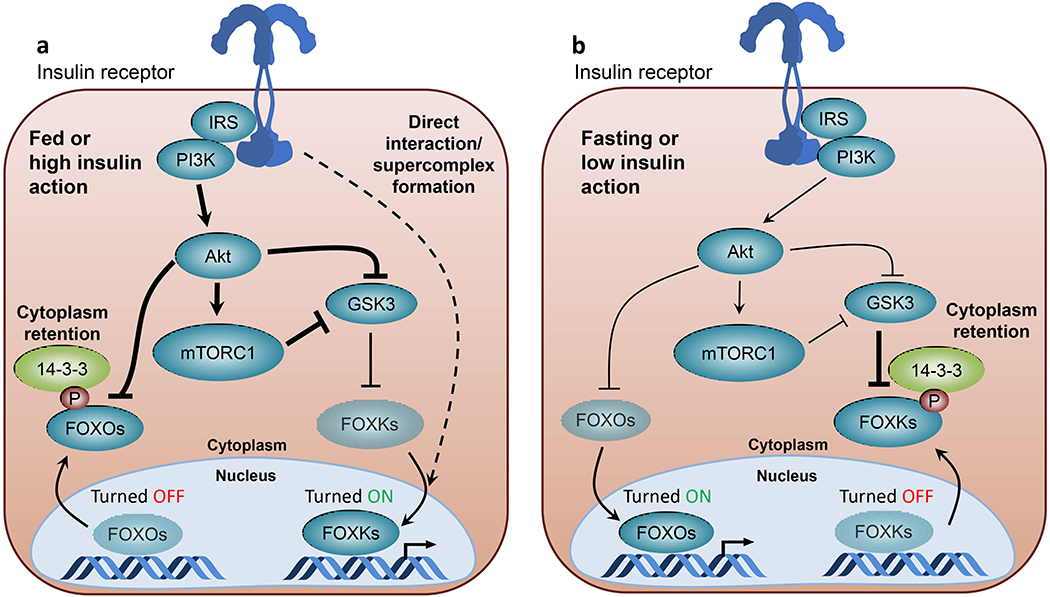
Reciprocal regulation of FOX transcription factors by insulin. (a) Under feeding or other conditions where insulin action is high, FOXOs are phosphorylated by Akt on serine residues, creating interaction sites for 14-3-3 proteins, leading to cytoplasmic retention and inhibited transcriptional activity. Under these conditions, increased Akt and mTORC1 activity inhibits GSK3 signalling and relieves FOXKs from inhibitory GSK3-mediated phosphorylation, leading to increased nuclear translocation and FOXK transcriptional activity. Since FOXKs and the insulin receptor co-immunoprecipitate together [21], it is possible that some of FOXK’s transcriptional effects are due to insulin receptor–FOXK complexes in the nucleus (dotted line). (b) Under fasting or other conditions of insulin deficiency or insulin resistance, low activity of PI3K/Akt/mTORC1 pathways results in FOXO hypophosphorylation leading to increased nuclear localisation and transcriptional activity. Under these conditions, increased GSK3 activity leads to increased FOXK phosphorylation and interaction with phosphoserine-binding 14-3-3 proteins, resulting in cytoplasmic retention and decreased transcriptional activity. Line thickness indicates strength of signalling activity, with thicker lines indicating stronger signalling activity. Faded shading of text boxes indicates lower FOXO/FOXK abundance at the nucleus or cytoplasm within in each state, and arrows between the nucleus and cytoplasm indicate the direction of translocation. This figure is available as part of a downloadable slideset.
Another emerging class of FOX proteins that act in insulin signalling are the FOXK1 and FOXK2 transcription factors [21, 52]. In contrast to FOXOs, which are turned off by insulin, FOXKs display increased nuclear localisation and transcriptional activity following insulin stimulation (Fig. 2a) and exhibit complex regulation. In the basal state, GSK3 phosphorylates FOXKs leading to increased interaction with 14-3-3 proteins and nuclear exclusion (Fig. 2b); this is reversed by insulin-induced Akt and mTORC1 activation [21, 52]. In hepatocytes, FOXKs regulate genes involved in the cell cycle, apoptosis and lipid metabolism [21], while in adipocytes and muscle, FOXKs promote glucose transport and lactate production by stimulation of glycolytic metabolism and inhibition of mitochondrial pyruvate oxidation [53].
In addition to phosphorylation, insulin also regulates the expression and processing of transcription factors. For example, sterol regulatory element binding proteins (SREBP) 1 and 2 are important regulators of triacylglycerol and cholesterol synthesis and are synthesised as precursors that reside in the endoplasmic reticulum (ER). Upon activation of Akt/mTOR pathways by insulin, these are transported to the Golgi apparatus where membrane-bound transcription factor protease, site 1/2 (SP1/SP2) mediate proteolytic cleavage, releasing the active forms of SREBP1/2, which then migrate into the nucleus and regulate the transcription of genes involved in lipid synthesis and transport [54].
A re-emerging concept in insulin control of gene expression is the possibility of direct effects of the insulin receptor itself. Studies from over 40 years ago showed binding of insulin to nuclear preparations [55]. The significance of such findings has only come to light by recent studies demonstrating interactions between the insulin receptor and FOXK1 [21] and interactions of the insulin receptor with RNA polymerase II (Pol II) on DNA in the nucleus [56]. Indeed, chromatin immunoprecipitation followed by sequencing (ChIP-seq) analysis of HepG2 hepatocytes revealed ~4000 peaks bound by the insulin receptor, many overlapping with Pol II sites on promoters. These occur in genes involved in a variety of cellular functions including lipid metabolism, translation and immunity, as well as genes involved in pathophysiological states, such as diabetes.
Insulin resistance as a central component of type 2 diabetes and the metabolic syndrome
Type 2 diabetes affects more than 400 million adults worldwide and its prevalence continues to increase at epidemic rates, thus posing one of the greatest public health challenges to society [57]. This is the result of both genetic and environmental factors. While it remains debated whether insulin resistance and relative beta cell failure constitute the primary defect in type 2 diabetes [58, 59], a 25 year prospective longitudinal study of people at high genetic risk of developing type 2 diabetes has demonstrated that insulin resistance precedes and predicts disease development [60]. Likewise, family studies have shown that glucose tolerant offspring of parents with type 2 diabetes show insulin resistance, while loss of first-phase insulin secretion was observed in those that developed impaired glucose tolerance [61]. Clamp and MRI studies have revealed skeletal muscle as a primary site of insulin resistance in the offspring of parents with type 2 diabetes, with the muscle of these individuals exhibiting reduced glucose uptake and reduced glycogen synthesis before hyperglycaemia develops [62]. This impaired glucose metabolism has been attributed to a number of defects, including decreased glucose transport [63], lower rates of insulin-induced ATP production [42] and reduced expression of genes involved in mitochondrial function [64, 65]. The major question that remains is what are the fundamental defects leading to insulin resistance and how do cell-intrinsic vs cell-extrinsic factors contribute to these defects? Cell-extrinsic factors include circulating or paracrine molecules (such as hormones, cytokines, lipids and metabolites) that are released from a cell or tissue other than the target cell/tissue, or absorbed by the intestine from the diet or microbiome action. Conversely, cell-intrinsic factors are those that persist after removal or normalisation of all extrinsic factors. These are most likely due to genetic or epigenetic effects, but may or may not be in the insulin signalling pathway itself. For example, a genetic defect that changes intracellular ATP or Mg2+ concentrations or membrane fluidity could affect insulin receptor- or kinase-mediated signalling, creating a state of insulin resistance. How each of these might contribute to insulin resistance in type 2 diabetes is discussed in the following sections.
Extrinsic factors in the pathogenesis of insulin resistance
In type 2 diabetes, most attention has focused on extrinsic factors contributing to insulin resistance, including the role of adipose tissue, circulating metabolites, inflammatory signals and the gut microbiome [66–68] (Fig. 3). In states of overnutrition, increased levels of circulating fatty acids and ectopic lipid accumulation in muscle and liver contribute to insulin resistance through the release of intermediate metabolites, such as diacylglycerols (DAG) and ceramides, that activate members of the novel PKC family (PKC δ, ε, θ), leading to increased Ser/Thr phosphorylation of the insulin receptor and IRS proteins and resulting in reduced tyrosine phosphorylation [69–73]. While IRS1 is the best studied substrate in the context of insulin resistance, Ser/Thr phosphorylation also impairs IRS2 signalling [74]. In addition, fatty acids activate Toll-like receptor 4 (TLR4) to promote activation of c-Jun N-terminal kinase (JNK) and inhibitor of κB kinase (IKK), which also increase Ser/Thr phosphorylation of IRS1, thus, reducing insulin action [75, 76]. Accumulation of ceramides can also activate protein phosphatase 2A (PP2A) and PKCζ, inhibiting Akt2. Adipose tissue expansion is also associated with increased adipose tissue inflammation and hypoxia [77], promoting recruitment of proinflammatory macrophages [78] that secrete cytokines, such as TNF-α and IL-6, which further worsen insulin resistance by activation of the TNF-α receptor (TNFR) and other cytokine receptors [79]. Cytokine signalling induces expression of suppressor of cytokine signalling (SOCS) proteins, such as SOCS1 and SOCS3, which directly bind to the insulin receptor via SH2 domains and prevent IRS1/2 tyrosine phosphorylation, as well as promoting their ubiquitination and proteasomal degradation [80]. Finally, increases in reactive oxygen species (ROS) and ER stress also occur in target tissues in states of insulin resistance, leading to activation of JNK, IKK isoforms and other Ser/Thr kinases [81, 82].
Fig. 3.
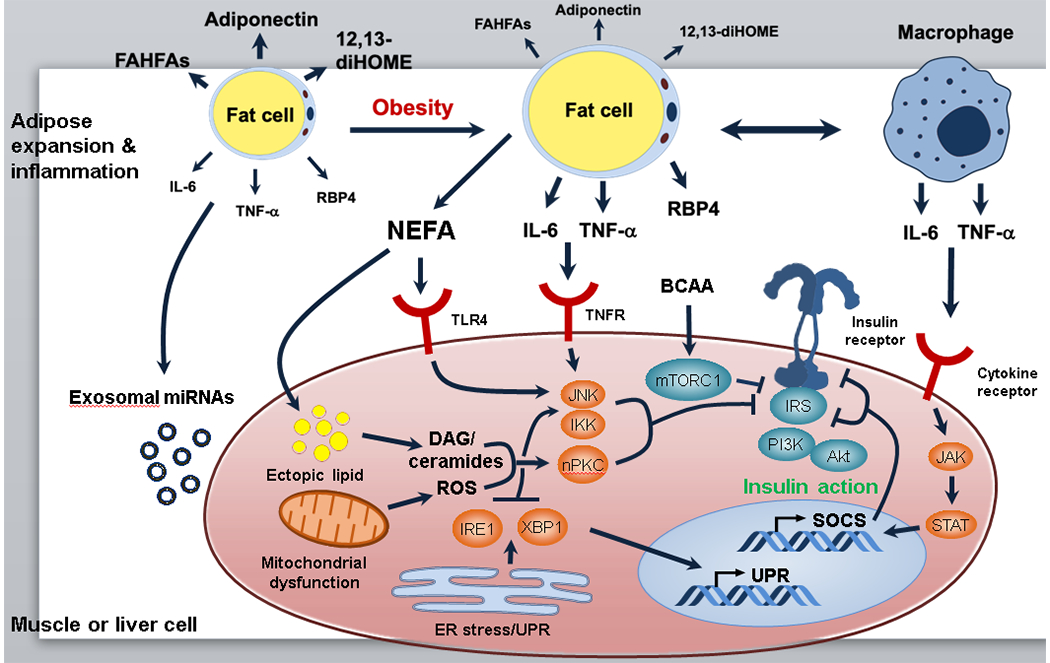
Extrinsic factors contributing to insulin resistance. Several environmental factors may lead to systemic changes affecting multiple tissues and contributing to impaired insulin signalling. Obesity negatively correlates with circulating levels of adiponectin [118] and signalling lipids with beneficial properties, such as 12,13-dihydroxy-9Z-octadecenoic acid (12,13-diHOME) [119] and branched fatty acid esters of hydroxy fatty acids (FAHFAs) [120]. Overnutrition leads to adipose tissue expansion and increased release of cytokines and other inflammatory mediators (e.g., retinol binding protein 4 [RBP4]) by macrophages and adipocytes themselves; these mediators bind to cytokine receptors on metabolic tissues and downregulate proximal insulin signalling due to activation of Ser/Thr kinases (e.g. JNK, IKK and novel PKCs [nPKCs]) and increased IRS serine phosphorylation, and due to increased transcription of SOCS proteins, which interfere with IRS tyrosine phosphorylation. Adipose tissue insulin resistance is associated with ectopic lipid accumulation, mitochondrial dysfunction and reactive oxygen species (ROS) generation, and ER stress in insulin-sensitive tissues. All of these mechanisms contribute to activation of Ser/Thr kinases and IRS serine phosphorylation. Adipose tissue expansion in obesity may also have an impact on systemic metabolism through altered release of exosomal miRNAs. Insulin signalling proteins are shown in blue and intracellular mediators of cytokine receptors and other stress signals are shown in orange. Changes in adipocyte lipid/cytokine release during obesity are indicated font/arrow size, with bigger font/thicker arrows representing increased release and smaller font/thinner arrows representing decreased release. DAG, diacylglycerol; IRE1, inositol-requiring enzyme 1; JAK, Janus kinasePI3K, phosphoinositide 3-kinase; STAT, signal transducer and activator of transcription; TLR4, Toll-like receptor 4; TNFR, TNF-α receptor; UPR, unfolded protein response; XBP1, X-box binding protein 1. This figure is available as part of a downloadable slideset.
Circulating branched-chain amino acids (BCAAs) and aromatic amino acids (isoleucine, leucine, valine, phenylalanine and tyrosine) are also associated with insulin resistance [67], and lowering BCAA levels can improve insulin sensitivity, at least in mice [83]. It is suggested that BCAAs exert these effects by activation of mTORC1, again altering Ser/Thr IRS1/2 phosphorylation [84]. Gut microbiota may also play a role in regulating BCAA supply, as well as the production of short-chain fatty acids and other metabolites, which, in turn, have an impact on systemic insulin sensitivity [85]. Thus, there are multiple potential extracellular mediators of insulin resistance, which appear to act through increasing IRS1/2 Ser/Thr phosphorylation.
Understanding how changes in Ser/Thr phosphorylation of IRS proteins regulate insulin signalling is, however, challenging because there are over 50 Ser/Thr sites on IRS1/2 and many kinases can be involved [86]. In any case, these extrinsic factors appear to act primarily as ‘progression factors’ in disease pathogenesis since none of these kinases has been linked genetically to human type 2 diabetes (although PKCδ has been linked to insulin resistance in mice [87]). Since insulin itself regulates many of these same Ser/Thr phosphorylation sites with both positive and negative effects, depending on dose and duration of stimulation, it is possible that IRS1/2 Ser/Thr phosphorylation is a normal feedback mechanism that is subsumed by these metabolic stresses to inhibit insulin signalling. Recently, we and others have shown that adipose tissue can also crosstalk with other tissues through secretion of exosomal microRNAs (miRNAs) [88, 89]; however, how this fits in the regulation of insulin sensitivity at a signalling level remains to be determined.
Intrinsic factors and cell-autonomous insulin resistance
In vitro approaches, where cells are cultured under controlled conditions, provides an opportunity to minimise the influence of extrinsic factors and isolate cell-autonomous determinants of insulin resistance, which are more closely linked to the genetic and epigenetic alterations underlying type 2 diabetes. Skeletal muscle biopsies and primary cultured myoblasts derived from people with type 2 diabetes show insulin resistance and several metabolic defects. These include impaired insulin signalling at the level of IRS1-associated PI3K activity [90] and Akt/GSK3 phosphorylation [91, 92] and decreased glucose uptake and glycogen synthesis rates [93, 94]. However, primary cell models have limited usefulness for the definition of molecular mechanisms underlying insulin resistance due to limits in expandability and ability for screening using RNA interference (RNAi), chemical genetics or CRISPR. Induced pluripotent stem cells (iPSCs) represent a step forward in this direction since these cells have unlimited potential for expansion and differentiation into multiple lineages, allowing cells from patients to be used for mechanistic studies, large-scale ‘omics’ and gene-editing approaches. Such iPSC modelling has been applied to severe insulin resistance caused by insulin receptor mutations [95–97] and other forms of genetically determined type 2 diabetes and obesity [98, 99].
Recently, we have applied the iPSC technology to study signalling defects that underlie skeletal muscle insulin resistance in type 2 diabetes [100]. Importantly, iPSC-derived myoblasts (iMyos) from individuals with type 2 diabetes show defects in insulin signalling at the level of Akt/GSK3/FOXO1 phosphorylation and decreased insulin-stimulated glucose uptake and mitochondrial respiration, similar to the defects observed in the muscle in type 2 diabetes. Global phosphoproteomics using LC-MS/MS revealed that these defects are part of a large multi-dimensional network of signalling changes involving ~1200 Ser/Thr phosphorylation sites on 725 proteins. Only a small proportion of these abnormalities are in classical insulin-regulated phosphorylations that define critical nodes in insulin action [101]. More importantly, type 2 diabetic iMyos show a large degree of perturbations in pathways outside of the canonical insulin signalling pathway and not regulated by insulin (Fig. 4). These include up- and downregulation of phosphorylation on several Rho GTPases, proteins involved in cytoskeleton remodelling and vesicle trafficking, and many nuclear proteins involved in gene transcription, mRNA splicing and/or processing and chromatin remodelling. These findings clearly open our view to a wider definition of mechanisms of insulin resistance at the molecular and cellular level that needs to be taken into account in understanding the pathogenesis of type 2 diabetes.
Fig. 4.
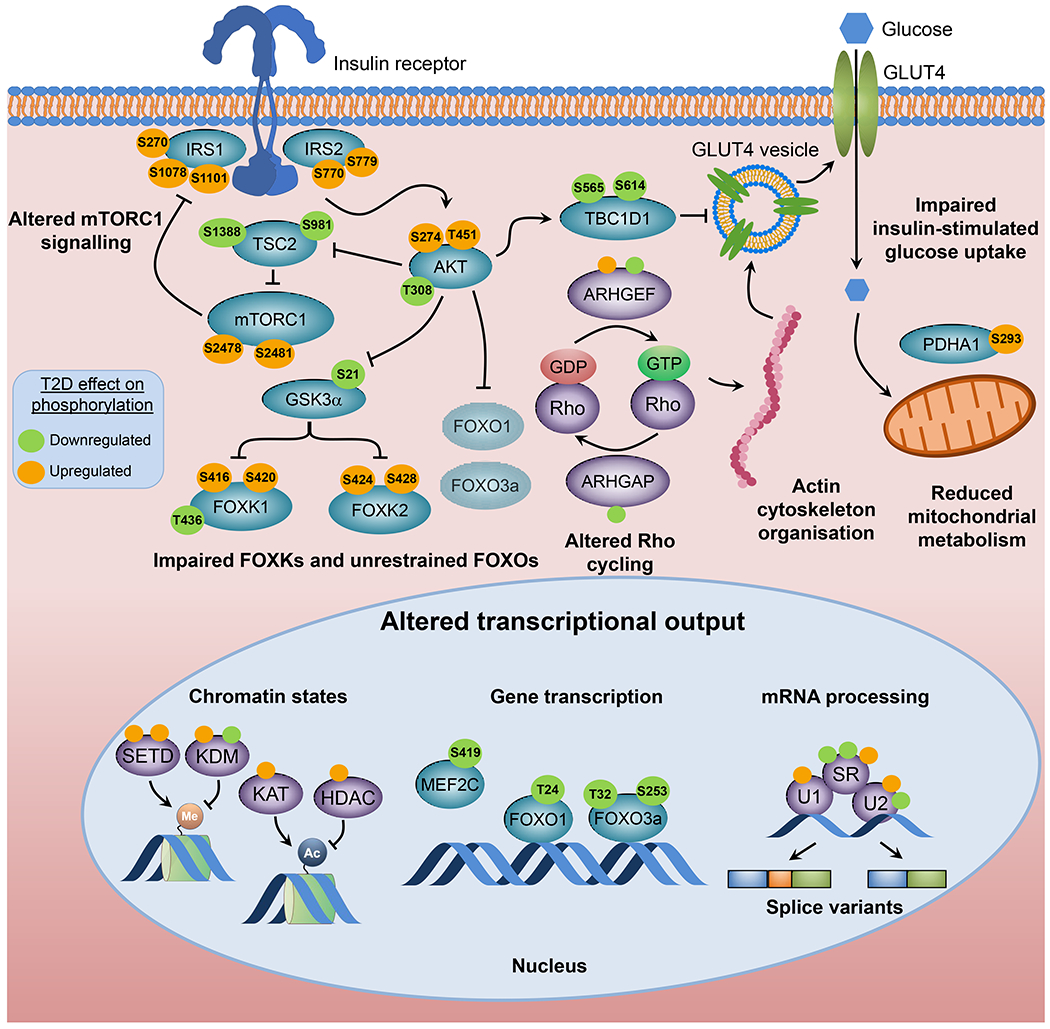
Intrinsic factors contributing to insulin resistance. Cell-autonomous insulin resistance is associated with defects in glucose transport, mitochondrial metabolism and insulin signalling. Global phosphoproteomics of iMyos from individuals with type 2 diabetes reveal a network of signalling defects that underlie skeletal muscle insulin resistance [100]. Proteins linked to insulin action and metabolism are indicated in blue and site-specific effects of type 2 diabetes evidenced by increased and decreased basal phosphorylation are shown in orange and green, respectively. Groups of multiple proteins of the same category are shown in purple and non-labelled circles indicate groups of up- or downregulated phosphosites. All signalling events are derived from MS-based phosphoproteomics except for AktT308, GSK3αS21 and FOXO1T24/FOXO3T32, which are from immunoblot analysis. Faded shading of text boxes indicates lower cytoplasm abundance. Ac, acetyl group; ARHGAP, Rho GTPase activating protein; ARHGEF, Rho guanine nucleotide exchange factor; HDAC, histone deacetylase; KAT, lysine acetyltransferase; KDM, lysine demethylase; Me, methyl group; MEF2C, myocyte enhancer factor 2C; PDHA1, pyruvate dehydrogenase E1 subunit alpha 1; SETD, SET domain containing histone lysine methyltransferase; SR, serine- and arginine-rich splicing factor; T2D, type 2 diabetes; TBC1D1, TBC1 domain family member 1; TSC2, tuberous sclerosis 2; U1, U1 small nuclear ribonucleoprotein complex; U2, U2 small nuclear ribonucleoprotein complex. This figure is available as part of a downloadable slideset.
A major challenge going forward will be to identify the molecular defect(s) that drive these signalling changes. Possibilities include kinases and phosphatases, a wide range of co-regulators of the activity of kinases and phosphatases, redox balance, ionic milieu, scaffolding proteins and other factors. While these alterations could represent some form of metabolic memory or epigenetic regulation due to altered DNA methylation [102, 103], this seems unlikely since genetic reprogramming of iPSCs is known to erase most epigenetic marks [104]. Likewise, while genome-wide association studies (GWAS) have collectively identified over 500 independent SNPs associated with type 2 diabetes [105], few of these are in proteins active in insulin signalling. Furthermore, although some SNPs may fall into regulatory regions acting on adjacent or even distant genes, most of these GWAS variants occur in non-coding regions of the genome [106, 107]. While there has been some progress in linking GWAS variants to alterations in beta cell function, insulin sensitivity and energy balance [108, 109], even taken together, all GWAS loci account for only a small fraction of the strong familial clustering of type 2 diabetes, leaving understanding the primary defect a major challenge.
Advances in profiling technologies have led to a greater appreciation of the potential role of non-coding RNAs, especially miRNAs and long non-coding RNAs (lncRNAs), in the control of cellular metabolism. Indeed, studies using in vitro models, as well as tissues from rodent models of and humans with obesity and type 2 diabetes, have revealed a network of altered miRNAs targeting the insulin receptor, as well as the IRS/PI3K/Akt pathways, thus, contributing to metabolic abnormalities [110]. For example, miR-29a and miR-29c are elevated in skeletal muscle of individuals with type 2 diabetes and ob/ob mice, and ectopic expression of miR-29a and miR-29c in muscle cells is sufficient to cause insulin resistance and impaired glucose transport [111]. While some miRNA changes in type 2 diabetes may result from tissue crosstalk through exosomal delivery [112], miRNA profiling of cultured myoblasts from donors with type 2 diabetes also revealed some modest, but significant, changes compared with control donors [113]. Similar to miRNAs, some lncRNAs are also regulated by insulin and other physiological cues [37, 114], and are dysregulated in type 2 diabetes [115], resulting in abnormal insulin signalling [116]. The finding of altered phosphorylation and gene expression of factors involved in mRNA splicing in iMyos [100] and skeletal muscle biopsies from individuals with type 2 diabetes [117] could provide another link between genetic regulation and the insulin resistance of type 2 diabetes.
Conclusions and perspectives
Insulin and IGF-1 signalling is present in virtually every cell of the body and plays a central role in the control of metabolism, growth and differentiation. In spite of significant progress, understanding the primary driver of altered insulin receptor signalling in type 2 diabetes, obesity and the metabolic syndrome represents a continuing challenge. Integrating multiple ‘omics’ layers into a unique disease signature and translating these findings into novel and personalised therapies is an important challenge for the next decade.
Funding
Work in the authors’ laboratories is supported by NIH grants R01DK031036, R01DK055545, R01DK082659 and R01DK121967 (to CRK), the Diabetes Research Center (DRC) P30DK036836 (to Joslin Diabetes Center), and the Mary K. Iacocca Professorship (to CRK).
Abbreviations
- AS160
Akt substrate of 160 kDa
- BCAA
Branched-chain amino acid
- BCL2
B cell lymphoma 2
- ER
Endoplasmic reticulum
- FOX
Forkhead box
- GSK3
Glycogen synthase kinase-3
- GWAS
Genome-wide association studies
- IGF1R
Insulin-like growth factor 1 receptor
- IKK
Inhibitor of κB kinase
- iMyo
Induced pluripotent stem cell-derived myoblast
- iPSC
Induced pluripotent stem cell
- IR-A
Insulin receptor isoform A
- IR-B
Insulin receptor isoform B
- JNK
c-Jun N-terminal kinase
- lncRNA
Long non-coding RNA
- miRNA/miR
MicroRNA
- mTORC
Mechanistic target of rapamycin complex
- PDK
3-Phosphoinositide dependent protein kinase
- PI3K
Phosphoinositide 3-kinase
- PIP3
Phosphatidylinositol (3,4,5)-triphosphate
- PKC
Protein kinase C
- Pol II
RNA polymerase II
- SGK
Serum- and glucocorticoid-induced protein kinase
- SH2
Src homology 2
- SOCS
Suppressor of cytokine signalling
- SREBP
Sterol regulatory element binding
Footnotes
Authors’ relationships and activities The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Tweet Celebrating the 100th birthday of insulin discovery by highlighting the underlying defect in #insulin action in type 2 diabetes. Read review by Thiago M. Batista, @nida.haider and C. Ronald Kahn from the @diabetesmaster lab. @JoslinDiabetes #Insulin100 #T2D
References
- [1].Macleod JJR (1922) Treatment of Diabetes Mellitus by Pancreatic Extracts. Can Med Assoc J 12(6): 425–426 [PMC free article] [PubMed] [Google Scholar]
- [2].Himsworth HP (2013) Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. 1936. Int J Epidemiol 42(6): 1594–1598. 10.1093/ije/dyt203 [DOI] [PubMed] [Google Scholar]
- [3].Freychet P, Roth J, Neville DM Jr. (1971) Insulin receptors in the liver: specific binding of ( 125 I)insulin to the plasma membrane and its relation to insulin bioactivity. Proc Natl Acad Sci U S A 68(8): 1833–1837. 10.1073/pnas.68.8.1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR (1982) Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 298(5875): 667–669. 10.1038/298667a0 [DOI] [PubMed] [Google Scholar]
- [5].Chevalier S, Burgess SC, Malloy CR, Gougeon R, Marliss EB, Morais JA (2006) The greater contribution of gluconeogenesis to glucose production in obesity is related to increased whole-body protein catabolism. Diabetes 55(3): 675–681. 10.2337/diabetes.55.03.06.db05-1117 [DOI] [PubMed] [Google Scholar]
- [6].Perry RJ, Camporez JP, Kursawe R, et al. (2015) Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160(4): 745–758. 10.1016/j.cell.2015.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seino S, Seino M, Nishi S, Bell GI (1989) Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A 86(1): 114–118. 10.1073/pnas.86.1.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Belfiore A, Malaguarnera R, Vella V, et al. (2017) Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev 38(5): 379–431. 10.1210/er.2017-00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cai W, Sakaguchi M, Kleinridders A, et al. (2017) Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat Commun 8: 14892. 10.1038/ncomms14892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Urso B, Cope DL, Kalloo-Hosein HE, et al. (1999) Differences in signaling properties of the cytoplasmic domains of the insulin receptor and insulin-like growth factor receptor in 3T3-L1 adipocytes. J Biol Chem 274(43): 30864–30873. 10.1074/jbc.274.43.30864 [DOI] [PubMed] [Google Scholar]
- [11].Scapin G, Dandey VP, Zhang Z, et al. (2018) Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature 556(7699): 122–125. 10.1038/nature26153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Uchikawa E, Choi E, Shang G, Yu H, Bai XC (2019) Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. eLife 8. 10.7554/eLife.48630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thirone AC, Huang C, Klip A (2006) Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab 17(2): 72–78. 10.1016/j.tem.2006.01.005 [DOI] [PubMed] [Google Scholar]
- [14].Araki E, Lipes MA, Patti ME, et al. (1994) Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372(6502): 186–190. 10.1038/372186a0 [DOI] [PubMed] [Google Scholar]
- [15].Kaburagi Y, Satoh S, Tamemoto H, et al. (1997) Role of insulin receptor substrate-1 and pp60 in the regulation of insulin-induced glucose transport and GLUT4 translocation in primary adipocytes. J Biol Chem 272(41): 25839–25844. 10.1074/jbc.272.41.25839 [DOI] [PubMed] [Google Scholar]
- [16].Withers DJ, Gutierrez JS, Towery H, et al. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391(6670): 900–904. 10.1038/36116 [DOI] [PubMed] [Google Scholar]
- [17].Rabiee A, Kruger M, Ardenkjaer-Larsen J, Kahn CR, Emanuelli B (2018) Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell Signal 47: 1–15. 10.1016/j.cellsig.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ussar S, Bezy O, Bluher M, Kahn CR (2012) Glypican-4 enhances insulin signaling via interaction with the insulin receptor and serves as a novel adipokine. Diabetes 61(9): 2289–2298. 10.2337/db11-1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maddux BA, Goldfine ID (2000) Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes 49(1): 13–19. 10.2337/diabetes.49.1.13 [DOI] [PubMed] [Google Scholar]
- [20].Batista TM, Dagdeviren S, Carroll SH, et al. (2020) Arrestin domain-containing 3 (Arrdc3) modulates insulin action and glucose metabolism in liver. Proc Natl Acad Sci U S A 117(12): 6733–6740. 10.1073/pnas.1922370117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sakaguchi M, Cai W, Wang CH, et al. (2019) FoxK1 and FoxK2 in insulin regulation of cellular and mitochondrial metabolism. Nat Commun 10(1): 1582. 10.1038/s41467-019-09418-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Choi E, Zhang X, Xing C, Yu H (2016) Mitotic Checkpoint Regulators Control Insulin Signaling and Metabolic Homeostasis. Cell 166(3): 567–581. 10.1016/j.cell.2016.05.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mora A, Komander D, van Aalten DM, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15(2): 161–170. 10.1016/j.semcdb.2003.12.022 [DOI] [PubMed] [Google Scholar]
- [24].Bilanges B, Posor Y, Vanhaesebroeck B (2019) PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol 20(9): 515–534. 10.1038/s41580-019-0129-z [DOI] [PubMed] [Google Scholar]
- [25].Manning BD, Toker A (2017) AKT/PKB Signaling: Navigating the Network. Cell 169(3): 381–405. 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Feng J, Park J, Cron P, Hess D, Hemmings BA (2004) Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem 279(39): 41189–41196. 10.1074/jbc.M406731200 [DOI] [PubMed] [Google Scholar]
- [27].Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378(6559): 785–789. 10.1038/378785a0 [DOI] [PubMed] [Google Scholar]
- [28].Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6(3): 208–216. 10.1016/j.cmet.2007.08.006 [DOI] [PubMed] [Google Scholar]
- [29].Nakae J, Barr V, Accili D (2000) Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J 19(5): 989–996. 10.1093/emboj/19.5.989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].O’Neill BT, Lee KY, Klaus K, et al. (2016) Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest 126(9): 3433–3446. 10.1172/JCI86522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sancak Y, Thoreen CC, Peterson TR, et al. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25(6): 903–915. 10.1016/j.molcel.2007.03.003 [DOI] [PubMed] [Google Scholar]
- [32].Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J (2002) Tuberous sclerosis complex-1 and −2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A 99(21): 13571–13576. 10.1073/pnas.202476899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H (2005) Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 54(6): 1692–1697 [DOI] [PubMed] [Google Scholar]
- [34].Sano H, Kane S, Sano E, et al. (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278(17): 14599–14602. 10.1074/jbc.C300063200 [DOI] [PubMed] [Google Scholar]
- [35].Humphrey SJ, Azimifar SB, Mann M (2015) High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol 33(9): 990–995. 10.1038/nbt.3327 [DOI] [PubMed] [Google Scholar]
- [36].Humphrey SJ, Yang G, Yang P, et al. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab 17(6): 1009–1020. 10.1016/j.cmet.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Batista TM, Garcia-Martin R, Cai W, et al. (2019) Multi-dimensional Transcriptional Remodeling by Physiological Insulin In Vivo. Cell reports 26(12): 3429–3443 e3423. 10.1016/j.celrep.2019.02.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Karlsson HK, Chibalin AV, Koistinen HA, et al. (2009) Kinetics of GLUT4 trafficking in rat and human skeletal muscle. Diabetes 58(4): 847–854. 10.2337/db08-1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bruss MD, Arias EB, Lienhard GE, Cartee GD (2005) Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 54(1): 41–50. 10.2337/diabetes.54.1.41 [DOI] [PubMed] [Google Scholar]
- [40].Huang X, Vaag A, Hansson M, Weng J, Laurila E, Groop L (2000) Impaired insulin-stimulated expression of the glycogen synthase gene in skeletal muscle of type 2 diabetic patients is acquired rather than inherited. J Clin Endocrinol Metab 85(4): 1584–1590. 10.1210/jcem.85.4.6535 [DOI] [PubMed] [Google Scholar]
- [41].Sears DD, Hsiao G, Hsiao A, et al. (2009) Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci U S A 106(44): 18745–18750. 10.1073/pnas.0903032106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS (2003) Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A 100(13): 7996–8001. 10.1073/pnas.1332551100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].O’Brien RM, Granner DK (1991) Regulation of gene expression by insulin. Biochem J 278 (Pt 3): 609–619. 10.1042/bj2780609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brunet A, Bonni A, Zigmond MJ, et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6): 857–868 [DOI] [PubMed] [Google Scholar]
- [45].Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21(3): 952–965. 10.1128/MCB.21.3.952-965.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Haeusler RA, Hartil K, Vaitheesvaran B, et al. (2014) Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun 5: 5190. 10.1038/ncomms6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sandri M, Sandri C, Gilbert A, et al. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3): 399–412. 10.1016/s0092-8674(04)00400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakae J, Cao Y, Oki M, et al. (2008) Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 57(3): 563–576. 10.2337/db07-0698 [DOI] [PubMed] [Google Scholar]
- [49].I OS, Zhang W, Wasserman DH, et al. (2015) FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun 6: 7079. 10.1038/ncomms8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lu M, Wan M, Leavens KF, et al. (2012) Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18(3): 388–395. 10.1038/nm.2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].O’Neill BT, Bhardwaj G, Penniman CM, et al. (2019) FoxO Transcription Factors Are Critical Regulators of Diabetes-Related Muscle Atrophy. Diabetes 68(3): 556–570. 10.2337/db18-0416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].He L, Gomes AP, Wang X, et al. (2018) mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol Cell 70(5): 949–960 e944. 10.1016/j.molcel.2018.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sukonina V, Ma H, Zhang W, et al. (2019) FOXK1 and FOXK2 regulate aerobic glycolysis. Nature 566(7743): 279–283. 10.1038/s41586-019-0900-5 [DOI] [PubMed] [Google Scholar]
- [54].Shimano H, Sato R (2017) SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 13(12): 710–730. 10.1038/nrendo.2017.91 [DOI] [PubMed] [Google Scholar]
- [55].Goldfine ID, Smith GJ (1976) Binding of insulin to isolated nuclei. Proc Natl Acad Sci U S A 73(5): 1427–1431. 10.1073/pnas.73.5.1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hancock ML, Meyer RC, Mistry M, et al. (2019) Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell 177(3): 722–736 e722. 10.1016/j.cell.2019.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14(2): 88–98. 10.1038/nrendo.2017.151 [DOI] [PubMed] [Google Scholar]
- [58].Roden M, Shulman GI (2019) The integrative biology of type 2 diabetes. Nature 576(7785): 51–60. 10.1038/s41586-019-1797-8 [DOI] [PubMed] [Google Scholar]
- [59].Zaharia OP, Strassburger K, Strom A, et al. (2019) Risk of diabetes-associated diseases in subgroups of patients with recent-onset diabetes: a 5-year follow-up study. Lancet Diabetes Endocrinol 7(9): 684–694. 10.1016/S2213-8587(19)30187-1 [DOI] [PubMed] [Google Scholar]
- [60].Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR (1992) Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340(8825): 925–929 [DOI] [PubMed] [Google Scholar]
- [61].Eriksson J, Franssila-Kallunki A, Ekstrand A, et al. (1989) Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med 321(6): 337–343. 10.1056/NEJM198908103210601 [DOI] [PubMed] [Google Scholar]
- [62].Rothman DL, Magnusson I, Cline G, et al. (1995) Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A 92(4): 983–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cline GW, Petersen KF, Krssak M, et al. (1999) Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med 341(4): 240–246. 10.1056/NEJM199907223410404 [DOI] [PubMed] [Google Scholar]
- [64].Mootha VK, Lindgren CM, Eriksson KF, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3): 267–273. 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- [65].Patti ME, Butte AJ, Crunkhorn S, et al. (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100(14): 8466–8471. 10.1073/pnas.1032913100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gregor MF, Hotamisligil GS (2010) Inflammatory Mechanisms in Obesity. Annu Rev Immunol 29:415–445 [DOI] [PubMed] [Google Scholar]
- [67].Newgard CB (2012) Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 15(5): 606–614. 10.1016/j.cmet.2012.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148(5): 852–871. 10.1016/j.cell.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dunaif A, Xia J, Book CB, Schenker E, Tang Z (1995) Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest 96(2): 801–810. 10.1172/JCI118126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 51(7): 2005–2011 [DOI] [PubMed] [Google Scholar]
- [71].Lyu K, Zhang Y, Zhang D, et al. (2020) A Membrane-Bound Diacylglycerol Species Induces PKC-Mediated Hepatic Insulin Resistance. Cell Metab 32(4): 654–664 e655. 10.1016/j.cmet.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Moeschel K, Beck A, Weigert C, et al. (2004) Protein kinase C-zeta-induced phosphorylation of Ser318 in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem 279(24): 25157–25163. 10.1074/jbc.M402477200 [DOI] [PubMed] [Google Scholar]
- [73].Weigert C, Hennige AM, Brischmann T, et al. (2005) The phosphorylation of Ser318 of insulin receptor substrate 1 is not per se inhibitory in skeletal muscle cells but is necessary to trigger the attenuation of the insulin-stimulated signal. J Biol Chem 280(45): 37393–37399 [DOI] [PubMed] [Google Scholar]
- [74].Paz K, Hemi R, LeRoith D, et al. (1997) A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 272(47): 29911–29918. 10.1074/jbc.272.47.29911 [DOI] [PubMed] [Google Scholar]
- [75].Glass CK, Olefsky JM (2012) Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab 15(5): 635–645. 10.1016/j.cmet.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Johnson AM, Olefsky JM (2013) The origins and drivers of insulin resistance. Cell 152(4): 673–684. 10.1016/j.cell.2013.01.041 [DOI] [PubMed] [Google Scholar]
- [77].Sun K, Kusminski CM, Scherer PE (2011) Adipose tissue remodeling and obesity. J Clin Invest 121(6): 2094–2101. 10.1172/JCI45887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117(1): 175–184. 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Osborn O, Olefsky JM (2012) The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 18(3): 363–374. 10.1038/nm.2627 [DOI] [PubMed] [Google Scholar]
- [80].Lebrun P, Van Obberghen E (2008) SOCS proteins causing trouble in insulin action. Acta Physiol 192(1): 29–36. 10.1111/j.1748-1716.2007.01782.x [DOI] [PubMed] [Google Scholar]
- [81].Liu J, Ibi D, Taniguchi K, et al. (2016) Inflammation Improves Glucose Homeostasis through IKKbeta-XBP1s Interaction. Cell 167(4): 1052–1066 e1018. 10.1016/j.cell.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Raciti M, Lotti LV, Valia S, Pulcinelli FM, Di Renzo L (2012) JNK2 is activated during ER stress and promotes cell survival. Cell death & disease 3: e429. 10.1038/cddis.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cummings NE, Williams EM, Kasza I, et al. (2018) Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol 596(4): 623–645. 10.1113/JP275075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Newgard CB, An J, Bain JR, et al. (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9(4): 311–326. 10.1016/j.cmet.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Saad MJ, Santos A, Prada PO (2016) Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 31(4): 283–293. 10.1152/physiol.00041.2015 [DOI] [PubMed] [Google Scholar]
- [86].Copps KD, White MF (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55(10): 2565–2582. 10.1007/s00125-012-2644-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Li M, Vienberg SG, Bezy O, O’Neill BT, Kahn CR (2015) Role of PKCdelta in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 64(12): 4023–4032. 10.2337/db14-1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Thomou T, Mori MA, Dreyfuss JM, et al. (2017) Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542(7642): 450–455. 10.1038/nature21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ying W, Riopel M, Bandyopadhyay G, et al. (2017) Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171(2): 372–384 e312. 10.1016/j.cell.2017.08.035 [DOI] [PubMed] [Google Scholar]
- [90].Bouzakri K, Roques M, Gual P, et al. (2003) Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes 52(6): 1319–1325 [DOI] [PubMed] [Google Scholar]
- [91].Cozzone D, Frojdo S, Disse E, et al. (2008) Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia 51(3): 512–521. 10.1007/s00125-007-0913-8 [DOI] [PubMed] [Google Scholar]
- [92].Gaster M, Brusgaard K, Handberg A, Hojlund K, Wojtaszewski JF, Beck-Nielsen H (2004) The primary defect in glycogen synthase activity is not based on increased glycogen synthase kinase-3alpha activity in diabetic myotubes. Biochem Biophys Res Commun 319(4): 1235–1240. 10.1016/j.bbrc.2004.05.109 [DOI] [PubMed] [Google Scholar]
- [93].Gaster M, Petersen I, Hojlund K, Poulsen P, Beck-Nielsen H (2002) The diabetic phenotype is conserved in myotubes established from diabetic subjects: evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes 51(4): 921–927. 10.2337/diabetes.51.4.921 [DOI] [PubMed] [Google Scholar]
- [94].McIntyre EA, Halse R, Yeaman SJ, Walker M (2004) Cultured muscle cells from insulin-resistant type 2 diabetes patients have impaired insulin, but normal 5-amino-4-imidazolecarboxamide riboside-stimulated, glucose uptake. J Clin Endocrinol Metab 89(7): 3440–3448. 10.1210/jc.2003-031919 [DOI] [PubMed] [Google Scholar]
- [95].Burkart AM, Tan K, Warren L, et al. (2016) Insulin Resistance in Human iPS Cells Reduces Mitochondrial Size and Function. Scientific reports 6: 22788. 10.1038/srep22788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Iovino S, Burkart AM, Kriauciunas K, et al. (2014) Genetic insulin resistance is a potent regulator of gene expression and proliferation in human iPS cells. Diabetes 63(12): 4130–4142. 10.2337/db14-0109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Iovino S, Burkart AM, Warren L, Patti ME, Kahn CR (2016) Myotubes derived from human-induced pluripotent stem cells mirror in vivo insulin resistance. Proc Natl Acad Sci U S A 113(7): 1889–1894. 10.1073/pnas.1525665113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gupta MK, Vethe H, Softic S, et al. (2020) Leptin Receptor Signaling Regulates Protein Synthesis Pathways and Neuronal Differentiation in Pluripotent Stem Cells. Stem cell reports 15(5): 1067–1079. 10.1016/j.stemcr.2020.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Teo AK, Windmueller R, Johansson BB, et al. (2013) Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J Biol Chem 288(8): 5353–5356. 10.1074/jbc.C112.428979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Batista TM, Jayavelu AK, Wewer Albrechtsen NJ, et al. (2020) A Cell-Autonomous Signature of Dysregulated Protein Phosphorylation Underlies Muscle Insulin Resistance in Type 2 Diabetes. Cell Metab 32:844–859. 10.1016/j.cmet.2020.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7(2): 85–96 [DOI] [PubMed] [Google Scholar]
- [102].Barres R, Osler ME, Yan J, et al. (2009) Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab 10(3): 189–198. 10.1016/j.cmet.2009.07.011 [DOI] [PubMed] [Google Scholar]
- [103].Ribel-Madsen R, Fraga MF, Jacobsen S, et al. (2012) Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS One 7(12): e51302. 10.1371/journal.pone.0051302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Frobel J, Hemeda H, Lenz M, et al. (2014) Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem cell reports 3(3): 414–422. 10.1016/j.stemcr.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Vujkovic M, Keaton JM, Lynch JA, et al. (2020) Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52(7): 680–691. 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Miguel-Escalada I, Bonas-Guarch S, Cebola I, et al. (2019) Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet 51(7): 1137–1148. 10.1038/s41588-019-0457-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Williams K, Ingerslev LR, Bork-Jensen J, et al. (2020) Skeletal muscle enhancer interactions identify genes controlling whole-body metabolism. Nat Commun 11(1): 2695. 10.1038/s41467-020-16537-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Claussnitzer M, Dankel SN, Kim KH, et al. (2015) FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373(10): 895–907. 10.1056/NEJMoa1502214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Manning AK, Hivert MF, Scott RA, et al. (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44(6): 659–669. 10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Nigi L, Grieco GE, Ventriglia G, et al. (2018) MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. International journal of molecular sciences 19(12):3705. 10.3390/ijms19123705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Massart J, Sjogren RJO, Lundell LS, et al. (2017) Altered miR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Skeletal Muscle. Diabetes 66(7): 1807–1818. 10.2337/db17-0141 [DOI] [PubMed] [Google Scholar]
- [112].Katayama M, Wiklander OPB, Fritz T, et al. (2018) Circulating Exosomal miR-20b-5p is Elevated in Type 2 Diabetes and Could Impair Insulin Action in Human Skeletal Muscle. Diabetes 68(3):515–526. 10.2337/db18-0470 [DOI] [PubMed] [Google Scholar]
- [113].Henriksen TI, Davidsen PK, Pedersen M, et al. (2017) Dysregulation of a novel miR-23b/27b-p53 axis impairs muscle stem cell differentiation of humans with type 2 diabetes. Mol Metab 6(7): 770–779. 10.1016/j.molmet.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Yang L, Li P, Yang W, et al. (2016) Integrative Transcriptome Analyses of Metabolic Responses in Mice Define Pivotal LncRNA Metabolic Regulators. Cell Metab 24(4): 627–639. 10.1016/j.cmet.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Carter G, Miladinovic B, Patel AA, Deland L, Mastorides S, Patel NA (2015) Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin 4: 102–107. 10.1016/j.bbacli.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Shi Y, Parag S, Patel R, et al. (2019) Stabilization of lncRNA GAS5 by a Small Molecule and Its Implications in Diabetic Adipocytes. Cell Chem Biol 26(3): 319–330 e316. 10.1016/j.chembiol.2018.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Pihlajamaki J, Lerin C, Itkonen P, et al. (2011) Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 14(2): 208–218. 10.1016/j.cmet.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Weyer C, Funahashi T, Tanaka S, et al. (2001) Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86(5): 1930–1935. 10.1210/jcem.86.5.7463 [DOI] [PubMed] [Google Scholar]
- [119].Lynes MD, Leiria LO, Lundh M, et al. (2017) The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat Med 23(5): 631–637. 10.1038/nm.4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yore MM, Syed I, Moraes-Vieira PM, et al. (2014) Discovery of a Class of Endogenous Mammalian Lipids with Anti-Diabetic and Anti-inflammatory Effects. Cell 159(2): 318–332 [DOI] [PMC free article] [PubMed] [Google Scholar]