

Abstract
The COVID-19 pandemic prompted many scientists to investigate remedies against SARS-CoV-2 and related viruses that are likely to appear in the future. As the main protease of the virus, MPro, is highly conserved among coronaviruses, it has emerged as a prime target for developing inhibitors. Using a combination of virtual screening and molecular modeling, we identified small molecules that were easily accessible and could be quickly diversified. Biochemical assays confirmed a class of pyridones as low micromolar non-covalent inhibitors of the viral main protease.
Keywords: Viral Main Protease, Small-Molecule Inhibitor, SARS-CoV-2, Coronavirus, Molecular Modeling
Graphical Abstract
Introduction:
Small molecules continue to play a crucial role in the fight against viral diseases. Examples for their success include medications for HIV and HCV, which have been made manageable or can be cured by inhibitors of proteases and RNA polymerases.1 This will likely be the case for the current COVID-19 pandemic as well. Although vaccines based on chemically modified mRNA packaged in lipid nanoparticles have proven to be highly effective, their future usefulness may be impeded by rapid mutations in the viral envelope.2 In addition, vaccines are less effective in the immunocompromised and are not embraced by a substantial portion of the population for a variety of reasons. Therefore, the development of easily applicable and stable small molecules that fight SARS-CoV-2 and related coronaviruses remains a priority.3
Amongst the limited set of viral target proteins, the SARS-CoV-2 main protease (MPro, also called 3CLPro) stands out. This enzyme, a cysteine protease, cleaves its substrate after a glutamine residue, which appears to be unknown for human cysteine proteases that could be responsible for off-target effects.4 Over the course of the COVID-19 pandemic, the amino acid sequence of MPro has remained remarkably conserved,5 although it has not been challenged yet by drugs on a large scale. Numerous X-ray structures of MPro with covalent and non-covalent inhibitors and small molecule fragments bound are available to aid computational designs.6 In addition, SARS-CoV-2 MPro is highly homologous to corresponding proteases of other coronaviruses and enteroviruses and likely to be related to proteases of harmful viruses that will emerge in the future.7
As such, efforts to develop MPro inhibitors have been launched on a large scale. To date, they have mostly been centered on covalent inhibitors derived from peptides that correspond to the natural cleavage site of the MPro substrate.8 Given the rapid advance of this approach, which had been pursued for coronaviruses well before the emergence of SARS-CoV-2, we decided to take an alternative one: the de novo design of molecules through virtual screening and further refinement of the best candidates with molecular docking. This was always done with an eye on synthetic accessibility and the ability to quickly diversify successful candidates. As an additional distinguishing criterium, we decided to work on molecules that bind non-covalently, at least in the first phase of our program. We now disclose series of small molecules that fulfill criteria for “druggability” and can be assembled in a few synthetic steps. Our most successful ones inhibit MPro at single digit micromolar concentrations in biochemical assays.
Results and Discussions:
Identification of a Small-Molecule lead scaffold
The ASINEX PPI non-macrocyclic screening library consists of 11870 fragments and compounds that were computationally docked into the catalytic site of the crystal structure of the SARS-CoV-2 main protease (PDB: 6LU7 and 6Y84; details can be found in the Supporting Information).9 This provided a virtual spectrum of docking score vs ligand efficiency (Figure 1a, blue) in which some of initial virtual hits were manually evaluated and optimized in terms of ligand efficiency, synthetic accessibility, and docking score (Figure 1a, red). Based on these parameters, we selected three distinct types of molecules, represented by compounds 1-3 for synthesis and biological testing (Figure 1b).
Figure 1.
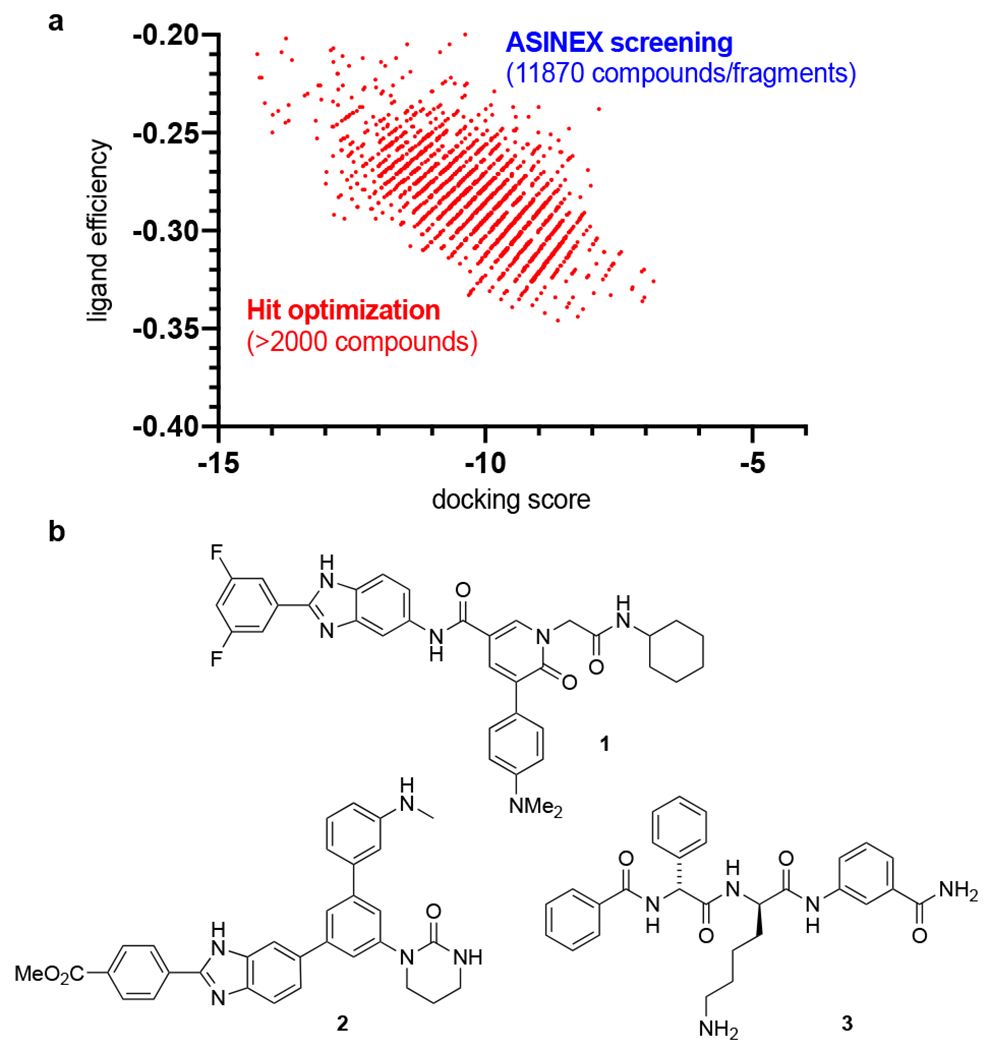
a) Computational lead identification (red) by optimizing initial hits in the ASINEX screening (blue). b) Representative target compounds 1-3 with excellent docking scores.
N-Alkyl-3-arylpyridone 5-carboxamides of type 1 were recognized as an easily accessible scaffold as well as m-teraryl linked cyclic ureas of type 2. In addition to these, short d-Phg-d-Lys peptides of type 3 were identified as potential inhibitors that should remain proteolytically stable and capable to disrupt the natural function of the protease. Methyl 6-hydroxynicotinate (4) is selectively N-alkylated with chloroacetamide 5a using K2CO3 (Scheme 1a). Bromination and Suzuki cross-coupling allow access to the arylated pyridone methyl ester 6a, which is subsequently hydrolyzed. The resulting carboxylic acid is coupled using HATU to the benzimidazole amine 7 to form N-alkyl-3-arylpyridone 5-carboxamide 1. The cyclic urea 2 was synthesized in similar efficiency (Scheme 1b). The initial isocyanate addition to 3,5-dibromoaniline (8) was followed by the cyclization of the urea moiety using NaH. Two sequential Suzuki cross-coupling reactions on the dibromide 9 with aryl boronic acid pinacol esters 10 and 11 rendered the desired m-teraryl linked urea 2. The short d-Phg-d-Lys peptide 3 was manually assembled starting from Fmoc-d-Lys(Boc)-OH 12 by a tailored sequence of EDCI/HOBt-based couplings and protecting group removals (Scheme 1c).
Scheme 1.
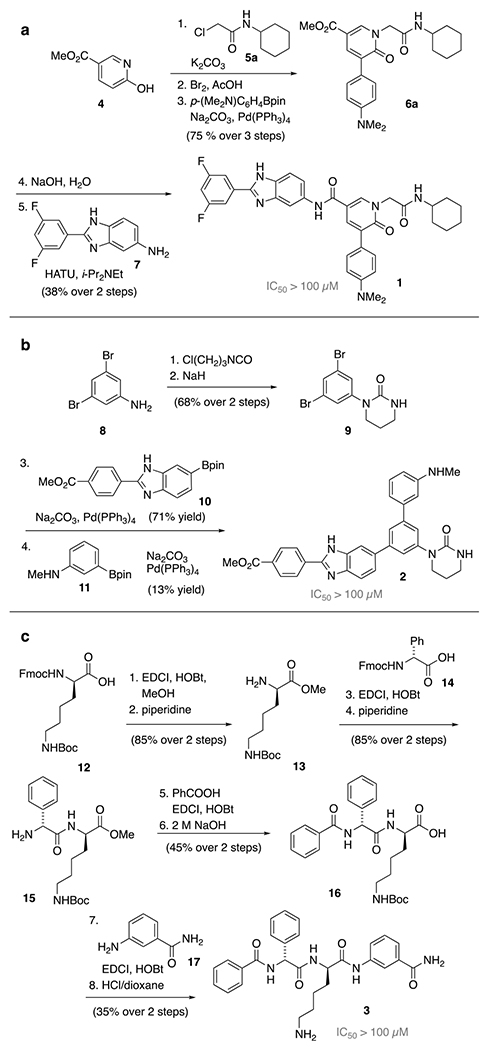
Assembly of initially identified compounds 1-3.
With our first target molecules in hand, we proceeded to test them in enzymatic activity assays that monitor the formation of fluorescent cleavage products (see Supporting Information).10 Despite their excellent docking scores, pyridone 1, the cyclic urea 2, and the d-peptide 3 failed to inhibit the viral main protease even at high concentrations. Gratifyingly, however, some synthetic intermediates that were also tested showed activities that were worth following up upon. For instance, the Boc-protected intermediate 16 inhibited MPro with an IC50 of 121 μM. The truncated pyridone methyl ester intermediate 6a showed more substantial inhibition (IC50 = 19 μM). Replacing the benzimidazole amide of 1 with a methyl ester not only reduces the molecular weight but also the lipophilicity (clogP lowered from ~4.4 to ~3.5). Therefore, this pyridone scaffold was identified as a promising lead for further optimization and our subsequent investigations focused on this class of compounds.
SAR Investigations
The succinct synthesis of pyridone methyl ester 6a, outlined in Scheme 1, was generally applicable to various other derivatives but also allowed a broad range of cross-coupling reactions (see Supporting Information for details). Initial structure-activity relationship (SAR) investigations showed that a substituent next to the pyridine carbonyl was critical. For instance, unsubstituted pyridone 4’ did not show any activity. Also the activities of the unsubstituted phenyl 6b and the more lipophilic fluoro aryls 6c-e were negligibly low. While the CF3 derivative 6f and the acetamide 6g showed improved IC50 values, the sulfonamide 6h remained inactive. In the absence of an X-ray structure, systematic synthesis and biological evaluation was required to improve our understanding of how the pyridone series interacts with the binding site. We anticipate that a favorable interaction (e.g. hydrogen bonding) very close to the 4-position of the aryl substituent is required to further lower the IC50. Therefore, the 4-pyridyl 6i and 6j was synthesized, showing increased activity at 14 and 12 μM, respectively. Also, 4-anisole 6k as well as methoxy-pyrimidine 6l supported this hypothesis with an increase in activity, whereas the corresponding aryloxy ether 6m is likely missing such a key interaction. Possibly due the orientation of the sulfur lone pairs the thiomorpholine 6n is less optimal. The more extended morpholine 6o, the sulfone analog 6p and the piperidine-4-carbonitrile 6q seem to be missing this favorable interaction as well. A stark increase in inhibition of the main protease was found with dihydropyrrolopyridine 6r having an IC50 of 3.2 μM. To probe the length of the substituent further, we examined ethynylpyridine 6s in comparison to ethynylbenzene 6t. Both had diminishing effects on the potency. Hence a more favorable interaction could be imagined with a smaller functional group. Whereas the alkyne 6u was ineffective, the nitrile 6v was inhibiting the main protease with an IC50 of 3.3 μM. In addition to the improved potency, the nitrile 6v had also a favorable calculated logP of 1.1 and a polar surface area of 101 Å, which promise good cell permeation.
During our SAR investigations, we realized that some of the pyridones, such as the acetylated derivative 6g showed strong fluorescence (λabs 318 nm; λem 420 nm in aq. PBS with 1% DMSO). This reaffirmed our selection of a bathochromically shifted rhodamine based fluorescent probe for the enzymatic assays that did not interfere with our inhibitors (see Supporting Information for details).
To further improve the activity of the pyridone scaffold we decided to explore variations in the acetamide side chain. Unfortunately, Suzuki cross-coupling reactions on the free 4-bromo-6-hydroxynicotinate core were met with limited success.
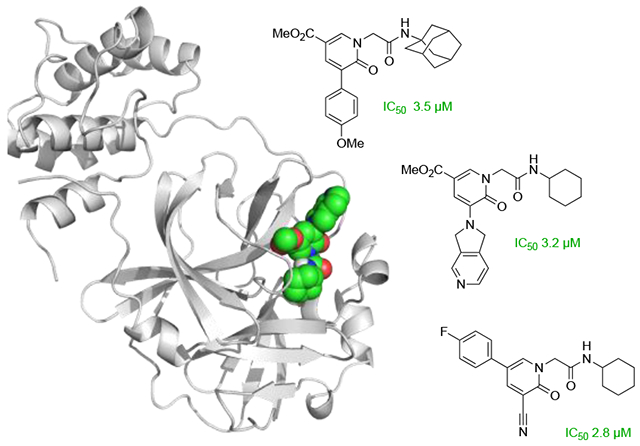
Based on reports by Gademann and co-workers the hydroxypyridine 18 was protected first using SEM-Cl (Scheme 2).11 This enabled derivatization like the nitrile substitution and Suzuki cross-coupling reaction. As anisole is a neutral, fairly stable moiety and showed an IC50 of 23 μM in derivative 6k, it was chosen together with the more active nitrile for further exploration. After deprotection of the SEM group, the acetamides were installed under the previously introduced conditions. For the nitrile, both the morpholine amide 20a and the adamantanyl amide 20b did not show any inhibition in the enzyme assay. Interestingly, for the anisole, a N,N-diethylamide 21a lost the activity completely. However, the adamantyl amide 21b and the morpholino amide 21c showed a nearly tenfold lower IC50 of 3.5 μM and 3.4 μM, respectively.
Scheme 2.
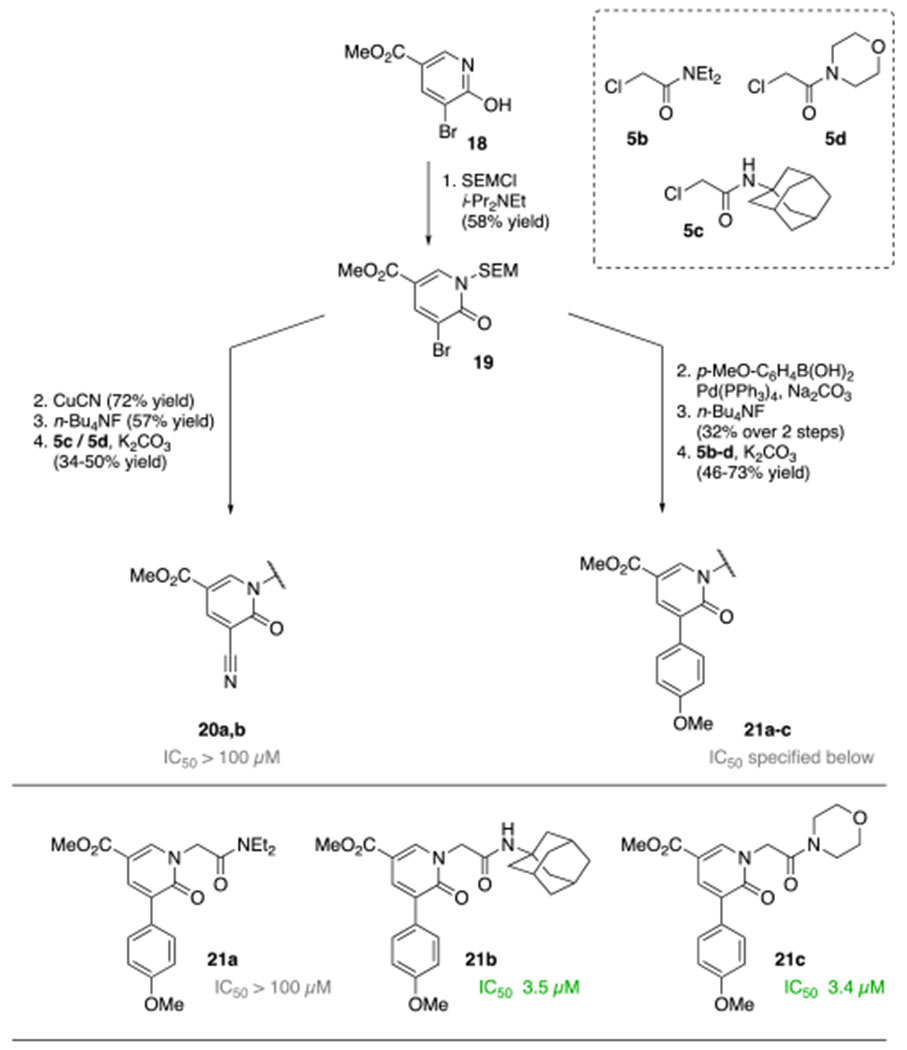
Derivatizing the pyridone side chain of the nitrile and anisole analog.
Our concern of the pyridone methyl ester hydrolysis as a serious liability grew, when we observed a significantly increased IC50 of the corresponding carboxylic acid of the anisole derivative 22a. The installation of dimethyl amides, morpholino amides and phenyl amides were examined for both 4-anisole and nitrile derivatives 22a-c and 23a,b (Scheme 3a). Disappointingly, all derivatives showed a complete loss of activity. In search of another functional group we decided to explore the installation of an aryl moiety instead. N-Alkylation of 5-bromopyridin-2-ol (25) using chloroacetamide 5a allowed the installation of the fluorophenyl via Suzuki cross-coupling (Scheme 3b). Installation of a nitrile at the 3-position leads to 28, a significantly more active SARS-CoV-2 main protease inhibitor at an IC50 of 2.8 μM. This is our strongest inhibitor to date and is in stark contrast to the activity of compound 27, which lacks the nitrile (IC50 ≈ 77 μM).
Scheme 3.
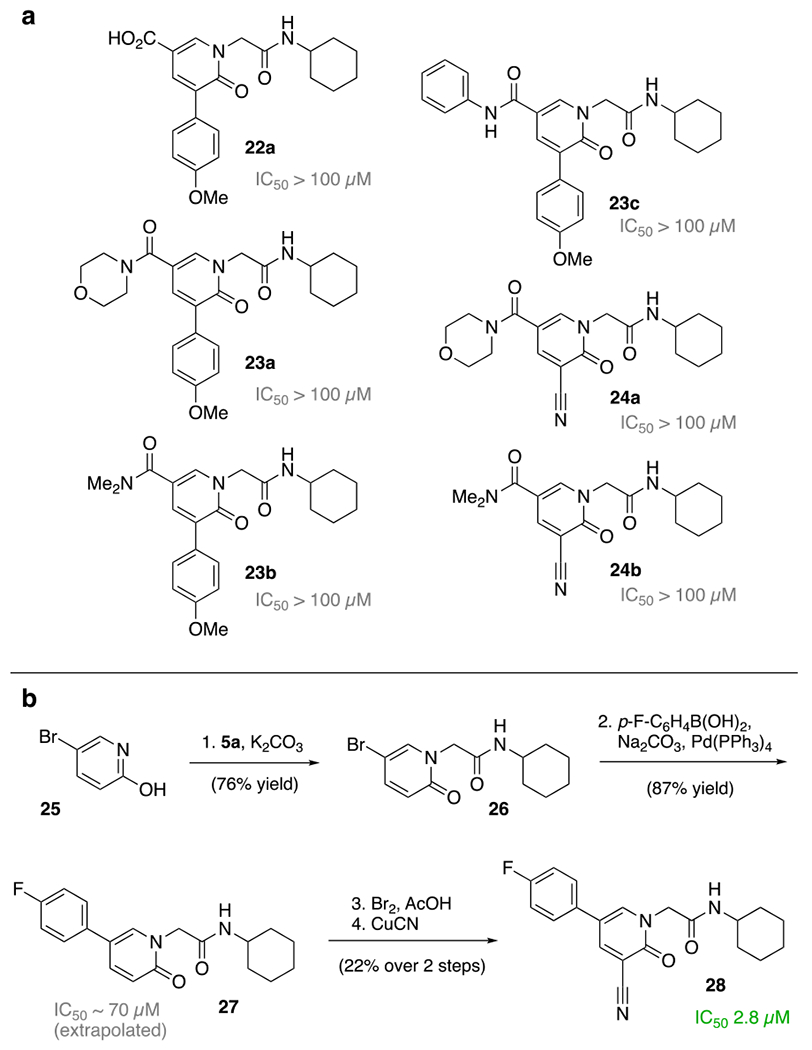
a) As the carboxylic acid 22a loses significant activity, amides 23a-c and 24a/b have been synthesized. b) Outline of the synthesis route to the highly active SARS-CoV-2 MPro inhibitor 28.
In sum, we have introduced small molecules that could potentially be developed into antivirals against SARS-CoV-2. Using a combination of virtual screening, molecular docking, and luck, we quickly were able to identify low micromolar inhibitors of the viral main protease MPro. Their further development will require X-ray crystallographic studies, which will provide insights into the binding site and pose of our inhibitors and can also serve to calibrate our docking results. Attempts in this direction are well underway and results will be reported in due course.
Supplementary Material
Figure 2.
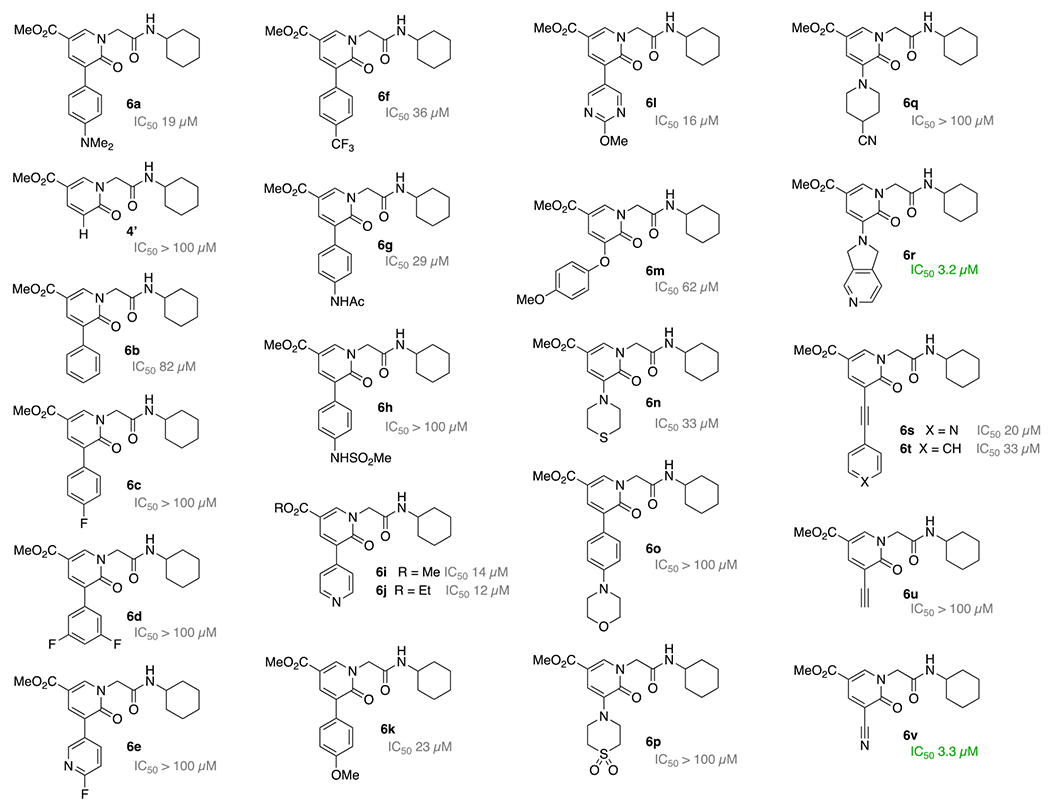
Synthesized pyridone esters 4’, 6a-v to inhibit the SARS-CoV-2 main protease and investigate the structure-activity relationship of the scaffold.
Funding Information
D.T. and his group are thankful for the COVID-19 Catalyst Grant by NYU. Y.Z. would like to acknowledge the support by the National Institutes of Health grant R35 GM127040. C.F. thanks the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship (178569). N.A.V. thanks the German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes) for a PhD Fellowship. Z.P. and K.P.R. are supported by the NYU MacCracken Fellowship.
Footnotes
Supporting Information
YES
Primary Data
NO.
Conflict of Interest
The authors declare no conflict of interest.
References and Notes
- (1).(a) De Clercq E; Li G Clin. Microbiol. Rev 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) de Leuw P; Stephan C Expert Opin. Pharmacother 2018,19, 577–587. [DOI] [PubMed] [Google Scholar]; (c) Flexner CN Engl. J. Med 1998,338,1281–1293. [DOI] [PubMed] [Google Scholar]
- (2).(a) Harvey WT; Carabelli AM; Jackson B; Gupta RK; Thomson EC; Harrison EM; Ludden C; Reeve R; Rambaut A; COVID-19 Genomics UK Consortium, Peacock SJ; Robertson DL Nat. Rev. Microbiol 2021, 19, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krammer F Nature 2020, 586, 516–527. [DOI] [PubMed] [Google Scholar]
- (3).(a) Press Release of the United States Department of Health and Human Services: June 17, 2021, “Biden Administration to Invest $3 Billion from American Rescue Plan as Part of COVID-19 Antiviral Development Strategy”, accessed June 25.2021.; (b) Tummino TA; Rezelj VV; Fischer B; Fischer A; O’Meara MJ; Monel B; Vallet T; White KM; Zhang Z; Alon A; et al. Science 2021, doi: 10.1126/science.abi4708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cannalire R; Cerchia C; Beccari AR; Di Leva FS; Summa VJ Med. Chem 2020, doi: 10.1021/acs.jmedchem.Oc01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Science 2020, 368, 409–412; see also Ref. 9a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang H; Yang J RSC Med. Chem 2021, doi: 10.1039/D1MD00066G. [DOI] [Google Scholar]; (c) Ullrich S; Nitsche C Bioorg. Med. Chem. Lett 2020,30,127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Zhu G; Zhu C; Zhu Y; Sun F Curr. Res. Microb. Sci 2020, 1, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hilgenfeld R FEBS J. 2014, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mengist HM; Dilnessa T; Jin T Front. Chem 2021, doi: 10.3389/fchem.2021.622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Kneller D; Philips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A Nat. Comm 2020, 11, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lockbaum GJ; Reyes AC; Lee MJ; Tilvawala R; Nalivaika EA; Ali A; Yilmaz NK; Thompson PR; Schiffer Celia A. Viruses 2021, 13, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Douangamath A; Fearon D; Gehrtz P; Krojer T; Lukacik P; Owen CD; Resnick E; Strain-Damerell C; et al. Nat. Commun 2020, 11, 5047. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Crystal structure for the SARS-Cov-1 main protease: Yang H; Yang M; Ding Y; Liu Y; Lou Z; Zhou L; Sun L; Mo L; Ye S; Pang H; Gao GF; Anand K; Bartlam M; Hilgenfeld R; Rao Z Proc. Natl. Acad. Sci. U.S.A 2003,100,13190–13195. As well as Ref. 4a and 9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Stoermer M ChemRxiv. 2020, doi: 10.26434/chemrxiv.11637294.v3. [DOI] [Google Scholar]; (b) Zhang L; Lin D; Kusov Y; Nian Y; Ma Q; Wang J; von Brunn A; Leyssen P; Lanko K; Neyts J; de Wilde A; Snijder EJ; Liu H; Hilgenfeld RJ Med. Chem 2020, 63,4562–4578. [DOI] [PubMed] [Google Scholar]
- (8).(a) Pillaiyar T; Manickam M; Namasivayam V; Hayashi Y; Jung S-H Med. Chem 2016,59,6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) non-covalent p-lactam inhibitors: Malla TR; Tumber A; John T; Brewitz L; Strain-Damerell C; Owen CD; Lukacik P; Chan HTH; Maheswaran P; Salah E; Duarte F; Yang H; Rao Z; Walsh MA; Schofield CJ Chem. Commun 2021, 57, 1430–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; et al. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- (10).Biering SB; Van Dis E; Wehri E; Yamashiro LH; Nguyenla X; Dugast-Darzacq C; Graham TGW; Stroumza JR; Golovkine GR; Roberts AW; Fines DM; Spradlin JN; Ward CC; Bajaj T; Dovala D; Schulze-Gamen U; Bajaj R; Fox DM; Ott M; Murthy N; Nomura DK; Schaletzky J; Stanley SA ACS Infect. Dis 2021, doi: 10.1021/acsinfecdis.1c00017. [DOI] [PubMed] [Google Scholar]
- (11).(a) Jessen HJ; Schumacher A; Shaw T; Pfaltz A; Gademann K Angew. Chem. Int. Ed 2011,50, 4222–4226 [DOI] [PubMed] [Google Scholar]; (b) Breugst M; Mayr H J. Amer. Chem. Soc 2010,132,15380–15389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.