
Figure 1: Schematic representation of the possible ER stress mechanisms that mediate diabetes-induced increase in ischemic cell death:
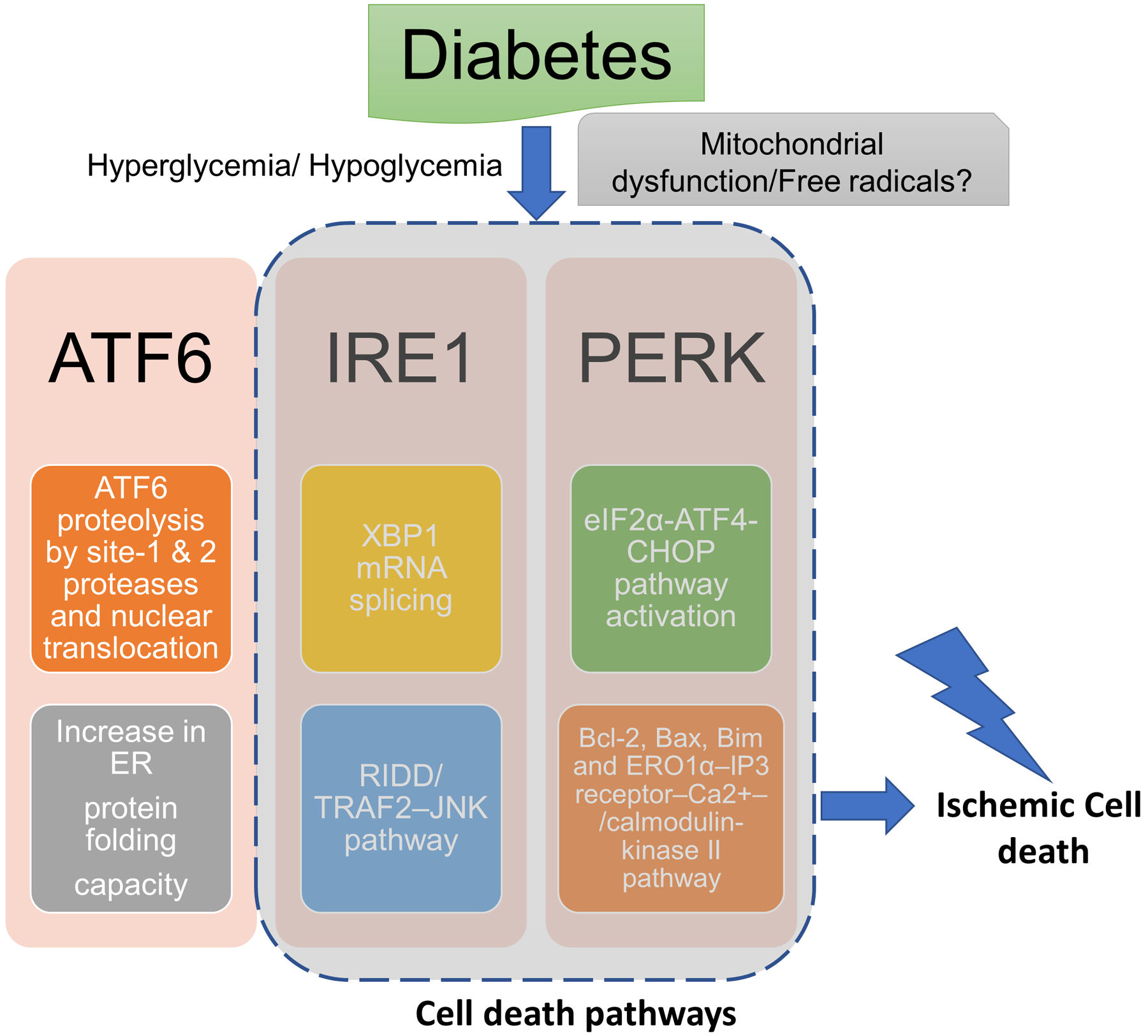
Diabetic hypoglycemia/ hyperglycemia may activate endoplasmic reticulum (ER) stress via mitochondrial dysfunction-induced free radical generation. The major possible pathways of ER stress that may mediate pronounced ischemic brain injury in diabetes: (1) activating transcription factor-6 (ATF6) translocates from the ER compartment of the cell to the Golgi apparatus where it is activated by site-1 and site-2 proteases via proteolysis. The activated ATF6 then moves to the nucleus and stimulates the production of UPR proteins via ER stress response element dependent changes in gene transcription. This binds to the ER stress response element and activates the transcription of UPR target genes which corrects ER stress. (2) inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1α) splices X-box protein 1 mRNA resulting in the activation of regulated IRE1-dependent decay (RIDD) and the IRE1α cytosolic domain activates tumor necrosis factor receptor-associated factor (TRAF)-TRAF2–JUN N-terminal kinase (JNK) signaling. (2) PERK causes phosphorylation of eukaryotic translation initiation factor 2 subunit-α (eIF2α) resulting in inhibition of protein synthesis by reducing mRNA translation initiation. Nevertheless, PERK-eIF2α pathway causes increase in the levels of ATF4 and C/EBP homologous protein (CHOP) and associated cell death pathways.