

Abstract
Alcohol consumption remains a leading cause of liver disease worldwide, resulting in a complex array of hepatic pathologies, including steatosis, steatohepatitis, and cirrhosis. Individuals who progress to a rarer form of alcohol-associated liver disease (ALD), alcohol-associated hepatitis (AH), require immediate life-saving intervention in the form of liver transplantation. Rapid onset of AH is poorly understood and the metabolic mechanisms contributing to the progression to liver failure remain undetermined. While multiple mechanisms have been identified that contribute to ALD, no cures exist and mortality from AH remains high. To identify novel pathways associated with AH, our group utilized proteomics to investigate AH-specific biomarkers in liver explant tissues. The goal of the present study was to determine changes in the proteome as well as epigenetic changes occurring in AH. Protein abundance and acetylomic analyses were performed utilizing nHPLC-MS/MS, revealing significant changes to proteins associated with metabolic and inflammatory fibrosis pathways. Here, we describe a novel hepatic and serum biomarker of AH, glycoprotein NMB (GPNMB). The anti-inflammatory protein GPNMB was significantly increased in AH explant liver and serum compared to healthy donors by 50-fold and 6.5-fold, respectively. Further, bioinformatics analyses identified an AH-dependent decrease in protein abundance across fatty acid degradation, biosynthesis of amino acids, and carbon metabolism. The greatest increases in protein abundance were observed in pathways for focal adhesion, lysosome, phagosome, and actin cytoskeleton. In contrast with the hyperacetylation observed in murine models of ALD, protein acetylation was decreased in AH compared to normal liver across fatty acid degradation, biosynthesis of amino acids, and carbon metabolism. Interestingly, immunoblot analysis found epigenetic marks were significantly increased in AH explants, including Histone H3K9 and H2BK5 acetylation. The increased acetylation of histones likely plays a role in the altered proteomic profile observed, including increases in GPNMB. Indeed, our results reveal that the AH proteome is dramatically impacted through unanticipated and unknown mechanisms. Understanding the origin and consequences of these changes will yield new mechanistic insight for ALD as well as identify novel hepatic and serum biomarkers, such as GPNMB.
Keywords: Alcohol-associated hepatitis, histone, acetylation, proteomics
Graphical Abstract
INTRODUCTION
Alcohol consumption remains a leading cause of hepatic pathology worldwide and is one of the greatest sources of preventable morbidity and mortality 1, 2. Alcohol-associated liver disease (ALD) comprises a spectrum of pathologies, including metabolic syndrome, steatosis, steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma 3. The etiology underlying the progression and development of ALD is multifactorial, including variables such as diet, exercise, genetics, microbiome, and comorbid conditions such as autoimmune disorders or diabetes. Inflammatory responses are also a central component of ALD, where hepatic infiltration by leukocytes is a hallmark of alcohol-associated hepatitis (AH) 4, 5. Critically, the onset of severe AH in some patients requires rapid action in the form of liver transplantation 6–8. The pathological hepatic responses are complicated and varied; therefore, the potential to diagnose and treat patients prior to the onset or progression to AH is a pressing need. Indeed, recent evidence suggests that over 1% of all U.S. hospital admissions annually are due to AH 9. Identifying factors involved in the acute onset of AH is of paramount importance toward more rapid diagnosis, development of therapeutic interventions, and reducing the financial and psychological costs of these hospital admissions 10, 11.
Among other effects, excessive alcohol consumption induces significant alterations in hepatic biochemistry through oxidative metabolism of ethanol to acetaldehyde and acetate 12. Persistent accumulation of these metabolites alters central carbon metabolism including glycolysis, lipid metabolism, and the TCA cycle which impact downstream processes such as anabolic metabolism and wound healing 13–16. Additional consequences include the activation of inflammatory responses; however, the interplay between metabolic and inflammatory mechanisms contributing to AH remain complex and wholly undefined 5. Furthermore, the generation of excess hepatic acetaldehyde and acetate has been shown to perturb proteomic and epigenetic regulation through the disruption of lysine acetylation 17–19. Over the last decade, numerous reports utilizing rodent models have shown that alcohol metabolism increases protein acetylation, including mitochondrial and cytosolic targets 19–28. Unfortunately, translating the consequences of these altered modifications to the human condition remains onerous. Lysine acetylation is thought to provide a snapshot into the active metabolic profile of the liver while also leaving an epigenetic imprint, altering chromatin structure and gene expression through histone acetylation 29, 30. Revealing how persistent and prolonged alcohol-induced lysine acetylation alters proteomic and epigenetic marks in AH patients may provide key data contributing to understanding the metabolic and inflammatory perturbations that occur during AH.
Given that novel biomarkers are lacking in the field, we took a collective approach using proteomic analyses to quantify changes in the proteome and associated PTMs in an unbiased way. Indeed, our proteomic investigation provides novel insight into alcohol-induced feedback mechanisms surrounding hepatocyte metabolism, inflammation, hepatocyte repair, and epigenetic marks. Several key metabolic pathways demonstrated an AH-induced decrease in protein abundance, while inflammatory and repair processes were found to be significantly increased. Comparing our proteomic results with previously published transcriptomic data revealed a significant level of overlap among biochemical processes and pathways even though the exact gene and protein identifications were quite different. Lysine acetylation was largely decreased throughout the proteome, indicating a diminished hepatic metabolic state. Interestingly, histone acetylation was significantly increased at two known regulatory sites, suggesting chronic alcohol consumption leads to persistent changes in the hepatic epigenome of patients suffering from AH. Finally, our proteomic analyses provide a novel catalogue of potential biomarkers for follow-up investigation, including glycoprotein NMB (GPNMB) whose expression is reported to be regulated by histone acetylation 31.
MATERIALS AND METHODS
Study Approval and Clinical Characterization
The clinical research was approved by the Human Ethics Committee of Johns Hopkins University Hospital (IRB 00107893). Written, informed consent was obtained from all participants and was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). Deidentified human samples and data were used in accordance with the NIH and institutional guidelines for human subject research. This study evaluated 5 normal donor liver tissues and 6 liver tissues from AH patients who were referred for liver transplantation at Johns Hopkins University Hospital 7, 8. No information is provided for liver tissue from normal patients as the center acquiring the normal donor livers did not have IRB approval to collect information on the donors. Serum from normal patients was obtained at the University of Colorado (IRB 10–0354) and were age, sex, and race matched to AH patients (Supplemental Data). Alcohol-associated hepatitis patients were comprised of ages ranging from 32 to 61 years old. Four patients were male and two were female. All patients were Caucasian. Five of the six alcohol-associated hepatitis patients had fibrosis indicated on their pathological report. One alcohol-associated hepatitis patient was currently smoking with the remainder of the cohort being former smokers. Current data and information obtained utilizing the clinical resource associated R24 NIH NIAAA grant (AA025017) can be found here: https://www.hopkinsmedicine.org/transplant/research/alcoholic-hepatitis.html
Immunohistochemistry of Healthy and AH Liver Sections
Tissue slides containing human liver explants from AH and healthy donor tissue were obtained from Johns Hopkins University 7, 8. Immunohistochemistry (IHC) was performed using a previously published citrate-based antigen retrieval method (Millipore Sigma) by incubating hepatic sections with primary antibody targeted for GPNMB (R&D systems, #AF2550) for 1 hour at room temperature 25. Detection utilized a biotinylated secondary goat antibody and diaminobenzidine as previously published 32. Immunohistochemical detection of CD68 was performed by the University of Colorado Cancer Center Pathology Shared Resource. The negative control, using no primary antibody, and positive control was performed on human hepatic end-stage liver disease or normal human liver for GPNMB and CD68, respectively. Images for the controls are included in the Supplemental Materials. IHC images were obtained using an Olympus IX73 (Waltham, MA) microscope and an Olympus 5 MP DP27 camera. Images were cropped for publication using Photoshop CS2 (Adobe Systems, Inc., San Jose, CA). Four individual images were taken from each slide and the average percent of positive pixels/field calculated, then the average of each group arrived at by averaging these values for each slide.
Sample Preparation for Protein Abundance and Acetylomic Quantitation by Mass Spectrometry
Adapted from a previously published protocol, protein (3.6 mg) was prepared from normal (n=5) and AH liver explants (n=4) and were ground by mortar and pestle under liquid nitrogen prior to homogenization 33. Protein samples were spiked with 50 ng of acetylated bovine serum albumin (BSA) as an internal standard, trypsin-digested overnight, acidified using trifluoroacetic acid, and purified via Sep-Pak® C18 Classic Cartridges (Waters, #WAT051910). An aliquot of 54 μg was evaporated to dryness and stored at −80°C for general protein quantitation (see below). Each remaining sample was frozen at −80°C for 4 hours and lyophilized for 48 hours. Peptides were then incubated for 2 hours at 4°C with immunoaffinity beads conjugated to acetyl-Lys antibody (Cell Signaling, #13416). After incubation, supernatants were removed and the beads were washed 2 times with IAP buffer (Cell Signaling, #9993) and 3 times with LC-MS grade water (Honeywell). Peptides were eluted from the beads with 0.15% trifluoroacetic acid twice and purified on Pierce® C18 Spin tips (Thermo Scientific, #84850), evaporated to dryness, and stored at −80°C.
Acetylation Quantitation
Chromatography, mass spectrometry, SpectrumMill database search, and Profinder data extraction parameters were performed as described previously 33. Briefly, immunoprecipitated acetyl-Lys peptides were analyzed via 6550 Q-TOF with a nano source (Agilent) using intensity-dependent CID MS/MS to generate peptide identifications and in MS-only mode for acetyl-Lys peptide quantitation. Pooled samples were used for generation of an accurate mass retention time library (AMRT) library, used for acetyl peptide identification in normal and AH samples run in MS-only mode for quantitation. Acetyl peptide extraction and alignment was exported to Mass Profiler Professional V.14.9 (Agilent) and peak area was used for quantitation. Acetyl peptides (quantified with 2 or more unique peptides) were normalized to the appropriate relative protein fold-change (FC) within each sample. Raw area abundances in each acetyl peptide sample were multiplied by the relevant FC normalization for the specific protein and sample. Statistical analysis was performed at the peptide level.
General Protein Quantitation
Aliquots from each sample (54 μg) were dissolved in 30 fmol/μL of retention time standard mix (PROCAL RT Mix) in 3% ACN, 0.1% formic acid in water for LC-MS/MS analysis. A BCA assay validated sample abundance and each sample was analyzed at a final concentration of 1.2 μg/μL. A total pooled sample was used for LC-MS/MS peptide identification and the remaining sample was used for MS-only peptide quantitation. Here, 15 μg of hepatic protein was resolved on-line using a 2.1 × 250 mm, 2.7μ Advancebio peptide mapping column (Agilent) using a 1290 Infinity II LC system (Agilent). Mobile phases consisted of water + 0.1% formic acid (A) and 90% aq. acetonitrile + 0.1% formic acid (B) at a flow rate of 0.2 mL/minute using a gradient holding 5% B over 1 minute, 5–8% B over 1 minute, 8–30% B over 77 minutes, and 30–40% B over 11 minutes for a total 90-minute gradient at 50°C followed by a column wash at 90% B for 5 minutes. Data was collected on a 6550 Q-TOF with dual AJS source (Agilent) and using intensity-dependent CID MS/MS for peptide identification and library, and in MS-only mode for protein quantitation. MS/MS data was collected in positive ion mode across 290–1700 m/z at a scan rate of 10 spectra/second for MS scans and 50–1700 m/z at a scan rate of 3 spectra/second for MS/MS scans. All charge states, excluding singly charged, were allowed during MS/MS acquisition. Charge states of 2 and 3 were given preference. MS-only data was collected in positive ion mode over mass ranges 290–1700 m/z at a scan rate of 1.5 spectra/second. SpectrumMill software (Agilent) was used to extract, search, and summarize peptide data. Spectra were searched against the SwissProt Homo sapiens database and a custom PROCAL RT mix database with up to 2 missed tryptic cleavages and fixed carbamidomethyl (C) and variable deamidated (NQ) and oxidation (M) modifications. The monoisotopic peptide mass tolerance was ± 20.0 ppm and MS/MS tolerance was ± 50 ppm. A minimum peptide score of 8, scored peak intensity of 50%, and protein score of 10 were used as cut-offs for the AMRT library.
Using the polynomial interpolation algorithm, MS-only quantitative data files were retention time warped to the normal liver pooled group MS/MS data file, extracted and aligned using a recursive workflow with Profinder V.B.10.00 software (Agilent) as previously described 33. Peptides were annotated using ID Browser software (Agilent) by matching the mass and retention time from aligned experimental data to peptide hits in the AMRT library generated from MS/MS data. A 15 ppm mass error and 0.4 minute RT drifts were allowed for initial matching. Annotations that had a score lower than 50 or mass ppm error greater than ±10 ppm were removed, and results were imported into Mass Profiler Professional for statistical analysis. Procal RT mix peptides were extracted and compared across sample files to ensure low technical platform variance.
Due to the limited explant sample size and quantity, specific chromatin or histone enrichments were not performed for nHPLC-MS/MS analysis via acetyl-Lys immunoprecipitation, but a targeted analysis of histone acetylation was performed utilizing antibody immunoblotting as detailed below.
Protein Pathway and KEGG Enrichment Analysis
Pathway analysis was performed as previously described using the Database for Annotation, Visualization, and Integrated Discovery (DAVID, version 6.8) 33. Volcano plot analysis and enrichment of biological pathways via KEGG was applied to proteins and acetyl peptides found in both normal and AH samples. A list of Uniprot IDs for proteomic and acetylomic results were uploaded to the database. The human proteome was the reference background. Enrichment analysis used both functional annotation charts and functional annotation clustering to cluster similar terms correlated with our protein list into groups organized by function. Significance was measured by p-value using Fisher’s exact test (EASE score). The Benjamini-Hochberg procedure was used to globally correct enrichment p-values of terms. A threshold of significance was set at p<0.05.
GPNMB ELISA
Human serum samples for AH patients were provided by Johns Hopkins University. Age, sex, and race matched normal patient serum was obtained at the University of Colorado. Human serum was diluted 1:40 with 1% bovine serum albumin in PBS. The concentration of GPNMB in the serum was quantified using a commercial sandwich ELISA kit from R&D Systems (#DY2550) according to the manufacturer’s instructions. The AH patients GPNMB levels are reported as a percentage of the normal patients’ serum average.
Proteomic Comparison with RNA-seq Data
The proteomic dataset was compared to a recently published transcriptomic dataset that was performed on this tissue cohort (GSE143318) 34. Proteins that had a FC greater than 1.25 and a p-value less than 0.05 were submitted for DAVID analysis and were compared to the genes from the RNA-seq dataset with an FDR less than 0.05 and log2FC greater than 0.06. The top twenty KEGG pathways and GO terms with the highest number of genes from the proteomics analysis were utilized to determine overlap with the previously reported transcriptomics dataset.
Chromatin Extraction
Chromatin was enriched from normal and AH liver explant tissue for analysis. Liver tissue was dounce homogenized in hypotonic lysis buffer (10 mM HEPES/KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5% Igepal (v/v), 5 mM sodium butyrate, protease and phosphatase inhibitors, 10 mM nicotinamide, 1 μM trichostatin A). Homogenates were incubated on ice for 30 minutes to lyse cells and subsequently centrifuged at 1000×g for 10 minutes at 4°C. The pellets were then washed thrice with hypotonic lysis buffer. The nuclear pellet was resuspended in high salt buffer (20 mM HEPES/KOH pH 7.9, 1.5 mM MgCl2, 420 mM KCl, 0.2 mM EDTA, 25% glycerol (v/v), 5 mM sodium butyrate, protease and phosphatase inhibitors, 10 mM nicotinamide, 1 μM trichostatin A) and rotated at 4°C overnight. The homogenates were spun at 4°C for 20 minutes at 1000×g. The resultant pellet was resuspended in water containing 0.2 mM EDTA, 5 mM sodium butyrate, protease and phosphatase inhibitors, and 1 μM trichostatin A.
Western Blotting
Lysates were separated using a reducing denaturing polyacrylamide gel and transferred to PVDF membrane. Sample loading was verified following protein transfer using stain free technology along with a trihalo compound included in the gel casting process. Membranes were blocked with Licor Odyssey Blocking Buffer for 1 hour at 25°C. Primary antibodies were purchased from Cell Signaling Technology and include Histone H3 acK9 (#9649), Histone H3 acK14 (#7627), Histone H3 acK18 (#13998), Histone H3 acK27 (#8173), Histone H3K ac56 (#4243), Histone H3 (#4499), Histone H2A (#12349), Histone H2B (12364), Histone H2B acK5 (#12799), Histone H4 acK8 (#2594), Histone H4 acK16 (#13534), Histone H4 (#13919) and acetyl-Lys (#9441). Membranes were incubated overnight at 4° C, washed thrice with phosphate buffered saline containing 1% Tween-20 (PBS-T), and subsequently incubated in IRDye 800CW goat anti-rabbit secondary antibody, washed 3x in PBS-T and imaged on a Bio-Rad ChemiDoc MP. The signal from the acetyl-Lys-specific antibody was normalized to the corresponding signal of the unmodified histone which were unchanged between the two patient groups.
Statistical Analysis
General protein and acetyl peptides were filtered to those found in 100% of 1 of 2 conditions for group-to-group comparisons. Compounds were filtered on volcano plots using a moderated t-test and peptides that had a FC ≤ −1.5 or ≥ 1.5 and p-value < 0.05 were considered significant. Benjamini-Hochburg multiple-testing correction was also applied to generate a list of acetyl site candidates or proteins that had differential abundance with high confidence that could be used to probe the effects of acetylation modifications in individual proteins. Error bars depict the standard error of the mean.
RESULTS
Clinical Characterization of AH Explant Tissue
As shown in Table 1, several serum parameters were monitored in these patients, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (AP), creatinine, albumin, and bilirubin. These clinical data were assessed alongside BMI, which ranged from 22.9 to 36.9, representing a range of normal to obese in patients with AH. This clinical data clearly showed the need for a biomarker due to these variable parameters.
Table 1.
Clinical characterization of alcohol-associated hepatitis explant patients.
| Patient | Age (Y) | Sex | Race | Listed MELD | WBC (K/cu mm) | Platelets (K/cu mm) | AST (U/L) | ALT (U/L) | AP (U/L) | Creatinine (mg/dL) | Albumin (g/dL) | Bilirubin (mg/dL) | BMI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| AH1 | 32 | M | Caucasian | 34 | 23.7 | 147 | 141 | 76 | 188 | 2.1 | 3.2 | 38.8 | 36.9 |
| AH2 | 34 | F | Caucasian | 34 | 5.7 | 119 | 144 | 49 | 87 | 1.5 | 3.7 | 27.7 | 26.7 |
| AH3 | 49 | F | Caucasian | 38 | 17.0 | 27 | 127 | 52 | 175 | 1 | 2.9 | 14.2 | 26.2 |
| AH4 | 48 | M | Caucasian | 45 | 8.7 | 66 | 121 | 41 | 110 | 2.7 | 4 | 48 | 30.1 |
| AH5 | 41 | M | Caucasian | 34 | 14.0 | 57 | 95 | 44 | 129 | 0.9 | 2.9 | 33.4 | 22.9 |
| AH6 | 61 | M | Caucasian | 38 | 7.9 | 45 | 71 | 36 | 98 | 1.5 | 3.4 | 35.6 | 26.3 |
|
| |||||||||||||
| Normal Range | 13.9–16.3 | 0–37 | 0–40 | 30–120 | 0.6–1.3 | 3.5–5.3 | 0–1.2 | 0.3–1.9 | 18.5–24.9 | ||||
Y: years; MELD: Model for End-stage Liver Disease; WBC: white blood cells; K/cu mm: thousand per cubic millimeter; U/L: units per liter; AST: aspartate aminotransferase; ALT: alanine aminotransferase; AP: alkaline phosphatase; BMI: body mass index
Protein Quantitation Revealed a Novel Biomarker in GPNMB
Overall, protein quantitation provided a confirmatory analysis of ALD-mediated proteome alterations and as a reference for quantifying changes in protein acetylation. Here, in the context of AH, our quantitative proteomic method identified 7,486 peptides corresponding to a total of 895 unique protein targets. Compared to normal explants, a total of 391 proteins were found to have significantly increased abundance (Table 2), while 504 proteins were significantly decreased (Table 3 & Figure 1B). Overall, the significantly altered proteins were enriched for KEGG pathways that have been previously associated with AH 35–37. Numerous metabolic processes were found to be depressed, while cirrhotic, fibrotic, and inflammatory processes were abundantly activated. Protein pathways found to have decreased abundance include metabolic pathways, biosynthesis of antibiotics, carbon metabolism, fatty acid degradation, drug metabolism, and biosynthesis of amino acids, among many other metabolically related pathways. Pathways where protein abundance was increased include focal adhesion, regulation of actin cytoskeleton, lysosome, PI3K-Akt signaling, phagosome, extracellular matrix receptor interaction, bacterial invasion of epithelial cells, amoebiasis, and alcoholism (Figure 1).
Table 2.
KEGG pathways and associated proteins found to be increased in AH.
| KEGG Pathway Enrichment | Uniprot Accession Gene ID for Increased Proteins |
|---|---|
| Focal adhesion | ACTB, ACTN1, ACTN4, CDC42, COL1A1, COL1A2, COL4A1, COL4A2, COL6A1, COL6A2, COL6A3, FLNA, FN1, ILK, ITGA1, ITGB1, LAMB1, MYL12A, MYL9, PARVA, PPP1CA, RHOA, TLN1, TNC, TNXB, THBS1, VCL, VTN |
| Regulation of actin cytoskeleton | ACTB, ACTN1, ACTN4, ARPC1A, ARPC1B, ARPC2, ARPC4, CDC42, CFL1, EZR, FN1, GSN, IQGAP1, ITGA1, ITGB1, MSN, MYL12A, MYL9, PFN1, PPP1CA, RHOA, VCL |
| PI3K-Akt signaling pathway | COL1A1, COL1A2, COL4A1, COL4A2, COL6A1, COL6A2, COL6A3, FN1, GNB1, ITGA1, ITGB1, LAMB1, TNC, TNXB, THBS1, YWHAB, YWHAQ, YWHAZ, VTN |
| Lysosome | ASAH1, AP1B1, CTSA, CTSB, CTSC, CTSD, CTSZ, CLTC, GAA, GLB1, HEXA, HEXB, LAMP1, LAMP2, LIPA, PSAP, SCARB2, TPP1 |
| Phagosome | ACTB, ATP6V1A, C3, CANX, CORO1A, DYNC1H1, HLA-DRA, ITGB1, LAMP1, LAMP2, MPO, RAB7A, THBS1, TUBA1A, TUBB1, TUBB2A, TUBB4B, TUBB |
| ECM-receptor interaction | COL1A1, COL1A2, COL4A1, COL4A2, COL6A1, COL6A2, COL6A3, FN1, HSPG2, ITGA1, ITGB1, LAMB1, THBS1, TNC, TNXB, VTN |
| Pathogenic Escherichia coli infection | ACTB, ARPC1A, ARPC1B, ARPC2, ARPC4, CDC42, EZR, ITGB1, NCL, RHOA, TUBA1A, TUBB1, TUBB2A, TUBB4B, TUBB |
| Proteoglycans in cancer | ACTB, CDC42, DCN, EZR, FLNA, FN1, HSPG2, IQGAP1, ITGB1, LUM, MSN, PPP1CA, RHOA, THBS1, VTN |
| Bacterial invasion of epithelial cells | ACTB, ARPC1A, ARPC1B, ARPC2, ARPC4, CDC42, CLTC, FN1, ILK, ITGB1, RHOA, SEPTIN11, SEPTIN2, VCL |
| Leukocyte transendothelial migration | ACTB, ACTN1, ACTN4, CDC42, EZR, GNAI1, GNAI2, ITGB1, MSN, MYL12A, MYL9, RHOA, VCL |
| Systemic lupus erythematosus | ACTN1, ACTN4, C3, C4A, ELANE, H2AFY, HIST1H2AA, HIST1H2AE, HIST1H2BA, HIST1H2BB, HIST1H3A, HIST1H4I, HLA-DRA |
| Viral carcinogenesis | ACTN1, ACTN4, C3, CDC42, GSN, HIST1H2BA, HIST1H2BB, HIST1H4I, PKM, RHOA, YWHAB, YWHAQ, YWHAZ |
| Endocytosis | APRC1B, ARPC1A, ARPC2, ARPC4, CAPZA1, CAPZB, CDC42, CLTC, EHD2, RAB11A, RAB7A, RHOA, VPS35 |
| Alcoholism | CALM1, GNAI1, GNAI2, GNB1, H2AFY, HIST1H2AA, HIST1H2AE, HIST1H2BA, HIST1H2BB, HIST1H3A, HIST1H4I, PPP1CA |
Table 3.
KEGG pathways and associated proteins found to be decreased in AH.
| KEGG Pathway Enrichment | Uniprot Accession Gene ID for Decreased Proteins |
|---|---|
| Metabolic pathways | AASS, ABAT, ACAA1, ACAA2, ACADM, ACADS, ACADSB, ACAT1, ACAT2, ACO1, ACOT1, ACOT1, ACOX1, ACOX2, ACSL1, ACSL5, ACSM2A, ACSM2B, ACSS3, ACY1, ADH1A, ADH1B, ADH1C, ADH4, ADH6, ADK, AGL, AGMAT, AGXT, AHCY, AK2, AK4, AKR1A1, AKR1C3, AKR1C4, AKR1D1, ALAD, ALDH1A1, ALDH1B1, ALDH2, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDH9A1, ALDOB, ALDOC, AMDHD1, ANPEP, AOX1, ARG1, ASL, ASS1, ATP5A1, ATP5B, ATP5C1, ATP5F1, ATP5H, ATP5J, ATP5O, BDH1, BHMT, CBR1, CBS, CES1, CMBL, COMT, CPS1, CRYL1, CTH, CYP1A2, CYP27A1, CYP2C8, CYP2C9, CYP2E1, CYP3A4, CYP4F2, CYP8B1, DCXR, DHCR7, DHRS4, DMGDH, DPYS, ECHS1, EHHADH, ENO3, EPHX2, FAH, FASN, FBP1, FDPS, FH, FTCD, GALK1, GATM, GBE1, GCDH, GFPT1, GLDC, GLUD1, GOT2, GPT, GRHPR, GSTZ1, H6PD, HAAO, HADH, HADHA, HADHB, HAO1, HGD, HIBADH, HIBCH, HMGCS1, HMGCS2, HPD, HSD11B1, HSD17B10, HSD17B6, HSD17B8, IDH1, IDH2, KHK, LAP3, LDHAL6B, MAOA, MAOB, MAT1A, MCCC2, MDH2, MGLL, MPST, MTHFD1, MUT, NADK2, NAMPT, NAPRT, NDUFA4, NDUFAB1, NDUFS3, OTC, PAH, PAICS, PC, PCCA, PCK1, PCK2, PGM1, PHGDH, PIPOX, PKLR, PNP, PRDX6, PRPS1, PSAT1, PYGB, PYGL, QDPR, QPRT, RDH16, RGN, RPN1, SARDH, SCP2, SDHA, SDS, SHMT1, SHMT2, SLC27A5, SORD, STT3A, SUCLG1, SUCLG2, TKFC, TST, UGDH, UGP2, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7, UPB1, UQCRC2 |
| Biosynthesis of antibiotics | AASS, ACAA1, ACAA2, ACADM, ACAT1, ACAT2, ACO1, ACY1, AGXT, AK2, AK3, AK4, AKR1A1, ALDH1B1, ALDH2, ALDH3A2, ALDH7A1, ALDH9A1, ALDOB, ALDOC, ARG1, ASL, ASS1, CAT, CBS, CMBL, CTH, ECHS1, EHHADH, ENO3, FBP1, FDPS, FH, GCDH, GFPT1, GLDC, GOT2, HADH, HADHA, HADHB, HAO1, HMGCS1, HMGCS2, HSD17B10, IDH1, IDH2, LDHAL6B, MDH2, OTC, PAICS, PCCA, PCK1, PCK2, PCYOX1, PGM1, PGM3, PHGDH, PKLR, PRPS1, PSAT1, RGN, SDHA, SDS, SHMT1, SHMT2, SUCLG1, SUCLG2, UGP2 |
| Carbon metabolism | ACADM, ACADS, ACAT1, ACAT2, ACO1, AGXT, ALDH6A1, ALDOB, ALDOC, CAT, CPS1, ECHS1, EHHADH, ENO3, FBP1, FH, GLDC, GLUD1, GOT2, GPT, H6PD, HADHA, HAO1, HIBCH, IDH1, IDH2, MDH2, MUT, PCCA, PCCA, PHGDH, PKLR, PRPS1, PSAT1, RGN, SDHA, SDS, SHMT1, SHMT2, SUCLG1, SUCLG2, TKFC |
| Fatty acid degradation | ACAA1, ACAA2, ACADM, ACADS, ACADSB, ACAT1, ACAT2, ACOX1, ACSL1, ACSL5, ADH1A, ADH1B, ADH1C, ADH4, ADH6, ALDH1B1, ALDH2, ALDH3A2, ALDH7A1, ALDH9A1, ECHS1, ECI1, ECI2, EHHADH, GCDH, HADH, HADHA, HADHB |
| Valine, leucine and isoleucine degradation | ABAT, ACAA1, ACAA2, ACADM, ACADS, ACADSB, ACAT1, ACAT2, ALDH1B1, ALDH2, ALDH3A2, ALDH6A1, ALDH7A1, ALDH9A1, AOX1, ECHS1, EHHADH, HADH, HADHA, HADHB, HIBADH, HIBCH, HMGCS1, HMGCS2, HSD17B10, MCCC2, MUT, PCCA |
| Metabolism of xenobiotics by cytochrome P450 | ADH1A, ADH1B, ADH1C, ADH4, ADH6, AKR1C1, AKR1C2, AKR1C4, AKR7A2, AKR7A3, CBR1, CYP1A2, CYP2C9, CYP2E1, CYP3A4, EPHX1, GSTA1, HSD11B1, MGST1, SULT2A1, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7 |
| Biosynthesis of amino acids | ACO1, ACY1, ALDOB, ALDOC, ARG1, ASL, ASS1, CBS, CPS1, CTH, ENO3, GOT2, GPT, IDH1, IDH2, MAT1A, OTC, PAH, PC, PHGDH, PKLR, PRPS1, PSAT1, SDS, SHMT1, SHMT2 |
| Drug metabolism - cytochrome P450 | ADH1A, ADH1B, ADH1C, ADH4, ADH6, AOX1, CYP1A2, CYP2C8, CYP2C9, CYP2E1, CYP3A4, FMO3, FMO5, GSTA1, MAOA, MAOB, MGST1, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7 |
| Chemical carcinogenesis | ADH1A, ADH1B, ADH1C, ADH4, ADH6, CBR1, CY2E1, CYP1A2, CYP2C8, CYP2C9, CYP3A4, EPHX1, GSTA1, HSD11B1, MGST1, SULT1A1, SULT2A1, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7 |
| Retinol metabolism | ADH1A, ADH1B, ADH1C, ADH4, ADH6, ALDH1A1, AOX1, CYP1A2, CYP2C8, CYP2C9, CYP3A4, DHRS4, HSD17B6, RDH16, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7 |
| Peroxisome | ACAA1, ACOX1, ACOX2, ACSL1, ACSL5, AGXT, CAT, CRAT, DHRS4, ECH1, ECI2, EHHADH, EPHX2, HAO1, IDH1, IDH2, PIPOX, PRDX1, SCP2, SLC27A2, SOD1 |
| Glycine, serine and threonine metabolism | AGXT, ALDH7A1, BHMT, CBS, CTH, DMGDH, GAMT, GATM, GLDC, GNMT, GRHPR, MAOA, MAOB, PHGDH, PIPOX, PSAT1, SARDH, SDS, SHMT1, SHMT2 |
| Steroid hormone biosynthesis | AKR1C1, AKR1C2, AKR1C3, AKR1C4, AKR1D1, COMT, CYP1A2, CYP2E1, CYP3A4, HSD11B1, HSD17B6, HSD17B8, UGT1A10, UGT1A3, UGT1A4, UGT1A6, UGT2A1, UGT2B15, UGT2B4, UGT2B7 |
| Glycolysis / Gluconeogenesis | ADH1A, ADH1B, ADH1C, ADH4, ADH6, AKR1A1, ALDH1B1, ALDH2, ALDH3A2, ALDH7A1, ALDH9A1, ALDOB, ALDOC, ENO3, FBP1, LDHAL6B, PCK1, PCK2, PGM1, PKLR |
| Protein processing in endoplasmic reticulum | HSP90B1, HSPA1A, HSPA5, HYOU1, LMAN1, LMAN2, P4HB, PDIA4, PDIA6, RPN1, RRBP1, SAR1A, SAR1B, SEC23A, SEC24C, SEC61A1, SSR1, STT3A, UGGT1 |
| Tryptophan metabolism | ACAT1, ACAT2, ALDH1B1, ALDH2, ALDH3A2, ALDH7A1, ALDH9A1, AOX1, CAT, CYP1A2, ECHS1, EHHADH, GCDH, HAAO, HADH, HADHA, MAOA, MAOB |
| Ribosome | RPL11, RPL12, RPL14, RPL24, RPL3, RPL30, RPL5, RPL9, RPLP0, RPS12, RPS16, RPS25, RPS3A, RPS4X, RPS6, RPS8, RPS9, RPSA |
Figure 1. Proteomic analysis of hepatic tissue from normal and alcohol-associated hepatitis explants.

(A) Schematic of general protein quantitation and acetylation quantitation by mass spectrometry. Normal, n=5; AH, n=4. (B) The pie graph depicts the number of proteins identified to be significantly changed in AH by protein quantitation. (C) The volcano plot represents the individual proteins found to be altered in the alcohol-associated hepatitis samples. The number of associated genes that are significantly changed in a particular pathway is detailed in the bar charts.
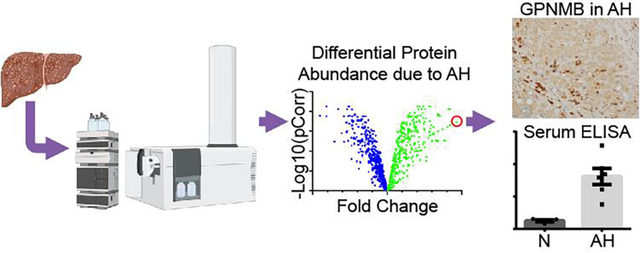
One of the most striking results from the proteomic analysis was that glycoprotein NMB (GPNMB) was found to have a 50-fold increase in AH tissue, the highest AH-induced increase of all identified proteins. Further, to confirm these findings, IHC analyses of GPNMB demonstrated an AH-dependent 13-fold increase in hepatocytes and a marked increase in cells found in the sinusoids (Figure 2A). CD68, a common surface marker of monocytes/macrophages, was utilized to better characterize the highly immunoreactive cells for GPNMB found in the sinusoids (Figure 2B). No differences were found in the number of CD68 immunostained cells between the groups (Figure 2C). A comparison of the images for GPNMB and CD68 from sequential slides made it apparent that the hepatocytes contributed most of the increase of GPNMB in AH. An ELISA was performed using age, gender, and race matched normal and AH patient serum. GPNMB abundance was significantly increased in AH serum by 647% of normal serum (Figure 2D).
Figure 2. GPNMB abundance is increased in AH explant liver and serum compared to normal donors.

Representative images from normal and alcohol-associated hepatitis slides probed for (A) GPNMB or (B) CD68. PT: portal triad; CV: central vein. The rectangle in the low power image denotes the region for the higher power image. Arrows point to immunopositive cells. (C) Quantitation of the immunohistochemistry for GPNMB shows a significant increase in alcohol-associated hepatitis samples. (n=3; * p<0.05) There is no change in CD68 positive cells between the two groups. (D) GPNMB abundance in serum of patients with alcohol-associated hepatitis is increased by 647% of normal serum via ELISA. * p<0.05; ** p<0.01.
RNA-seq Comparison Revealed Differences between Proteomic and Transcriptomic Analyses
Recently, these same AH liver tissues had been analyzed using RNA sequencing (RNA-seq) to identify differentially expressed genes, creating a publicly available RNA-seq dataset. Therefore, we next compared our proteomic results with this previously published transcriptomic dataset 34. The comparison of RNA-seq and proteomic data identified 47 transcripts significantly increased (Figure 3A) while 91 transcripts were significantly decreased in AH liver explants (Figure 3B). Further interrogation of both data sets indicated many commonalities between them despite finding minimal overlap between transcript and protein (Figure 3C&D). Increased KEGG pathways included focal adhesion, actin cytoskeleton, PI3K-Akt signaling, ECM receptor interaction, leukocytes migration, and phagosome. Decreased KEGG pathways shared similarities across metabolic pathways, biosynthesis of antibiotics, carbon metabolism, fatty acid degradation, amino acid metabolism, and drug metabolism. This similarity between the proteomics and transcriptomics extended to GO terms as well (Supplemental Data). These findings demonstrated very little overlap in actual mRNA and proteomic targets and indicated a need to include both analyses in a thorough characterization of patient samples.
Figure 3. Comparative analysis of proteomic and RNA-seq alterations due to AH.

Overall analysis of our proteomic changes in AH with a previously reported RNA-seq analysis on this AH tissue cohort 34. (A) Protein to gene comparison for hits significantly increased in AH explant tissue found 47 genes overlapping between the proteomics and RNA-seq data sets, (B) while 91 genes overlap which are found significantly decreased. DAVID analysis for KEGG pathways related to significantly (C) increased and (D) decreased proteins identified in our proteomic survey were compared with the RNA-seq analysis and revealed a remarkable overlap in KEGG pathways. The dataset for these analyses can be found in Supplemental Data.
Protein Acetylation is Decreased in the AH Liver
Quantitative acetylomic analysis comparing normal and AH explant tissues revealed that 316 acetyl peptides were significantly decreased in AH tissue, after adjusting for specific protein abundance, with a Log2(Fold Change) from −0.35 to −4.21 (Figure 4). Intriguingly, zero acetyl peptides were found to be significantly increased. Western blotting for acetyl-Lys confirmed the acetylomics results with a pattern of decreased acetylation found in the AH lysates (Figure 4A&B). Functional enrichment of the significant acetylated proteins identified a range of metabolically related pathways impacted by decreased acetylation (Table 4). These include biosynthesis of antibiotics, peroxisome, fatty acid metabolism, glycolysis, glyoxylate metabolism, and the TCA cycle. A myriad of metabolically related sites of acetylation were significantly decreased and provide an interesting lead-hit target list for examining altered protein activity, particularly glucose and lipid metabolism.
Figure 4. Acetylomic analysis of hepatic tissue from normal and alcohol-associated hepatitis explants.

(A) Western blotting for global protein acetylation on cytoplasmic-enriched liver lysates revealed a consistent decrease in AH tissue. Normal, n=4; AH, n=5. N: normal; MW: molecular weight ladder (B) Quantitation demonstrated a trend of decreased acetyl-Lys in AH samples. (C) Protein acetylation was found to be significantly decreased by quantitative mass spectrometry in AH versus normal explant tissue, as illustrated in the volcano plot. Normal, n=5; AH, n=4. (D) The bar chart highlights the top 20 KEGG pathways associated with the observed AH-induced decrease in protein acetylation. Overall, analysis of AH explant tissue not enriched for chromatin revealed a decrease in protein acetylation across metabolic, antioxidant, and disease-associated pathways.
Table 4.
KEGG pathways and associated acetylated proteins found to be decreased in AH.
| KEGG Pathway Enrichment | Uniprot Accession Gene ID for Decreased Acetylated Proteins |
|---|---|
| Metabolic pathways | AASS, ABAT, ACAA2, ACADVL, ACAT1, ACO2, ACOT1, ACOX1, ACSM2A, ACSS3, ADH1A, ADH1B, AGMAT, AHCY, AKR1C3, ALDH1A1, ALDH2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDH9A1, ALDOB, AOX1, ASL, ATP5A1, ATP5F1, ATP5H, ATP5O, BAAT, CBR1, CMPK1, COX4I1, COX5B, CPS1, DCXR, DLD, DLST, ECHS1, EHHADH, ENO1, FAHD1, FASN, FH, GAPDH, GLUD1, GOT2, GPI, GRHPR, HADH, HADHA, HADHB, HAO1, HIBCH, HMGCL, HMGCS2, HSD17B4, IDH1, IDH2, IVD, LDHA, MDH1, MDH2, MUT, NADK2, NNT, OTC, PCCB, PCK2, PNPO, PRDX6, PYGL, SCP2, SDHA, SHMT1, SHMT2, SUCLG1, TALDO1, TST, UGP2 |
| Biosynthesis of antibiotics | AASS, ACAA2, ACAT1, ACO2, ALDH2, ALDH7A1, ALDH9A1, ALDOB, ASL, CAT, DLD, DLST, ECHS1, EHHADH, ENO1, FH, GAPDH, GOT2, GPI, HADH, HADHA, HADHB, HAO1, HMGCS2, IDH1, IDH2, LDHA, MDH1, MDH2, OTC, PCCB, PCK2, SDHA, SHMT1, SHMT2, SUCLG1, TALDO1, UGP2 |
| Carbon metabolism | ACAT1, ACO2, ALDH6A1, ALDOB, CAT, CPS1, DLD, DLST, ECHS1, EHHADH, ENO1, FH, GAPDH, GLUD1, GOT2, GPI, HADHA, HAO1, HIBCH, IDH1, IDH2, MDH1, MDH2, MUT, PCCB, SDHA, SHMT1, SHMT2, SUCLG1, TALDO1 |
| Valine, leucine and isoleucine degradation | ABAT, ACAA2, ACAT1, ALDH2, ALDH6A1, ALDH7A1, ALDH9A1, AOX1, DLD, ECHS1, EHHADH, HADH, HADHA, HADHB, HIBCH, HMGCL, HMGCS2, IVD, MUT, PCCB |
| Peroxisome | ACOX1, BAAT, CAT, CRAT, ECH1, EHHADH, GSTK1, HAO1, HMGCL, HSD17B4, IDH1, IDH2, PRDX1, SCP2, SOD1, SOD2 |
| Fatty acid degradation | ACAA2, ACADVL, ACAT1, ACOX1, ADH1A, ADH1B, ALDH2, ALDH7A1, ALDH9A1, ECHS1, ECI1, EHHADH, HADH, HADHA, HADHAB |
| Biosynthesis of amino acids | ACO2, ALDOB, ASL, CPS1, ENO1, GAPDH, GOT2, IDH1, IDH2, OTC, SHMT1, SHMT2, TALDO1 |
| Glyoxylate and dicarboxylate metabolism | ACAT1, ACO2, CAT, DLD, GRHPR, HAO1, MDH1, MDH2, MUT, PCCB, SHMT1, SHMT2 |
| Propanoate metabolism | ABAT, ACAT1, ACSS3, ALDH6A1, ECHS1, EHHADH, HADHA, HIBCH, LDHA, MUT, PCCB, SUCLG1 |
| Glycolysis / Gluconeogenesis | ADH1A, ADH2B, ALDH2, ALDH7A1, ALDH9A1, ALDOB, DLD, ENO1, GAPDH, GPI, LDHA, PCK2 |
| Huntington’s disease | APT5A1, ATP5F1, ATP5H, ATP5J, ATP5O, COX4I1, COX5B, SDHA, SLC25A5, SOD1, SOD2, TGM2 |
| Citrate cycle (TCA cycle) | ACO2, DLD, DLST, FH, IDH1, IDH2, MDH1, MDH2, PCK2, SDHA, SUCLG1 |
| Pyruvate metabolism | ACAT1, ALDH2, ALDH7A1, ALDH9A1, DLD, FH, GRHPR, LDHA, MDH1, MDH2, PCK2 |
| Butanoate metabolism | ABAT, ACAT1, ACSM2A, ALDH5A1, ECHS1, EHHADH, HADH, HADHA, HMGCL, HMGCS2 |
| Tryptophan metabolism | ACAT1, ALDH2, ALDH7A1, ALDH9A1, AOX1, CAT, ECHS1, EHHADH, HADH, HADHA |
| Fatty acid metabolism | ACAA2, ACADVL, ACAT1, ACOX1, ECHS1, EHHADH, FASN, HADH, HADHA, HADHB |
| Lysine degradation | AASS, ACAT1, ALDH7A1, ALDH9A1, ALDK2, DLST, ECHS1, EHHADH, HADH, HADHA |
| Parkinson’s disease | APT5O, ATP5A1, ATP5F1, ATP5H, ATP5J, COX4I1, COX5B, PARK7, SDHA, SLC25A5 |
A broad interpretation of the regulatory nature of protein acetylation reveals that decreased acetylation will enhance protein activity while increased acetylation is inhibitory through charge masking and cofactor blocking, among others. This suggests hepatocyte-specific responses to the progression of ALD may be to activate numerous proteomic pathways through decreased acetylation. Comparing proteomic and epigenetic acetylation profiles would reveal if the overall hepatocyte responses activated gene expression and protein activity among specific pathways or globally.
Histone Acetylation is Increased in AH Tissue
Specific chromatin or histone enrichments were not performed for nHPLC-MS/MS analysis via acetyl-Lys immunoprecipitation due to the limited sample size and quantity. Since histone acetylation is a central regulator of gene expression, a targeted analysis of histone acetylation was performed. Notably, while chromatin was not explicitly enriched or isolated for MS analysis, our acetylomics data identified lysine acetylation on three histones: H2B, H3 and H4 (Supplemental Data). Three lysines were found to have significantly decreased acetylation in AH samples: Histone H2B acK109, Histone H3 acK79, Histone H4 acK77. However, all three lysine sites are not regionally part of histone tails, a region where PTMs are known to influence chromatin compaction, nucleosome dynamics, and transcription 38. Three lysine residues on Histone H3 (H3K14, H3K18, and H3K23) and four lysine residues on Histone H4 (H4K5, H4K8, H4K12, and H4K16) were found acetylated in AH liver (Supplemental Data). Unfortunately, each tryptic peptide attributed to these histones contained multiple acetylated lysine residues, which confounds the accurate quantitation of each individual lysine residue; therefore, they could not be quantified by our MS analysis.
Given the preponderance of lysine acetylation sites and the commercially available site-specific antibodies several histone acetylation sites were quantified via immunoblotting (Figure 5). Histone H3 acK9 and Histone H2B acK5 were significantly increased by 88% and 59%, respectively, compared to normal liver explants. The remaining four lysines of Histone H3 (H3K14, H3K18, H3K27, and H3K56) demonstrated higher variability, but trended toward increased acetylation from 50% to 110% when compared to normal explant tissue. This trend of increased lysine acetylation of histones in AH chromatin continued with the two lysine residues located in the tail of Histone H4 (H4K8 and H4K16), albeit to a lesser extent than Histone H3. The acetylomics decrease was likely due to the absence of histones in our acetyl-Lys immunoprecipitation enriched acetylome analysis. In aggregate, increased histone acetylation and enhanced gene expression appears to be a long-term consequence of alcohol abuse and a characteristic of AH.
Figure 5. Altered histone acetylation profiles in patients with alcohol-associated hepatitis.

The acetylation of histone proteins is known to alter gene expression through epigenetic signaling. Patients diagnosed with alcohol-associated hepatitis demonstrated significant alterations in the acetylation of Histone H2B and H3 specifically at sites that are known to be key epigenetic regulators of numerous metabolic, immunologic, and antioxidant pathways. Normal, n=4; AH, n=5. N:normal; * p<0.05.
DISCUSSION
The pathological mechanisms leading to the progression of AH remain poorly defined. Two recent review articles provide an in-depth analysis of the current state of research regarding the diagnosis and treatment of AH 5, 6. With a particular focus on protein abundance and lysine acetylation, our work significantly adds quantitative and qualitative information regarding AH-mediated changes in the proteome and acetylome to enhance our understanding of mechanisms of ALD. Presumptions regarding how acetylation could impact end-stage ALD pathology are confounding since 1) increased acetylation is a known consequence of active alcohol metabolism in rodent models and 2) decreased acetylation is associated with diminished cytosolic and mitochondrial metabolism, which is likely overrepresented in failing hepatic pathology 19–28. Protein acetylation is a dynamic biomarker of hepatocyte function, providing a link between central carbon metabolism, oxidative stress, and inflammation 29, 39, 40. Therefore, examining this unique post-translational modification in AH explant tissue provides an informative snapshot regarding alcohol-associated liver disease. Proteomic pathways significantly impacted were focused similarly in areas related to inflammation, central carbon metabolism, and oxidative stress. Overall, protein quantitation provides: 1) a holistic readout of up- and down-regulated protein abundance in ALD, 2) a baseline adjustment for the protein acetylation quantitation, and 3) a highlight of proteins involved in physiological processes underlying AH that provide a clear biomarker or therapeutic target. Most targets identified by the proteomic and acetylomic analysis demonstrate poignant changes in the hepatic proteome that provide an association between histone acetylation and protein abundance, revealing novel biomarkers while also defining new areas of interest.
The most significant finding from the proteomic analysis is that glycoprotein NMB (GPNMB) had the highest increase in abundance (50-fold and 13-fold by MS and IHC quantitation, respectively) of all proteins in AH tissue. Surprisingly, even though a dramatic increase was quantified in the AH explant tissue, it was not identified as an acetylated protein. GPNMB has not previously been reported to be increased in the liver of patients or in rodent models of ALD. The merit of protein quantitation is further justified as GPNMB was identified with this technique, but not by previous RNA-seq analysis. This highlights the value of integrating proteomic analysis in biomarker screens for ALD. ELISA analysis of serum also found that GPNMB serves as a unique biomarker of AH with over a 6-fold increase compared with normal serum. Interestingly, increased abundance of serum GPNMB in patients was recently reported as a promising biomarker and potential therapeutic target for fatty liver disease 41. That study found that the amount of GPNMB in hepatic macrophages and stellate cells plays a key role in antioxidant capacity and steatosis, suggesting a role in tissue repair. The authors also found that overexpression of GPNMB ameliorated hepatic steatosis and fibrosis in a diet-induced obesity model of NASH 41. A number of different research groups identified GPNMB as a regulator of M2 macrophage polarization and a key marker of renal inflammation, injury, and tissue repair 42–44. Additionally, GPNMB expression was highly upregulated in ischemic kidney injury in a rodent model, with a 15-fold increase in kidney tissue and 10-fold in macrophages, revealing Gpnmb as a novel pro-repair gene necessary for integrating the macroautophagic degradation pathway and phagocytosis 43. One report found that increasing histone acetylation through HDAC inhibition with TSA led to an increased expression of GPNMB 31, providing a direct link between the acetyl-directed regulation of histones and GPNMB expression. Together, these data suggest AH-induced histone acetylation may activate hepatic repair processes involving GPNMB. Likely, the upregulation of GPNMB is necessary, but insufficient, to heal the liver at this point. Our results support the need for further investigation into epigenetic regulatory pathways impacting GPNMB activity and expression. Here, our findings show that GPNMB is of particular relevance as a hepatic and serum biomarker of AH and may be a therapeutic target for ameliorating the early onset and progression of AH.
Not surprisingly, many inflammatory and fibrotic protein biomarkers were largely increased, including actins, collagens, integrins, and fibronectin, reflecting stage 3 or stage 4 fibrosis often observed in these patients 5. Fibronectin demonstrated a 2.7-fold increase in AH tissue and has long been known to be an early predictor of the development of ALD and cirrhosis 45. As a marker of tissue damage due to alcohol abuse, fibronectin remains an interesting target for mechanistic studies and validates our methodology and analysis; however, it was not found to be acetylated 46. One novel target identified is gamma-interferon-inducible lysosomal thiol reductase (GILT), a lysosomal enzyme regulating autophagy that reduces protein disulfides and plays a role in MHC class I and II recognition. GILT was found to be increased in AH tissue (5.6-fold, Supplemental Data). Interestingly, in fibroblasts GILT has been reported to regulate autophagy by altering glutathione redox systems, where a decrease in GILT results in an oxidized GSSG/GSH ratio and the accumulation of dysfunctional mitochondria 47. Cathepsin B (2.62-fold) and cathepsin D (5.15-fold) were found to be increased in AH tissue, supporting a link between AH and TNF-dependent cell death, autophagy, and hepatocyte pyroptosis 48 and the activation of hepatic stellate cells and liver fibrosis 49, 50. Previous studies have identified cathepsin D overexpressed in alcohol-associated cirrhotic liver 51 and increased serum activity in acute hepatic necrosis 52.
By comparing our proteomic analysis with RNA-seq data previously published by Trépo, et al, and by Khanova and colleagues on similar AH tissues, but not this same cohort, we discovered an overlap of only two proteins 53, 54. The two proteins found to be in common across all three datasets were annexin A3 (ANXA3) and macrophage-capping protein (CAPG). Interestingly, ANXA3 is an inhibitor of phospholipase A2, previously shown to be decreased in a rat model combining alcohol abuse with olive oil and pyrazole, an ALDH inhibitor 55. Our analysis revealed that AH liver explants had a 10-fold increase in ANXA3. This discrepancy could be a result of examining two different time points along the continuum of ALD: end-stage versus a middle stage demonstrating minor fibrosis. Assessing both rodent and human data, one conclusion could be that a switch from decreasing to increasing ANXA3 may play a role in disease progression and hepatocyte repair processes. The second protein identified is CAPG, which performs a blocking function at the barbed ends of actin. CAPG has been found upregulated in hepatocellular carcinoma 56 and associates with CBP/p300 in the promoter region of PIK3R1/P50 resulting in increased Histone H3 acK27 57. Both ANXA3 and CAPG were found to be increased in AH liver explants at the transcript level and similarly in our proteomics analysis. Unsurprisingly, an increase in mRNA of these two genes is part of a larger genomic signature that has been shown to identify patients with poor prognosis and provides a link between histone acetylation and gene expression 53.
Regulation of protein acetylation is achieved through both non-enzymatic and enzymatic mechanisms. This regulation occurs by altering cellular acetyl-CoA concentrations, as well as the activity of NAD+-dependent sirtuin deacetylases 58. In general, healthy mitochondria and a functioning TCA cycle are required to produce acetyl-CoA, which is critical for protein and histone acetylation 58, 59. Our analyses revealed a global decrease in the acetylation of proteins involved in a host of anabolic pathways. Interestingly, we identified significant increases in Histone H3 acK9 and H2B acK5 in AH patients. Many other sites of histone acetylation trended higher, suggesting that an increase in histone acetylation is a persistent consequence of alcohol abuse in AH tissue. Consequences for these changes remain undefined, as histone acetylation is an activating mark for gene expression and can impact a plethora of gene targets. In vitro and in vivo studies have demonstrated increased Histone H3 acK9 with a single dose of alcohol 21, 24. Indeed, alcohol-derived carbon units have been found to contribute to acetylation in rodent models, as research has demonstrated direct changes in alcohol-induced histone acetylation in as little as 1 hour post exposure with the increased acetylation resolved by 24 hours after exposure 20, 21. A chronic alcohol model in rodents also demonstrated increased Histone H3 acK9, as well as increased Histone H3K9 dimethylation 60. Another study in rodents fed a chronic ethanol diet revealed that Histone H3 acK9 associated in both the promoter and coding region of ADH1 suggesting that this epigenetic modification increases ADH1 expression 61. However, transcriptomic data for ADH1 remained unchanged in AH patients 62 and our proteomic data from AH patients demonstrated a 5-fold decrease in ADH1 abundance. Additionally, alcohol-induced hyperacetylation of Histone H3 and Histone H4 have been demonstrated to result in increased inflammation through elevated IL-6 and TNF-a 63. Critically, IL-6 is negatively regulated by GPNMB and expression of both is associated with HDAC activity and Histone H3/H4 epigenetic marks 31, 41. These findings suggest a complicated landscape of protein acetylation exists across multiple rodent and human model tissues. It also emphasizes the urgent need for integrated “OMICS” research in relevant ALD explant tissues and biopsies across the span of ALD progression to determine if data produced in rodent model systems are concordant 62.
A recent analysis of hepatic mRNA expression of AH explant tissue revealed that hexokinase domain containing 1 (HKDC1) was upregulated 100-fold in AH compared to normal and serum levels of HKDC1 were significantly increased by over 2-fold 62. Our data support the previous finding showing HKDC1 is increased in AH tissue, as we found HKDC1 upregulated 4.4-fold at the protein level (Supplemental Data). Importantly, a recent report has shown that several histone marks, including Histone H3 acK27, may contribute to the induction of HKDC1 expression which is elevated in AH explant tissue 62. This is another example of alcohol-induced histone acetylation that may result in epigenetic changes impacting metabolism, as well as inflammation and oxidative stress, which suggests a coordinated hepatic response to hepatocyte damage across proteomic and epigenetic processes 62, 64. Much less is known regarding the consequences of Histone H2B acK5. Histone H2BK5 has been shown to be a site with acetylation stoichiometry between 0.2 – 1.6% in an analysis of acetylated peptides in HeLa cells 65. Histone H2BK5 is also known to be primarily acetylated by CBP/p300 and to have an extremely fast turnover rate (~20 minutes) suggesting this histone modification tightly regulates the expression of specific genes 66. Further examining the acetylation and regulation of Histone H2BK5 may provide additional insight regarding the pathogenesis of AH.
Overall, our findings are the first to take a robust proteomic and acetylomic approach to better identify and understand unique changes occurring in AH. Critically, our study reveals GPNMB as a hepatic and serum biomarker for AH. Expansion of these analytical approaches in other stages of ALD will be informative. Furthermore, a robust data-intensive approach is needed to better interpret proteomic and epigenetic factors leading to the initiation and progression of ALD. These efforts will also require an expansive tissue-bank with many explant tissues across a diverse and inclusive population. A relatively deep and thorough analysis would include the methods applied above, as well as ChIP-seq, ATAC-seq, and metabolomics, among others 67. Our findings, along with previously reported analyses suggest that long term alcohol consumption alters the epigenetic regulation of gene expression in humans 62. These data are additive to the current body of work examining AH and suggest that altered hepatic metabolism due to alcohol toxicity impacts the hepatic acetylome and the downstream consequences of these changes are still under investigation.
STUDY LIMITATIONS
Several factors contributed to limiting the overall interpretation of our proteomics results. These include sample size and diversity, the end-stage condition of the hepatic tissue, and possible life-saving drug interventions. The tissue biobank is also a precious resource, limiting the amount of tissue available for analysis.
Supplementary Material
Highlights.
Proteomic analysis of alcohol-associated hepatitis explant tissue reveals biochemical pathways associated with metabolic dysfunction and inflammation.
GPNMB was identified as a hepatic and serum biomarker of alcohol-associated hepatitis.
Dysregulated protein acetylation was observed in AH and suggests an impaired metabolic state in hepatocytes.
Epigenetic marks, Histone H2BK5 and H3K9 acetylation, are significantly increased in alcohol-associated hepatitis explant tissue.
ACKNOWLEDGEMENTS
Tissue and slides from normal donor and alcohol-associated hepatitis liver explants were received from the Johns Hopkins University biobank ZS (NIAAA AA025017). The authors appreciate the contribution to this research made by E. Erin Smith, HTL (ASCP)CM QIHC; Allison Quador, HTL(ASCP)CM; and Jessica Arnold HTL(ASCP)CM of the University of Colorado Cancer Center Pathology Shared Resource. This resource is supported in part by the Cancer Center Support Grant (P30CA046934). MS analysis was performed in collaboration with Dr. Richard Reisdorph and the Mass Spectrometry Core in the Skaggs School of Pharmacy and Pharmaceutical Sciences. This work was funded in part by a National Institute on Alcohol Abuse and Alcoholism grant by KSF (NIAAA AA026928). Contents are the author’s sole responsibility.
Footnotes
COMPETING INTERESTS
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.World Health Organization., Global status report on alcohol and health 2014. World Health Organization: Geneva, 2014; p xiv–376 p. [Google Scholar]
- 2.Seitz HK; Lieber CS; Stickel F; Salaspuro M; Schlemmer HP; Horie Y, Alcoholic liver disease: from pathophysiology to therapy. Alcoholism, clinical and experimental research 2005, 29 (7), 1276–81. [DOI] [PubMed] [Google Scholar]
- 3.Seitz HK; Bataller R; Cortez-Pinto H; Gao B; Gual A; Lackner C; Mathurin P; Mueller S; Szabo G; Tsukamoto H, Alcoholic liver disease. Nat Rev Dis Primers 2018, 4 (1), 16. [DOI] [PubMed] [Google Scholar]
- 4.Lieber CS, Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol (Fayetteville, N.Y.) 2004, 34 (1), 9–19. [DOI] [PubMed] [Google Scholar]
- 5.Hosseini N; Shor J; Szabo G, Alcoholic Hepatitis: A Review. Alcohol Alcohol 2019, 54 (4), 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandrekar P; Bataller R; Tsukamoto H; Gao B, Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology (Baltimore, Md.) 2016, 64 (4), 1343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee BP; Chen PH; Haugen C; Hernaez R; Gurakar A; Philosophe B; Dagher N; Moore SA; Li Z; Cameron AM, Three-year Results of a Pilot Program in Early Liver Transplantation for Severe Alcoholic Hepatitis. Ann Surg 2017, 265 (1), 20–29. [DOI] [PubMed] [Google Scholar]
- 8.Weeks SR; Sun Z; McCaul ME; Zhu H; Anders RA; Philosophe B; Ottmann SE; Garonzik Wang JM; Gurakar AO; Cameron AM, Liver Transplantation for Severe Alcoholic Hepatitis, Updated Lessons from the World’s Largest Series. J Am Coll Surg 2018, 226 (4), 549–557. [DOI] [PubMed] [Google Scholar]
- 9.Thompson JA; Martinson N; Martinson M, Mortality and costs associated with alcoholic hepatitis: A claims analysis of a commercially insured population. Alcohol (Fayetteville, N.Y.) 2018, 71, 57–63. [DOI] [PubMed] [Google Scholar]
- 10.Forrest EH; Storey N; Sinha R; Atkinson SR; Vergis N; Richardson P; Masson S; Ryder S; Thursz MR; Allison M; Fraser A; Austin A; McCune A; Dhanda A; Katarey D; Potts J; Verma S; Parker R; Hayes PC; Group SN, Baseline neutrophil-to-lymphocyte ratio predicts response to corticosteroids and is associated with infection and renal dysfunction in alcoholic hepatitis. Aliment Pharmacol Ther 2019, 50 (4), 442–453. [DOI] [PubMed] [Google Scholar]
- 11.Singal AK; Shah VH, Current trials and novel therapeutic targets for alcoholic hepatitis. J Hepatol 2019, 70 (2), 305–313. [DOI] [PubMed] [Google Scholar]
- 12.Ceni E; Mello T; Galli A, Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World journal of gastroenterology 2014, 20 (47), 17756–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantena SK; King AL; Andringa KK; Landar A; Darley-Usmar V; Bailey SM, Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World journal of gastroenterology 2007, 13 (37), 4967–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey SM; Cunningham CC, Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med 2002, 32 (1), 11–6. [DOI] [PubMed] [Google Scholar]
- 15.Cahill A; Cunningham CC; Adachi M; Ishii H; Bailey SM; Fromenty B; Davies A, Effects of alcohol and oxidative stress on liver pathology: the role of the mitochondrion. Alcoholism, clinical and experimental research 2002, 26 (6), 907–15. [PMC free article] [PubMed] [Google Scholar]
- 16.Han D; Johnson HS; Rao MP; Martin G; Sancheti H; Silkwood KH; Decker CW; Nguyen KT; Casian JG; Cadenas E; Kaplowitz N, Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free radical biology & medicine 2017, 102, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doody EE; Groebner JL; Walker JR; Frizol BM; Tuma DJ; Fernandez DJ; Tuma PL, Ethanol metabolism by alcohol dehydrogenase or cytochrome P450 2E1 differentially impairs hepatic protein trafficking and growth hormone signaling. Am J Physiol Gastrointest Liver Physiol 2017, ajpgi 00027 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zakhari S, Alcohol metabolism and epigenetics changes. Alcohol research : current reviews 2013, 35 (1), 6–16. [PMC free article] [PubMed] [Google Scholar]
- 19.Pandey SC; Bohnsack JP, Alcohol Makes Its Epigenetic Marks. Cell metabolism 2020, 31 (2), 213–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mews P; Egervari G; Nativio R; Sidoli S; Donahue G; Lombroso SI; Alexander DC; Riesche SL; Heller EA; Nestler EJ; Garcia BA; Berger SL, Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574 (7780), 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriss CL; Gregory-Lott E; Storey AJ; Tackett AJ; Wahls WP; Stevens SM Jr., In Vivo Metabolic Tracing Demonstrates the Site-Specific Contribution of Hepatic Ethanol Metabolism to Histone Acetylation. Alcoholism, clinical and experimental research 2018, 42 (10), 1909–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restrepo RJ; Lim RW; Korthuis RJ; Shukla SD, Binge alcohol alters PNPLA3 levels in liver through epigenetic mechanism involving histone H3 acetylation. Alcohol (Fayetteville, N.Y.) 2017, 60, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assiri MA; Roy SR; Harris PS; Ali H; Liang Y; Shearn CT; Orlicky DJ; Roede JR; Hirschey MD; Backos DS; Fritz KS, Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation. Alcoholism, clinical and experimental research 2017, 41 (10), 1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla SD; Restrepo R; Fish P; Lim RW; Ibdah JA, Different mechanisms for histone acetylation by ethanol and its metabolite acetate in rat primary hepatocytes. J Pharmacol Exp Ther 2015, 354 (1), 18–23. [DOI] [PubMed] [Google Scholar]
- 25.Harris PS; Roy SR; Coughlan C; Orlicky DJ; Liang Y; Shearn CT; Roede JR; Fritz KS, Chronic ethanol consumption induces mitochondrial protein acetylation and oxidative stress in the kidney. Redox biology 2015, 6, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghezzi A; Krishnan HR; Lew L; Prado FJ 3rd; Ong DS; Atkinson NS, Alcohol-induced histone acetylation reveals a gene network involved in alcohol tolerance. PLoS genetics 2013, 9 (12), e1003986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shepard BD; Tuma PL, Alcohol-induced protein hyperacetylation: mechanisms and consequences. World journal of gastroenterology 2009, 15 (10), 1219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Picklo MJ Sr., Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochemical and biophysical research communications 2008, 376 (3), 615–9. [DOI] [PubMed] [Google Scholar]
- 29.Choudhary C; Weinert BT; Nishida Y; Verdin E; Mann M, The growing landscape of lysine acetylation links metabolism and cell signalling. Nature reviews. Molecular cell biology 2014, 15 (8), 536–50. [DOI] [PubMed] [Google Scholar]
- 30.Ali I; Conrad RJ; Verdin E; Ott M, Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem Rev 2018, 118 (3), 1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong X; Sawalha AH, Takayasu arteritis risk locus in IL6 represses the anti-inflammatory gene GPNMB through chromatin looping and recruiting MEF2-HDAC complex. Ann Rheum Dis 2019, 78 (10), 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali HR; Assiri MA; Harris PS; Michel CR; Yun Y; Marentette JO; Huynh FK; Orlicky DJ; Shearn CT; Saba LM; Reisdorph R; Reisdorph N; Hirschey MD; Fritz KS, Quantifying Competition among Mitochondrial Protein Acylation Events Induced by Ethanol Metabolism. Journal of proteome research 2019, 18 (4), 1513–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali HR; Michel CR; Lin YH; McKinsey TA; Jeong MY; Ambardekar AV; Cleveland JC; Reisdorph R; Reisdorph N; Woulfe KC; Fritz KS, Defining decreased protein succinylation of failing human cardiac myofibrils in ischemic cardiomyopathy. Journal of molecular and cellular cardiology 2020, 138, 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyun J; Sun Z; Ahmadi AR; Bangru S; Chembazhi UV; Du K; Chen T; Tsukamoto H; Rusyn I; Kalsotra A; Diehl AM, Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. The Journal of clinical investigation 2020, 130 (4), 2129–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M; Dou Y; Sun R; Zhang Y; Liu Y, Molecular mechanisms for alcoholic hepatitis based on analysis of gene expression profile. Hepat Mon 2015, 15 (5), e27336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y; Chen SH; Jin X; Li YM, Analysis of differentially expressed genes and microRNAs in alcoholic liver disease. Int J Mol Med 2013, 31 (3), 547–54. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S; Maras JS; Das S; Hussain S; Mishra AK; Shasthry SM; Sharma CB; Weiss E; Elkrief L; Rautou PE; Gilgenkrantz H; Lotersztajn S; Paradis V; de la Grange P; Junot C; Moreau R; Sarin SK, Pre-therapy liver transcriptome landscape in Indian and French patients with severe alcoholic hepatitis and steroid responsiveness. Scientific reports 2017, 7 (1), 6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrence M; Daujat S; Schneider R, Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet 2016, 32 (1), 42–56. [DOI] [PubMed] [Google Scholar]
- 39.Carrico C; Meyer JG; He W; Gibson BW; Verdin E, The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell metabolism 2018, 27 (3), 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao S; Xu W; Jiang W; Yu W; Lin Y; Zhang T; Yao J; Zhou L; Zeng Y; Li H; Li Y; Shi J; An W; Hancock SM; He F; Qin L; Chin J; Yang P; Chen X; Lei Q; Xiong Y; Guan KL, Regulation of cellular metabolism by protein lysine acetylation. Science (New York, N.Y.) 2010, 327 (5968), 1000–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katayama A; Nakatsuka A; Eguchi J; Murakami K; Teshigawara S; Kanzaki M; Nunoue T; Hida K; Wada N; Yasunaka T; Ikeda F; Takaki A; Yamamoto K; Kiyonari H; Makino H; Wada J, Beneficial impact of Gpnmb and its significance as a biomarker in nonalcoholic steatohepatitis. Scientific reports 2015, 5, 16920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel-Chamberlin M; Wang Y; Satirapoj B; Phillips LM; Nast CC; Dai T; Watkins RA; Wu X; Natarajan R; Leng A; Ulanday K; Hirschberg RR; Lapage J; Nam EJ; Haq T; Adler SG, Hematopoietic growth factor inducible neurokinin-1 (Gpnmb/Osteoactivin) is a biomarker of progressive renal injury across species. Kidney Int 2011, 79 (10), 1138–48. [DOI] [PubMed] [Google Scholar]
- 43.Li B; Castano AP; Hudson TE; Nowlin BT; Lin SL; Bonventre JV; Swanson KD; Duffield JS, The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J 2010, 24 (12), 4767–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou L; Zhuo H; Ouyang H; Liu Y; Yuan F; Sun L; Liu F; Liu H, Glycoprotein non-metastatic melanoma protein b (Gpnmb) is highly expressed in macrophages of acute injured kidney and promotes M2 macrophages polarization. Cell Immunol 2017, 316, 53–60. [DOI] [PubMed] [Google Scholar]
- 45.Junge J; Bentsen KD; Christoffersen P; Orholm M; Sorensen TI; Horn T, Fibronectin as predictor of cirrhosis in men who abuse alcohol. Br Med J (Clin Res Ed) 1988, 296 (6637), 1629–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz-Seible RS; Casey CA, Fibronectin: functional character and role in alcoholic liver disease. World journal of gastroenterology 2011, 17 (20), 2482–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang HS; Maric M, Lysosomal thiol reductase negatively regulates autophagy by altering glutathione synthesis and oxidation. Free radical biology & medicine 2011, 51 (3), 688–99. [DOI] [PubMed] [Google Scholar]
- 48.Guo H; Xie M; Zhou C; Zheng M, The relevance of pyroptosis in the pathogenesis of liver diseases. Life sciences 2019, 223, 69–73. [DOI] [PubMed] [Google Scholar]
- 49.Yang L; Jin GH; Zhou JY, The Role of Ceramide in the Pathogenesis of Alcoholic Liver Disease. Alcohol Alcohol 2016, 51 (3), 251–7. [DOI] [PubMed] [Google Scholar]
- 50.Li Y; Zhu M; Huo Y; Zhang X; Liao M, Anti-fibrosis activity of combination therapy with epigallocatechin gallate, taurine and genistein by regulating glycolysis, gluconeogenesis, and ribosomal and lysosomal signaling pathways in HSC-T6 cells. Exp Ther Med 2018, 16 (6), 4329–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohhira Motoyuki and Ono Minoru and Masumi Ohhira and Hitoyoshi Ohta and Akinori Matsumoto and Chihiro Sekiya and Masayoshi, N., Unique properties of cathepsin D purified from alcoholic cirrhotic liver. International Hepatology Communications 1993, 1 (3), 152–157. [Google Scholar]
- 52.Gove CD; Wardle EN; Williams R, Circulating lysosomal enzymes and acute hepatic necrosis. J Clin Pathol 1981, 34 (1), 13–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trepo E; Goossens N; Fujiwara N; Song WM; Colaprico A; Marot A; Spahr L; Demetter P; Sempoux C; Im GY; Saldarriaga J; Gustot T; Deviere J; Thung SN; Minsart C; Serste T; Bontempi G; Abdelrahman K; Henrion J; Degre D; Lucidi V; Rubbia-Brandt L; Nair VD; Moreno C; Deltenre P; Hoshida Y; Franchimont D, Combination of Gene Expression Signature and Model for End-Stage Liver Disease Score Predicts Survival of Patients With Severe Alcoholic Hepatitis. Gastroenterology 2018, 154 (4), 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khanova E; Wu R; Wang W; Yan R; Chen Y; French SW; Llorente C; Pan SQ; Yang Q; Li Y; Lazaro R; Ansong C; Smith RD; Bataller R; Morgan T; Schnabl B; Tsukamoto H, Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology (Baltimore, Md.) 2018, 67 (5), 1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia X; Yin L; Feng Y; Peng X; Ma F; Yao Y; Liu X; Zhang Z; Yuan Z; Zhang L, A dynamic plasma membrane proteome analysis of alcohol-induced liver cirrhosis. Proteome Sci 2012, 10 (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hass HG; Vogel U; Scheurlen M; Jobst J, Gene-expression Analysis Identifies Specific Patterns of Dysregulated Molecular Pathways and Genetic Subgroups of Human Hepatocellular Carcinoma. Anticancer Res 2016, 36 (10), 5087–5095. [DOI] [PubMed] [Google Scholar]
- 57.Chi Y; Xue J; Huang S; Xiu B; Su Y; Wang W; Guo R; Wang L; Li L; Shao Z; Jin W; Wu Z; Wu J, CapG promotes resistance to paclitaxel in breast cancer through transactivation of PIK3R1/P50. Theranostics 2019, 9 (23), 6840–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papanicolaou KN; O’Rourke B; Foster DB, Metabolism leaves its mark on the powerhouse: recent progress in post-translational modifications of lysine in mitochondria. Front Physiol 2014, 5, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Reyes I; Diebold LP; Kong H; Schieber M; Huang H; Hensley CT; Mehta MM; Wang T; Santos JH; Woychik R; Dufour E; Spelbrink JN; Weinberg SE; Zhao Y; DeBerardinis RJ; Chandel NS, TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Molecular cell 2016, 61 (2), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aroor AR; Restrepo RJ; Kharbanda KK; Shukla SD, Epigenetic histone modifications in a clinically relevant rat model of chronic ethanol-binge-mediated liver injury. Hepatol Int 2014, 8 Suppl 2, 421–30. [DOI] [PubMed] [Google Scholar]
- 61.Park PH; Lim RW; Shukla SD, Gene-selective histone H3 acetylation in the absence of increase in global histone acetylation in liver of rats chronically fed alcohol. Alcohol Alcohol 2012, 47 (3), 233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Massey V; Parrish A; Argemi J; Moreno M; Mello A; Garcia-Rocha M; Altamirano J; Odena G; Dubuquoy L; Louvet A; Martinez C; Adrover A; Affo S; Morales-Ibanez O; Sancho-Bru P; Millan C; Alvarado-Tapias E; Morales-Arraez D; Caballeria J; Mann J; Cao S; Sun Z; Shah V; Cameron A; Mathurin P; Snider N; Villanueva C; Morgan TR; Guinovart J; Vadigepalli R; Bataller R, Integrated Multi-Omics Reveals Glucose Use Reprogramming and Identifies a Novel Hexokinase in Alcoholic Hepatitis. Gastroenterology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kendrick SF; O’Boyle G; Mann J; Zeybel M; Palmer J; Jones DE; Day CP, Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology (Baltimore, Md.) 2010, 51 (6), 1988–97. [DOI] [PubMed] [Google Scholar]
- 64.Choudhury M; Park PH; Jackson D; Shukla SD, Evidence for the role of oxidative stress in the acetylation of histone H3 by ethanol in rat hepatocytes. Alcohol (Fayetteville, N.Y.) 2010, 44 (6), 531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hansen BK; Gupta R; Baldus L; Lyon D; Narita T; Lammers M; Choudhary C; Weinert BT, Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat Commun 2019, 10 (1), 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weinert BT; Narita T; Satpathy S; Srinivasan B; Hansen BK; Scholz C; Hamilton WB; Zucconi BE; Wang WW; Liu WR; Brickman JM; Kesicki EA; Lai A; Bromberg KD; Cole PA; Choudhary C, Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174 (1), 231–244 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan F; Powell DR; Curtis DJ; Wong NC, From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol 2020, 21 (1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.