

Abstract
Methods for rapid preparation of densely functionalized and stereochemically complex N-heterocyclic scaffolds are in demand for exploring potential bioactive chemical space. This work describes experimental and computational studies to better understand the features of aziridinium ylides as intermediates for the synthesis of highly substituted dehydromorpholines. The development of this chemistry has enabled the extension of aziridinium ylide chemistry to the concomitant formation of both a C–N and a C–O bond in a manner that preserves the stereochemical information embedded in the substrate. Additionally, we have uncovered several key insights that describe the importance of steric effects, rotational barriers around the C–N bond of the aziridinium ylide, and non-covalent interactions (NCIs) on the ultimate reaction outcome. These critical insights will assist in the further development of this chemistry to generate N-heterocycles that will further expand complex amine chemical space.
Keywords: aziridine, aziridinium ylide, carbene, morpholine, tunability, non-covalent interactions
Graphical Abstract
INTRODUCTION
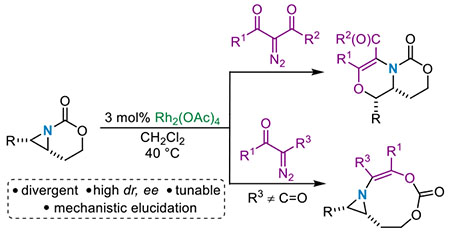
Onium ylides1–5, including sulfur6a–d, oxonium7 and ammonium,8a,b are common intermediates employed in the syntheses of complex molecules. They are typically generated from the reaction of a heteroatom with a metal-supported carbene to furnish reactive zwitterionic intermediates that engage in a diverse set of reaction pathways, including 1,2-Stevens rearrangements,8b 2,3-sigmatropic rearrangements,9 and N–H insertion reactions (Scheme 1A).3 The aziridinium ylides10 represent a unique subclass of ammonium ylides whose reactivity is both poorly understood and underexplored, despite the potential for these reactive species to serve as key intermediates in the transformation of simple precursors to densely functionalized, stereochemically rich N-heterocycles. We are interested in applying the ability to shuttle aziridinium ylides along divergent pathways to explore diverse new amine chemical space, particularly in the context of using our new methods for the preparation of DNA-encoded libraries (DEL) to explore their potential bioactivity.11a–c
Scheme 1.
Prior work in aziridinium ylide chemistry and new divergent reactivity.
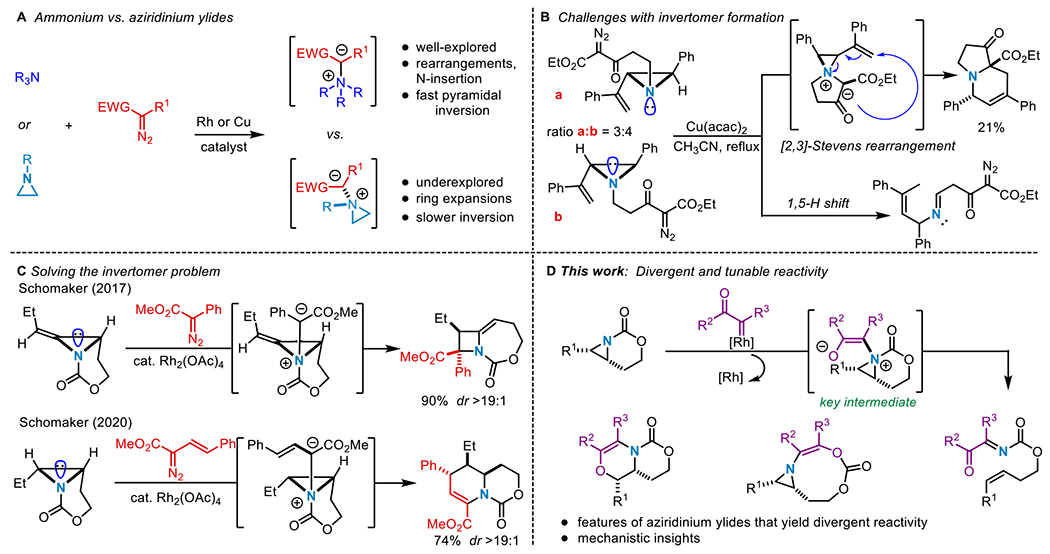
Manipulating the reactivity of aziridinium ylides to achieve a desired reaction outcome can be challenging as compared to their acyclic ammonium counterparts. Aziridinium ylides are embedded in a highly strained ring, where the bonding constraints of the aziridine increase the energy required for pyramidal inversion of the nitrogen; often this rate is slow enough to measure using variable temperature (VT) or dynamic NMR spectroscopy. 12a–c The presence of two invertomers hinders the ability to selectively form and engage aziridinium ylides in secondary reactions and has limited their applications in the synthesis of complex N-heterocycles. For example, in 2001 and 2004 respectively, Clark13 and Rowlands14 observed intramolecular [2,3]-Stevens rearrangements of aziridinium ylides to deliver dehydropiperidines in low 21-24% yields (Scheme 1B). In the former case, the low yield was attributed to the instability of the starting material which decomposed in one day when stored under argon at −30 °C.13 In Rowlands’ work, the alkyl aziridine substrate existed as two N-invertomers a and b in a ratio of 3:4, with an estimated barrier to N-inversion at room temperature of ~16-20 kcal/mol. Only a possessed the required geometry for the desired rearrangement; as a result, the major invertomer b underwent an unproductive 1,5-hydride shift at a faster rate than N-inversion to generate the invertomer required for the desired reactivity.14 In addition to the formation of invertomers of the aziridinium ylide, competitive cheletropic extrusion renders the use of these intermediates in selective transformations particularly challenging.15
We used rigid bicyclic aziridines (Scheme 1C), generated by silver-catalyzed nitrene transfer of homoallenic and homoallylic carbamates, as precursors to aziridinium ylides.16a–c The nitrogen of the carbamate tether in the bicyclic aziridine possesses a hybridization of ~sp3, where the nitrogen lone pair is unable to engage in effective orbital overlap with the π-system of the carbonyl group. As a result, the carbamate behaves as a σ-electron-withdrawing group and raises the barrier to N-pyramidal inversion.17 The geometric constraint imposed by the bicyclic nature of the tether also raises the nitrogen inversion barrier;17 a similar effect is observed in rigid cyclic amines such as sparteine and Tröger’s base.18ab,19ab Both of these unique structural characteristics can be exploited to generate diastereomerically enriched aziridinium ylides that ultimately furnish highly substituted methyleneazetidines20a–b and dehydropiperidines21 in excellent yield and dr (Scheme 1C). This strategy effectively circumvents competing pathways that have been proposed in previous attempts to leverage the reactivity of aziridinium ylides. However, despite this progress, the future development of productive chemistry of aziridinium ylides requires a more detailed understanding of how the identity of the carbene precursor, potential dynamic behavior in the ylide intermediate and non-covalent interactions influence the ultimate outcome of the reaction. In this work, we describe our mechanistic findings to explain divergent reaction pathways from aziridinium ylides generated by leveraging ketone-containing carbenes, derived from Rh catalysis, to furnish fully substituted dehydromorpholines (Scheme 1D).22–24 Both experimental and computational studies are presented that will inform future efforts of our and other groups’ work on these fascinating and versatile reactive species.
Results and discussion
Initial explorations of diverse carbene precursors.
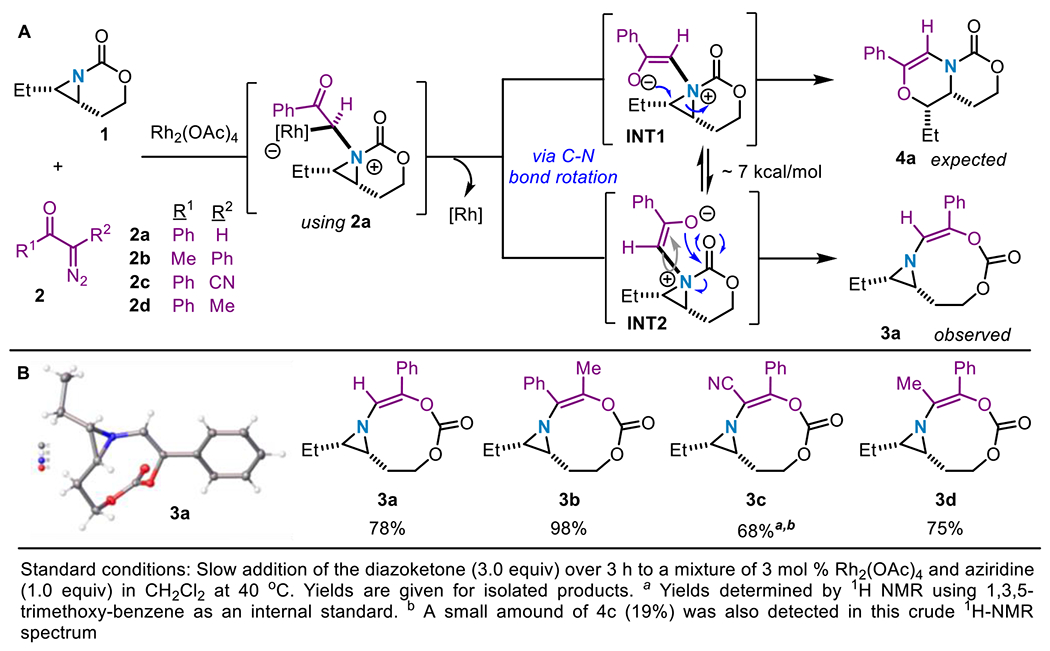
We first investigated whether the carbonyl oxygen of a donor-acceptor diazoketone, such as 2a, could serve as a competent nucleophile to open the aziridinium ylide en route to a dehydromorpholine (Scheme 2A). Only one nucleophilic ketone group is present in 2a; thus, we expected reaction of 2a with 1 to furnish 4a in good yield via conformer INT1. Surprisingly, treatment of 1 with 2a under Rh2(OAc)4 catalysis gave an unusual [3,9]-aziridine product 3a in 78% yield. Presumably, the 7 kcal/mol barrier to interconversion of aziridinium ylide INT1 to INT2 by rotation around the C–N bond (Scheme 2) enables nucleophilic attack of the ketone oxygen on the electrophilic carbonyl of the carbamate tether to furnish 3a. This result was unexpected, as nucleophilic addition to the carbamate tether was not observed in previous syntheses of azetidines or dehydropiperidines from bicyclic aziridines.20ab,21
Scheme 2.
Ring expansions of 1 with donor-acceptor diazoketone carbene precursors.
We were curious as to the origin of 3a and hypothesized that the aryl group may be important in biasing the reaction towards this unexpected product. However, exchanging the Ph group of 2a to a Me group in 2b still furnished 3b in an excellent 98% yield (Scheme 2B). Altering the electronics of R2 had no effect on product selectivity. Both electron-withdrawing and donating substituents (e.g. CN in 2c, Me in 2d) furnished the [3,9]-aziridine in 75% (3d) and 68% (3c) yields, respectively (Scheme 2B). The consistent product outcome suggested that neither R1 nor R2 influences the preferred formation of the [3,9]-aziridine product.
To gain more insight into the formation of 3a at the expense of the expected 4a, Density Functional Theory (DFT) calculations were carried out at the dispersion-corrected SMD(CH2Cl2)-B3LYP-D3/def2-SVP level (see computational details in the Supporting Information). Figure 1A shows the computed reaction profiles leading to the formation of the 3a or 4a from the corresponding free aziridinium ylides, which according to our previous calculations on related systems,20b,21 derives from the nucleophilic addition of the aziridine 2a to the Rh2-carbenoid intermediate (formed upon reaction of the diazo-compound and Rh2(OAc)4). From the data in Figure 1A, it becomes apparent that metal-free ylide INT2 involved in the [3,9]-pathway is lower in energy than its analogous intermediate INT1 involved in the alternative [6,6]-pathway (ΔΔG = −4.2 kcal/mol). A similar finding was observed in their metal-ylide counterparts (ΔΔG = −5.7 kcal/mol). According to the NCIPLOT method, this is in part ascribed to the occurrence of a stabilizing NCI involving the nucleophilic C–O lone pair of the ketone moiety and the electrophilic π*(C=O) molecular orbital of the carbamate, as confirmed for INT2 (Figure 1B; see also Figure S-14 in the Supporting Information for the metal-ylide intermediate INT2-Rh). The presence of this stabilizing NCI is also supported by the computed short O…C(=O) distance in INT2 of 2.530 Å, which is markedly shorter than the sum of the van der Waals radii (3.22 Å); this was also confirmed by the associated stabilization energy (ΔE(2) = −6.63 kcal/mol) computed for the lone pair (LP)(O)→π*(C=O) interaction using the Second-Order Perturbation Theory (SOPT) of the NBO method (Figure 1B). Furthermore, the barrier associated with the formation of 3a from INT2 is 5.3 kcal/mol lower than that associated with the formation of 4a from INT1 (Figure 1A). This noticeable difference in the calculated activation energies for the two potential reaction pathways and the occurrence of the NCIs, which show significant stabilization of the key ylide intermediate, leading to the preferred formation of 3a over 4a, despite the fact that the latter species is significantly favored thermodynamically. The barrier to interconversion between INT1 and INT2 (7.6 kcal/mol, Figure 1A) is reinforced by the lower barrier (ΔGG≠ = 1.5 kcal/mol) computed for the generation of the observed product 3a.
Figure 1.
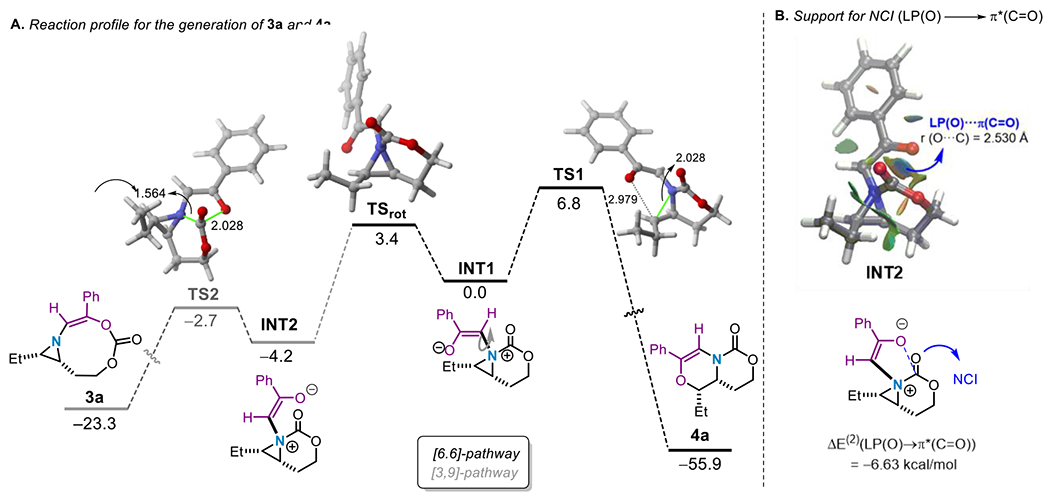
A. Computed reaction energy profiles for formation of 3a and 4a. Relative free energies (ΔG298, at 298 K) and bond distances are given in kcal/mol and angstroms, respectively. B. Contour plots (left) of the reduced density gradient isosurfaces (density cutoff of 0.04 a.u.) for intermediate INT2. The green surfaces indicate attractive noncovalent interactions. Associated SOPT-NBO stabilizing energy (right). All data have been computed at the SMD(CH2Cl2)B3LYP-D3/def2-SVP level of theory.
Controlling product outcomes with temperature.
Changing the carbene precursor to the CN-containing diazoketone 2c provided the first example of divergence between dehydromorpholine 4c and the [3,9]-aziridine 3c. Standard reaction conditions furnished 3c in 68% yield (by NMR), accompanied by 19% of the dehydromorpholine 4c (Scheme 3A, right). The 3.6:1 ratio of 3c:4c led us to employ computations to probe whether an NCI was also present in the corresponding aziridinium ylide and to estimate the barrier to C–N bond rotation. Computational analysis confirmed the presence of an NCI, where the O…C(=O) distance of INT4 is predicted to be 2.619 Å (Scheme 3B compared to 2.530 Å, Figure 1B), with a stabilizing interaction energy of ΔE(2) = −4.44 kcal/mol. The balance between the computed barrier to C–N bond rotation between INT3 and INT4 of 12.5 kcal/mol and the presence of an NCI leads to a divergent reaction where both 3c and 4c are accessible. These computational observations are reflected in the calculated reaction profile (Scheme 3B), where both the activation energy barriers leading to 3c and 4c are accessible (ΔG≠ ~8.5 kcal/mol), but slightly higher than that of 3a (Figure 1, see above). The higher barrier to C–N bond rotation (compared to the analogous ylide INT2) is likely a result of charge delocalization across the CN group (as compared to the ketone in INT2); nonetheless, it is still low enough to ultimately give rise to the product distribution based on temperature.
Scheme 3.
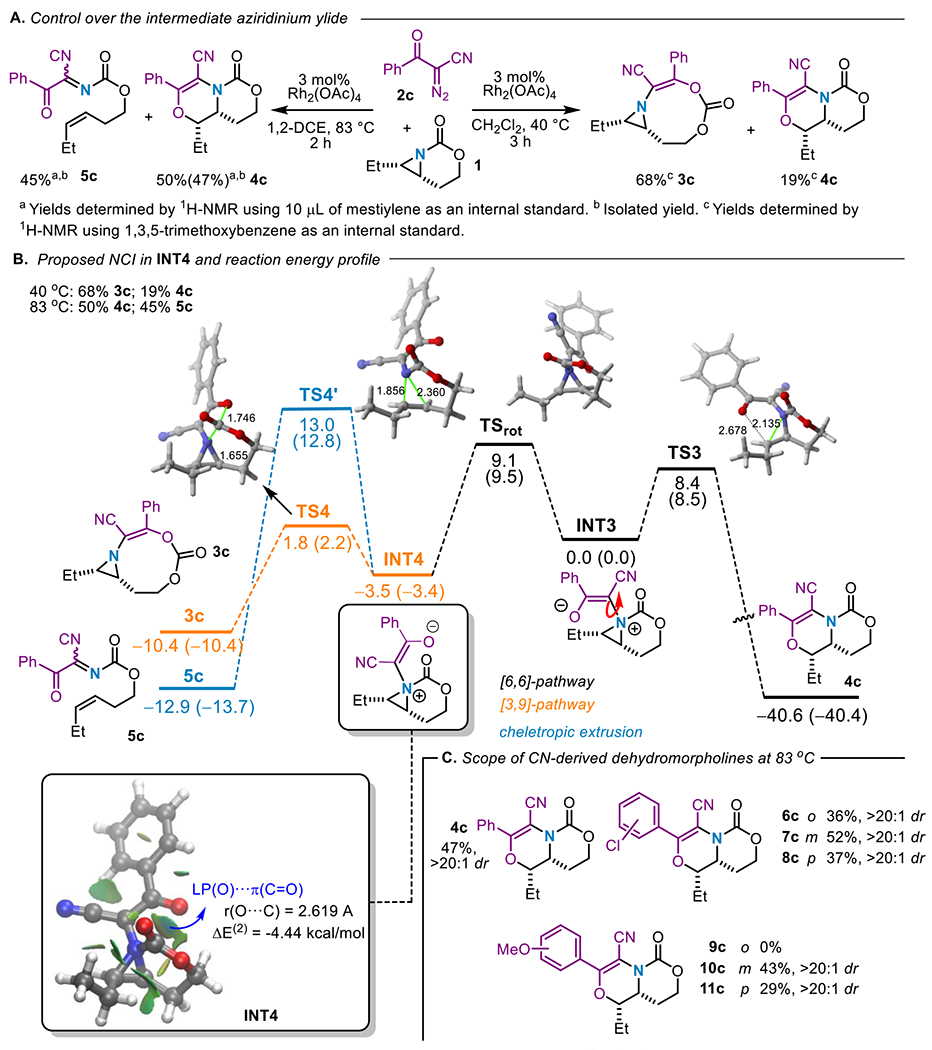
A. Controlling product distribution using temperature. B. C. Computed reaction profile (relative free energies are given in kcal/mol) at the SMD(CH2Cl2)B3LYP-D3/def2-SVP level at 25 °C and 83 °C (values within parentheses) and contour plots of the reduced density gradient isosurfaces (density cutoff of 0.04 a.u.) for intermediate INT4. D. Scope of the process.
The computed reaction profile depicted in Scheme 3C also shows that the NCI-stabilized ylide INT4 can undergo a cheletropic extrusion reaction to form the alkene 5c. We hypothesized running the reaction at higher temperatures would: (i) enable facile bond rotation and lead to a greater preference for 4c over the [3,9]-aziridine product 3c and (ii) allow access to the cheletropic extrusion product. To our delight, repeating this reaction at 83 °C in 1,2-dichloroethane (Scheme 3A, left) favored dehydromorpholine 4c formation in 50% yield by 1H-NMR (47% isolated), with the remaining mass balance (45% yield) accounted for by formation of 5c; no [3,9]-aziridine 3c formation was observed. Although full selectivity for 4c would be optimal, this reaction represents the first example where the fate of the aziridinium ylide can be controlled by the reaction conditions and not just the identity of the carbene precursor.
Employing higher temperatures with other cyano-derived diazoketones also proved fruitful (Scheme 3C). Ketones bearing ortho-, meta- and para-chlorosubstituted benzenes yielded dehydromorpholines 6c-8c in moderate yields and excellent dr of >20:1.. Ketones with meta- and para-methoxysubstituted benzenes were tolerated to furnish dehydromorpholines 10c and 11c in 43% and 29% yield, respectively. The ortho-substituted 9c was not observed under these conditions, possibly due to steric congestion around the reactive site that prevents effective ylide formation.
Based on our computational analysis of [3,9]-aziridine formation in Figure 1A, strategies to prohibit NCI formation in the key ylide intermediate were envisaged to promote formation of the desired dehydromorpholines. We hypothesized that raising the barrier to C–N bond rotation in the aziridinium ylide intermediate could ‘trap’ the kinetically formed conformer(s) and prevent formation of the NCI, potentially favoring a conformer similar to INT1 (Scheme 1A). To this end, the barriers for C–N bond rotation in aziridinium ylides generated from reaction of 1 with donor-acceptor and acceptor-acceptor carbene precursors 2a and 2d-f were calculated and compared (Figure 2). The results suggest that aziridinium ylides arising from reaction of 1 with 2a (which experimentally gave [3,9]-aziridines 3a, Scheme 1B) display a low barrier of ca. 7 kcal/mol to rotation about the C–N bond (black line). The reaction of 1 with 2d revealed a higher barrier of 12 kcal/mol (green line) for C–N bond rotation. In contrast, aziridinium ylides formed from reaction of 1 with dicarbonyl-containing diazo compounds 2e and 2f show significantly higher barriers to rotation around the C–N bond at ~23 kcal/mol (red line), and 20 kcal/mol (blue line), respectively. The increased rotational barrier may be attributed to the ability of the enolate of the ylide to be delocalized over both carbonyl-containing groups, providing additional structural rigidity and steric bulk that hinders C–N bond rotation.
Figure 2.
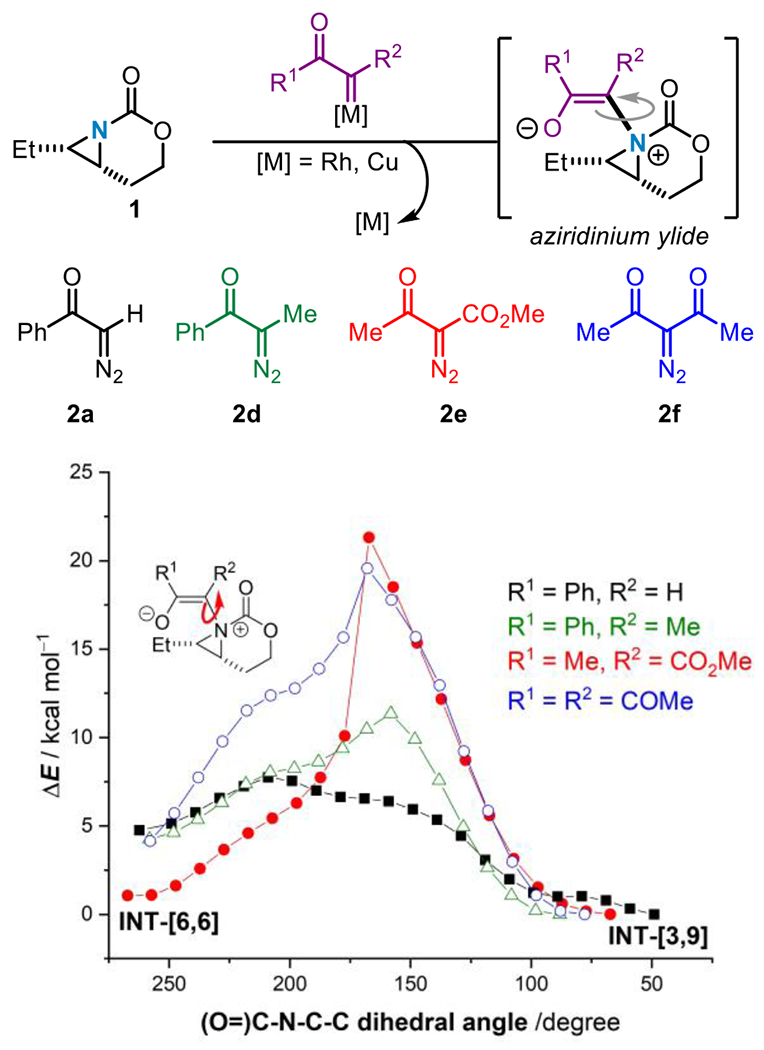
Computed barriers for C–N bond rotation in aziridinium ylides from diverse carbene precursors.
To test this hypothesis, aziridine 1 was treated with methylacetoacetate-derived 2e and catalytic Rh2(OAc)4 under optimized conditions (Scheme 4A, see Supporting Information for details). The dehydromorpholine 4e was obtained in 38% yield (48% by NMR), accompanied by the corresponding cheletropic extrusion product 5e (42% by NMR). Interestingly, no competing [3,9]-aziridine 3e was observed in the crude mixture. Computational analysis of this reaction (Scheme 4B and 4D) revealed the O…C(=O) distance is much longer for INT6 (3.085 Å) than for INT1 (2.530 Å), a distance close to the limit of the sum of the van der Waals radii (3.22 Å). The longer distance is presumably required to accommodate the second C=O from the acceptor-acceptor carbene. This is reflected in the low computed stabilization energy (ΔE(2) (LP(O)→π*(C=O)) = −0.50 kcal/mol) which can be considered negligible. This effectively prevents formation of the undesired NCI; as a result, INT6 and INT5 are nearly degenerate (ΔΔG ~ 1 kcal/mol), a markedly different scenario from that involving INT1/INT2 ylides (Figure 1A, see above). Although this computational data presents a reasonable scenario for the 1:1 product mixture, we were puzzled why no 3e was observed, considering the predicted energy barrier is only 5.3 kcal/mol compared to 15.6 kcal/mol for 5e (Scheme 4D). A variety of potential pathways were considered. One possibility invoked a retro-hetero-Diels-Alder reaction of 4e to give 5e; however, re-exposing 4e under the standard reaction conditions (Scheme 4C) gave only recovered 4e. Alternatively, 5e might arise from a retro-hetero-Diels-Alder of a product formed by attack of the ester carbonyl on the aziridinium ylide by delocalization of the negative charge in INT6. However, computational analysis argues against this pathway, as it shows an energy barrier of 18.9 kcal/mol (see Figure S-15 in the Supporting Information).
Scheme 4.
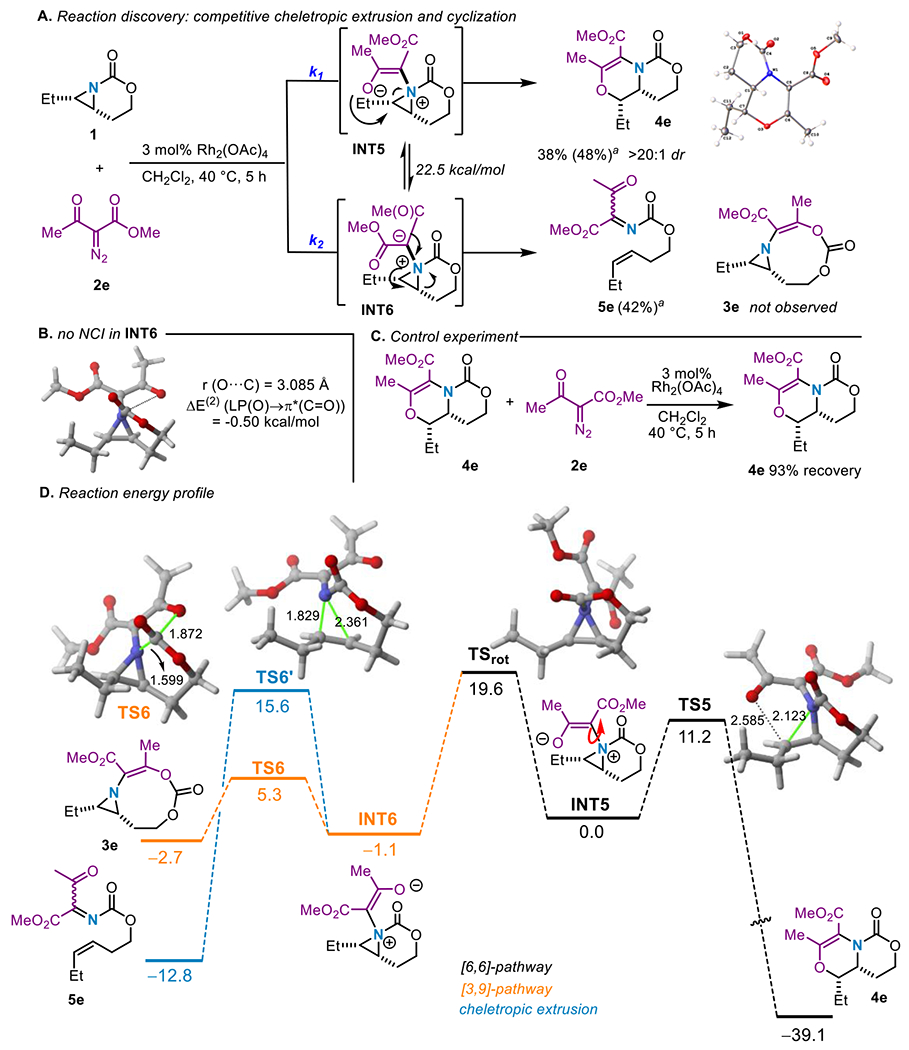
Competing cheletropic extrusion/ring expansion to dehydromorpholines. See caption to Figure 1 for computational details.
To experimentally interrogate the origin of 5e, NMR time-course data was collected to identify long-lived reaction intermediates or other products not observed in the final crude mixture. The data (Figure 3) showed a consistent increase in product 4e over the 4 h reaction period. Interestingly, a small amount of 3e was noted, which increased from 1-2 h, then decreased from 2-4 h, at which time the cheletropic extrusion product 5e was observed. The 2 h reaction aliquot was further analyzed by 1D TOCSY (see Figure S-9 in the Supporting Information), which revealed a unique spin system corresponding to 3e. These data are the first experimental evidence supporting the conclusion that the reaction is under thermodynamic control (see above), where the [3,9]-aziridine 3e gives rise to 5e. This result is further supported by the computational data, which show that the formation of 3e from INT-6 is reversible in view of the low computed reaction and barrier energies (1.6 and 8.0 kcal/mol, respectively, see Scheme 4D).
Figure 3.
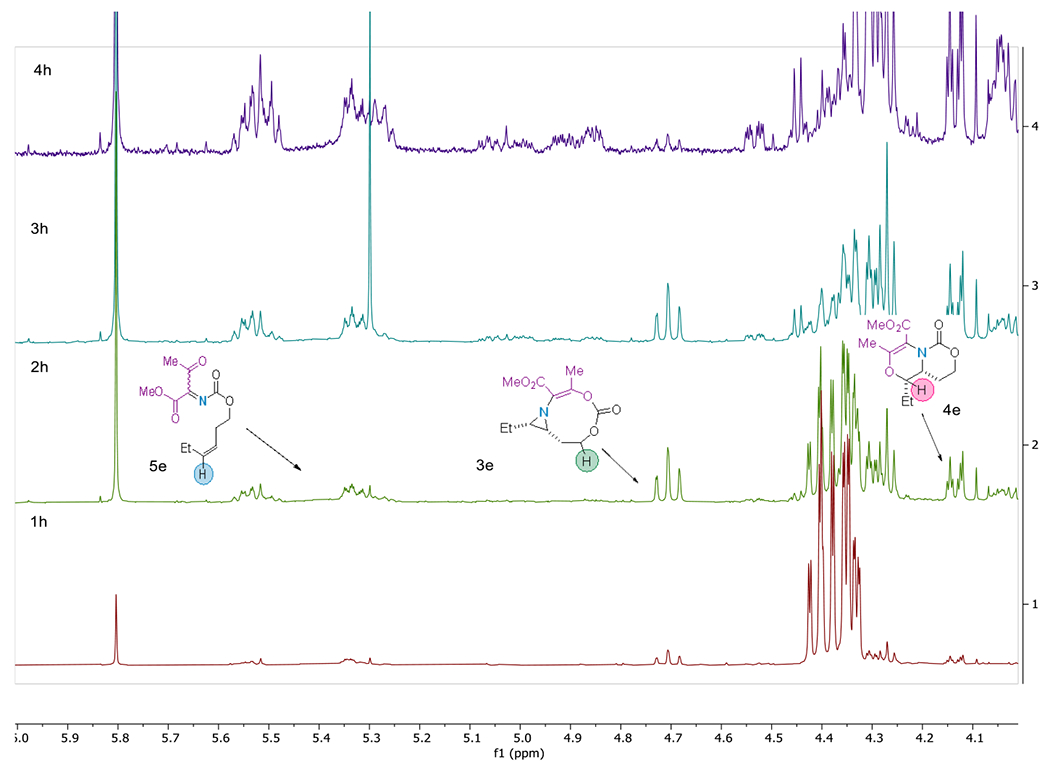
1H NMR time course data for the reaction of 1 and 2e.
Our combined computational and experimental evidence suggests that divergent reactivity to give a ~1:1 mixture of 4e and 5e is due to the non-Curtin-Hammett kinetic formation of aziridinium ylide conformers INT5 and INT6 (Scheme 4A). INT5 contains a syn-relationship between the enolate of the methyl ketone and the external aziridine C–N bond, which represents the correct geometry for dehydromorpholine formation. The other potential aziridinium ylide INT6 does not display the correct orientation, and thus initially forms 3e, which is ultimately converted to 5e.
Our attempts to use non-symmetric acceptor-acceptor diazo carbene precursors to form dehydromorpholines highlighted the importance of steric effects, rotational barriers around the C–N bond of the aziridinium ylide and NCIs in determining the reaction outcome. Symmetric diketone-containing carbene precursors, such as 2f (Scheme 5A), mitigated these complications by yielding only a single ylide intermediate that engages in nucleophilic ring-opening of the aziridinium ylide to form the dehydromorpholine. A series of cis-substituted aziridines bearing linear alkyl chains, including a primary alkyl chloride, furnished 4f and 12f-14f as the sole products in moderate yields and high dr of >20:1. Isopropyl-substituted aziridine 14 was tolerated to furnish 14f, albeit in moderate yield, indicating the sensitivity of the reaction to steric effects. Small amounts of cheletropic extrusion product (~8%) were also observed in reactions of branched aziridines. Enantioenriched benzyl-substituted aziridine (S,R)-15, obtained in 94% ee, was subjected to the reaction conditions; the product 15f was obtained in 39% yield and an excellent >20:1 dr. Importantly, the 97% ee of 15f (Scheme 5B) highlighted our ability to effectively transfer stereochemical information from the aziridine into the products to furnish enantioenriched N-heterocycles. This experimental result also excludes the possibility of a [4+2] hetero-Diels-Alder reaction being the operative mechanism that leads to the dehydromorpholine products 4f and 12f-15f (Schemes 5A–B).
Scheme 5.
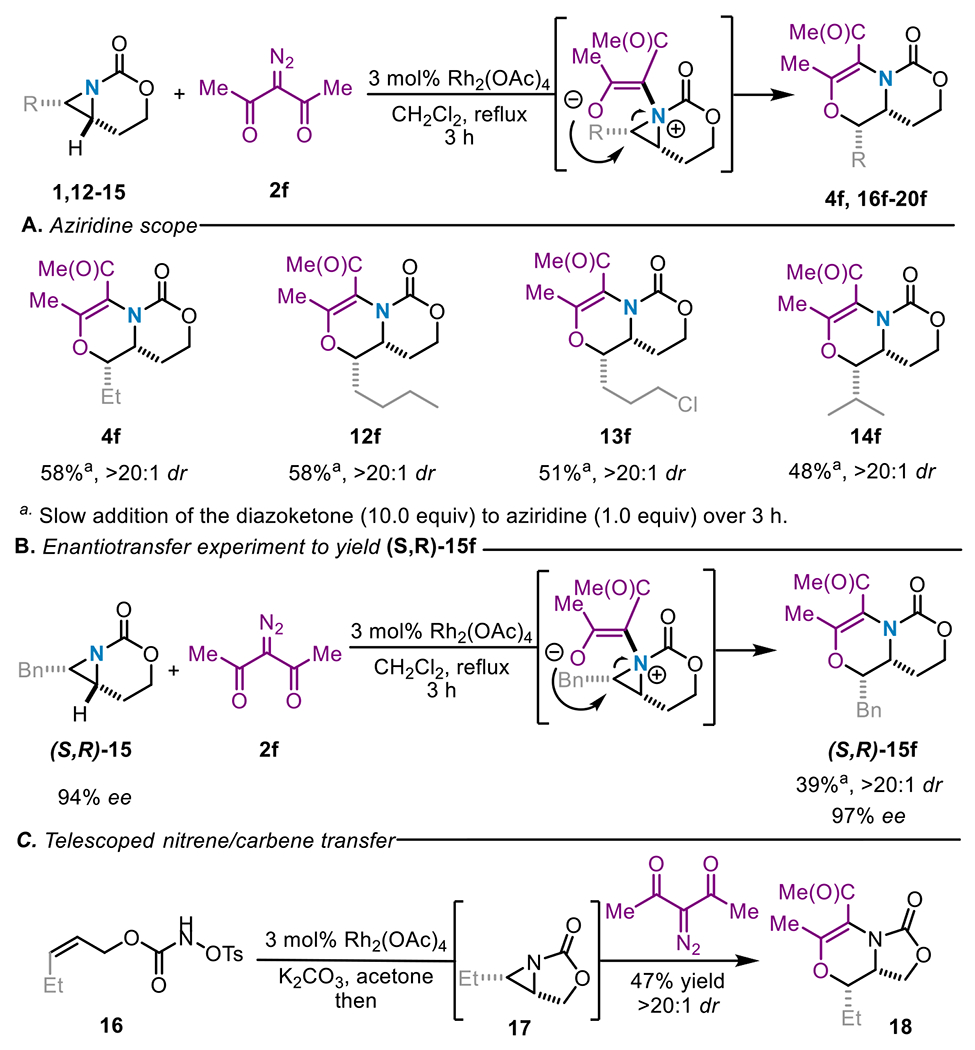
Dehydromorpholine formation from symmetric carbene precursors.
This chemistry was not restricted to aziridines bearing a 6-membered bicyclic tether; a [3,5]-bicyclic aziridine 17 was capable of yielding the substituted dehydromorpholine 18 (Scheme 5C). A nice feature of this chemistry was the ability to carry out the reaction in a telescoped fashion directly from the pre-oxidized nitrene precursor 16 to deliver 18 in 47% yield and >20:1 dr over both the aziridination and the ring expansion steps. Expansion of the telescoped reaction to more sterically demanding symmetric and unsymmetric carbene precursors with 1 proved challenging, highlighting the sensitivity of this transformation to sterics (see the Supporting Information for further details). Efforts are ongoing to address this limitation.
CONCLUSION
In conclusion, we have demonstrated a new method for the synthesis of highly substituted dehydromorpholines through the intermediacy of aziridinium ylides. The development of this chemistry has enabled the extension of aziridinium ylide chemistry to the concomitant formation of both a C–N and a C–O bond in a manner that preserves the stereochemical information embedded in the substrate aziridine. In addition, we have uncovered several key insights that describe the importance of steric effects, rotational barriers around the C–N bond of the aziridinium ylide, and NCIs on the ultimate reaction outcome. These critical insights will assist in the further development of this chemistry to generate novel and complex N-heterocycles that will further expand complex amine chemical space.
Supplementary Material
ACKNOWLEDGMENT
Dr. Charlie Fry and Dr. Heike Hoffstetter of the University of Wisconsin-Madison are thanked for assistance with NMR spectroscopy, while Dr. Martha Vestling of UW-Madison is thanked for her assistance with collecting MS data. Amelia M. Wheaton is thanked for help with processing X-ray crystallographic data. Professor Robert Bergman of the University of California-Berkeley is thanked for helpful comments during the preparation of this manuscript.
Funding Sources
J.M.S. thanks the NIH 1R01GM132300-01 for support of this work. The NMR facilities at UW-Madison are funded by the National Science Foundation (NSF; CHE-9208463, CHE-9629688) and National Institutes of Health (NIH; RR08389-01). The Q-Exactive mass spectrometer was acquired from an NM-S10 award (NIH-1S10OD020022-1). The Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF Award #CHE-1919350 to the UW–Madison Department of Chemistry. Bruker Quazar APEX2 was purchased by UW–Madison Department of Chemistry with a portion of a generous gift from Paul J. and Margaret M. Bender. I.F. acknowledges financial support from the Spanish MCIN/AEI/10.13039/501100011033 (Grants PID2019-106184GB-I00 and RED2018-102387-T).
ABBREVIATIONS
- dr
diastereomeric ratio
- ee
enantiomeric excess
- INT
intermediate
- NCI
non-covalent interaction
- DFT
density functional theory
- VT
variable temperature
- LP
lone pair
- SOPT
second order perturbation theory
- NBO
natural bond orbital
- TOCSY
total correlation spectroscopy
Footnotes
Supporting Information. Experimental procedures, computational details, and characterization data for all new compounds are available in the Supporting Information. X-ray crystallographic information is available for 3a (CDCC Deposition Number 2124058) and 4e (2124059). This material is available free of charge via the Internet at http://pubs.acs.org.
The following files are available free of charge: Supplementary Information (PDF)
REFERENCES
- 1.Dequina HJ; Nicastri KA; Schomaker JM Chapter One - Additions of N, O, and S Heteroatoms to Metal-Supported Carbenes: Mechanism and Synthetic Applications in Modem Organic Chemistry. In Advances in Organometallic Chemistry, Pérez PJ, Ed.; Academic Press, 2021; Vol. 76, pp 1–100. 10.1016/bs.adomc.2021.04.001. [DOI] [Google Scholar]
- 2.Doyle MP 5.2 - Transition Metal Carbene Complexes: Diazodecomposition, Ylide, and Insertion. In Comprehensive Organometallic Chemistry II, Abel EW, Stone FGA, Wilkinson G, Eds.; Elsevier: Oxford, 1995; pp 421–468. 10.lQ16/B978-008046519-7.00117-9. [DOI] [Google Scholar]
- 3.Gillingham D; Fei N Catalytic X–H Insertion Reactions Based on Carbenoids. Chem. Soc. Rev 2013, 42, 4918–4931. 10.1039/C3CS35496B. [DOI] [PubMed] [Google Scholar]
- 4.Jana S; Guo Y; Koenigs RM Recent Perspectives on Rearrangement Reactions of Ylides via Carbene Transfer Reactions. Chemistry – A European Journal 2021, 27, 1270–1281. 10.1002/chem.2020Q2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney JB SigmatropicRearrangements of ‘Onium’ Ylids. Chem. Soc. Rev 2009, 38, 1027–1038. 10.1039/B604828P. [DOI] [PubMed] [Google Scholar]
- 6a.Kaiser D; Klose I; Oost R; Neuhaus J; Maulide N Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev 2019, 119, 8701–8780. 10.1021/acs.chemrev,9b00111. [DOI] [PMC free article] [PubMed] [Google Scholar]; 6b. Zhang Y; Wang J Catalytic [2,3]-Sigmatropic Rearrangement of Sulfur Ylide Derived from Metal Carbene. Coordination Chemistry Reviews 2010, 254, 941–953. 10.1016/j.ccr.2009.12.005. [DOI] [Google Scholar]; 6c. N. Lakeev S; O. Maydanova I; Z. Galin F; A. Tolstikov G Sulfur Ylides in the Synthesis of Heterocyclic and Carbocyclic Compounds. Russ. Chem. Rev 2001, 70, 655–672. 10.1070/RC2001vQ70n08ABEH000645. [DOI] [Google Scholar]; 6d. Magdesieva NN; Sergeeva TA Use of Sulfonium Ylides in the Synthesis of Heterocyclic Systems (Review). Chemistry of Heterocyclic Compounds 1990, 26, 123–145. 10.1007/BF004994Q5. [DOI] [Google Scholar]
- 7.Murphy GK; West FG Oxonium Ylide Rearrangements in Synthesis. In Molecular Rearrangements in Organic Synthesis; John Wiley & Sons, Ltd, 2015; pp 497–538. 10.1002/9781118939901.chl6. [DOI] [Google Scholar]
- 8a.Roiser L; Zielke K; Waser M Ammonium Ylide Mediated Cyclization Reactions. Asian Journal of Organic Chemistry 2018, 7, 852–864. 10.1002/ajoc.201800Q91. [DOI] [PMC free article] [PubMed] [Google Scholar]; 8b. Vanecko JA; Wan H; West FG Recent Advances in the Stevens Rearrangement of Ammonium Ylides. Application to the Synthesis of Alkaloid Natural Products. Tetrahedron 2006, 62, 1043–1062. 10.1016/j.tet.2005.09.123. [DOI] [Google Scholar]
- 9.Sheng Z; Zhang Z; Chu C; Zhang Y; Wang J Transition Metal-Catalyzed [2,3]-Sigmatropic Rearrangements of Ylides: An Update of the Most Recent Advances. Tetrahedron 2017, 73, 4011–4022. 10.1016/j.tet.2016.11.045. [DOI] [Google Scholar]
- 10a.Dequina HJ; Schomaker JM Aziridinium Ylides: Underused Intermediates for Complex Amine Synthesis. Trends in Chemistry 2020, 2, 874–887. 10.1016/j.trechm.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; 10b. Ranjith J; Ha H-J Synthetic Applications of Aziridinium Ions. Molecules 2021, 26, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Gironda-Martínez A; Donckele EJ; Samain F; Neri D DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci 2021, 4, 1265–1279. 10.1021/acsptsci.1c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]; 11b. Kleiner RE; Dumelin CE; Liu DR Small-Molecule Discovery from DNA-Encoded Chemical Libraries. Chem. Soc. Rev 2011, 40, 5707–5717. 10.1039/C1CS15076F. [DOI] [PMC free article] [PubMed] [Google Scholar]; 11c. Martín A; Nicolaou CA; Toledo MA Navigating the DNA Encoded Libraries Chemical Space. Communications Chemistry 2020, 3, 127. 10.1038/s42004-020-00374-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.de Loera D; Liu F; Houk KN; Garcia-Garibay MA Aziridine Nitrogen Inversion by Dynamic NMR: Activation Parameters in a Fused Bicyclic Structure. J. Org. Chem 2013, 78, 11623–11626. 10.1021/jo4022315. [DOI] [PubMed] [Google Scholar]; 12b. Anet FAL; Osyany JM Nuclear Magnetic Resonance Spectra and Nitrogen Inversion in 1-Acylaziridines. J. Am. Chem. Soc 1967, 89, 352–356. 10.1021/ja00978a032. [DOI] [Google Scholar]; 12c. Bottini AT; Roberts JD Nuclear Magnetic Resonance Spectra. Nitrogen Inversion Rates of N-Substituted Aziridines (Ethylenimines)1. J. Am. Chem. Soc 1958, 80, 5203–5208. 10.1021/ja01552a048. [DOI] [Google Scholar]
- 13.Clark JS; Hodgson PB; Goldsmith MD; Blake AJ; Cooke PA; Street LJ Rearrangement of Ammonium Ylides Produced by Intramolecular Reaction of Catalytically Generated Metal Carbenoids. Part 2. Stereoselective Synthesis of Bicyclic Amines. J. Chem. Soc., Perkin Trans. 1 2001, 24, 3325–3337. 10.1039/B108182A. [DOI] [Google Scholar]
- 14.Rowlands GJ; Kentish Barnes W Studies on the [2,3]-Stevens Rearrangement of Aziridinium Ions. Tetrahedron Lett. 2004, 45, 5347–5350. 10.1016/j.tetlet.2004.05.087. [DOI] [Google Scholar]
- 15.Hata Y; Watanabe M Fragmentation Reaction of Aziridinium Ylids. II. Tetrahedron Lett. 1972, 13, 4659–4660. 10.1016/S0040-4039(01)94391-6. [DOI] [Google Scholar]
- 16a.Ju M; Weatherly CD; Guzei IA; Schomaker JM Chemo- and Enantioselective Intramolecular Silver-Catalyzed Aziridinations. Angew. Chem. Int. Ed 2017, 56, 9944–9948. 10.1002/anie.201704786. [DOI] [PMC free article] [PubMed] [Google Scholar]; 16b. Rigoli JW; Weatherly CD; Alderson JM; Vo BT; Schomaker JM Tunable, Chemoselective Amination via Silver Catalysis. J. Am. Chem. Soc 2013, 135, 17238–17241. 10.1021/ja406654y. [DOI] [PMC free article] [PubMed] [Google Scholar]; 16c. Rigoli JW; Weatherly CD; Vo BT; Neale S; Meis AR; Schomaker JM Chemoselective Allene Aziridination via Ag(I) Catalysis. Org. Lett 2013, 15, 290–293. 10.1021/ol303167n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rauk A; Allen LC; Mislow K Pyramidal Inversion. Angew. Chem. Int. Ed 1970, 9, 400–414. 10.1002/anie.197004001. [DOI] [Google Scholar]
- 18a.(initial discovery) Couch JF Isolation of Sparteine from Lupinus Barbiger J.Am. Chem. Soc 1932, 54, 1691–1692. 10.1021/ja01343a504. [DOI] [Google Scholar]; 18b. (crystal structure of sulfate salt) Phillips DC Crystallographic Data for Certain Alkaloids. IV. Acta. Crystallogr 1954, 7, 603. 10.1107/S0365110X54002010. [DOI] [Google Scholar]
- 19a.(initial discovery) Tröger J Ueber Einige Mittelst Nascirenden Formaldehydes Entstehende Basen. Journal für Praktische Chemie 1887, 36, 225–245. 10.1002/prac.1887036Q123. [DOI] [Google Scholar]; 19b. (structural elucidation) Larson SB; Wilcox CS Structure of 5,11-Methano-2,8-Dimethyl-5,6,11,12-Tetrahydrodibenzo[It b,It f][1,5]Diazocine (Tröger’s Base) at 163 K. Acta Crystallogr. C 1986, 42, 224–227. 10.1107/S0108270186Q96713. [DOI] [Google Scholar]
- 20a.Schmid SC; Guzei IA; Schomaker JM A Stereoselective [3+1] Ring Expansion for the Synthesis of Highly Substituted Methylene Azetidines. Angewandte Chemie International Edition 2017, 56, 12229–12233. 10.1002/anie.201705202. [DOI] [PMC free article] [PubMed] [Google Scholar]; 20b. Schmid SC; Guzei IA; Fernández I; Schomaker JM Ring Expansion of Bicyclic Methyleneaziridines via Concerted, Near-Barrierless [2,3]-Stevens Rearrangements of Aziridinium Ylides. ACS Catal. 2018, 8, 7907–7914. 10.1021/acscatal.8b02206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eshon J; Nicastri KA; Schmid SC; Raskopf WT; Guzei IA; Fernández I; Schomaker JM Intermolecular [3+3] Ring Expansion of Aziridines to Dehydropiperidines through the Intermediacy of Aziridinium Ylides. Nature Communications 2020, 11, 1273. 10.1038/s41467-020-15134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.L.; Liu Q-B; Wang D-S; Li X; Han X-W; Xiao W-J; Zhou Y-G Tandem Ring-Opening/Closing Reactions of N-Ts Aziridines and Aryl Propargyl Alcohols Promoted by t-BuOK. Org. Lett 2009, 11, 1119–1122. 10.1021/ol802862p. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S; Shan C; Zhang S; Yuan L; Wang J; Tung C-H; Xing L-B; Xu Z Breaking Aziridines to Construct Morpholines with a Gold(I)-Catalyzed Tandem Ring-Opening and Cycloisomerization Reaction. Org. Biomol. Chem 2016, 14, 10973–10980. 10.1039/C6QB02284G [DOI] [PubMed] [Google Scholar]
- 24.Fang S; Zhao Y; Li H; Zheng Y; Lian P; Wan X [3 + 3]-Cycloaddition of α-Diazocarbonyl Compounds and N-Tosylaziridines: Synthesis of Polysubstituted 2H-1,4-Oxazines through Synergetic Catalysis of AgOTf/Cu(OAc)2. Org. Lett 2019, 27, 2356–2359. 10.1021/acs.orglett.9b00632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.