

Abstract
Alzheimer’s disease (AD) is one of the most devastating neurodegenerative diseases without effective therapies. Immunotherapies using antibodies to lower assembled Aβ provide a promising approach and have been widely studied. Anti-amyloid antibodies are often selective to amyloid conformation, and the lack of amyloid-antibody structural information limits our understanding of these antibodies’ conformation selection. Gantenerumab and crenezumab are two anti-Aβ antibodies that bind multiple forms of Aβ with different Aβ epitope preferences. Here, using molecular dynamic (MD) simulations, we study the binding of these two antibodies to the Aβ1–40 fibril, whose conformation is derived from an AD patient’s brain tissue. We find that gantenerumab recognizes the Aβ1–11 monomer fragment only at slightly lower pH than the physiological environment where His6 of Aβ1–11 is protonated. Both gantenerumab and crenezumab bind with integrated Aβ fibril rather than binding to monomers within the fibril. Gantenerumab preferentially binds to the N-terminal region of the Aβ1–40 fibril, and the binding is driven by aromatic interactions. Crenezumab can recognize the N-terminal region, as well as the cross-section of the Aβ1–40 fibril, indicating its multiple binding modes in Aβ fibril recognition. These results demonstrate conformation-dependent interactions of antibody–amyloid recognition.
Keywords: Alzheimer’s disease, Aβ amyloid, antibody, gantenerumab, crenezumab, MD simulations
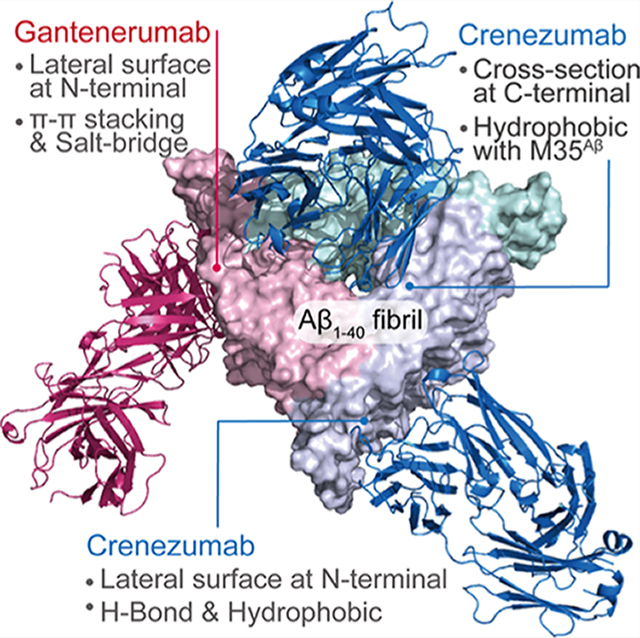
Graphical Abstract
INTRODUCTION
Alzheimer’s disease (AD) is a common neurodegenerative disease that causes impairment in brain functions such as memory and cognition.1 The amyloid hypothesis and tau hypothesis2,3 argue that amyloid-β (Aβ) senile plaques, formed by aggregated Aβ, and neuron fibril tangles, formed by highly phosphorylated tau proteins, are two main driving forces of AD.4 Aβ aggregates include dimers, oligomers, and fibrils according to their sizes and structures.5 Both Aβ oligomer and fibril contribute to the pathological process, while oligomeric Aβ species are reported to have stronger cytotoxicity.6–8 Therefore, clearing aggregated Aβ, especially oligomers, has emerged as a major therapeutic goal. The candidates for anti-Aβ drugs9,10 include nanoparticles,11 peptides,12 small molecules,13,14 and antibodies.15
Immunotherapies using antibodies to reduce assembled Aβ provide a promising approach and have been widely investigated since 1999.15 Both active and passive16 anti-Aβ immunotherapies are able to clear Aβ plaques17–20 with a possible mechanism of first antibody binding to Aβ followed by receptor-mediated efflux or Fc receptor-mediated phagocytosis of antibody–Aβ complexes.19,21–23 While for a long time no success was achieved in clinical trials because of the unsatisfactory curative effect24–28 or several side effects,29 an anti-amyloid antibody aducanumab30 recently showed modest but significant efficacy in a phase 3 trial. Even though there is an ongoing debate about the results,31,32 it nevertheless provided important validation of amyloid as a therapeutic target.33
There are at least two factors causing the inconsistent results with antibody drugs against Aβ: the limited brain penetration and lack of selectivity for the soluble Aβ oligomers.33 Amyloids are known for their polymorphic conformations.34 As expected, as an intrinsically disordered protein, Aβ forms multiple structures of oligomers and fibrils under different conditions.35–40 Different soluble Aβ aggregates can give rise to cellular toxicity through different mechanisms.41 Because an amyloid-directed antibody may target only one or a few related potentially toxic protein variants, this could explain why such therapeutics have had little clinical impact.42 The situation is complicated further by the lack of structural details of antibody–amyloid recognition. The related crystal structures resolved so far are antibodies binding monomeric Aβ. There may be different characteristics between assembled Aβ and highly flexible monomeric Aβ when interacting with antibodies. Complementary to experiments, molecular dynamics (MD) simulations can describe the Aβ-antibody interaction at an atomic level of detail. Zhao et al.43 simulated the recognition of monomeric, oligomeric, and fibril Aβ by three homologous antibodies (solanezumab, crenezumab (C-mab), and CreneFab) and successfully identified stable complexes of C-mab with Aβ pentamer (oligomer model) and 16mer (fibril model). MD simulation showed that C-mab recognizes the oligomer’s lateral and edge residues, with CDR loops effectively binding to the Aβ fibril lateral surface around the same region spanning residues 13–16. A representative three-layered model of Aβ oligomer and fibril were used in Zhao’s study (PDB ID 2MXU40). While Zhao’s pioneering study provided insights into the antibody–amyloid interaction, further investigations are needed to cross-examine different amyloid conformations with different antibody interactions for better understanding the mechanisms of Aβ-based immunotherapy.
In this work, we focus on two antibodies, gantenerumab (G-mab) and crenezumab (C-mab) and their interactions with the amyloid conformers derived from the patienťs brain tissue (PDB ID 2M4J37). As the first full human anti-Aβ antibody, G-mab was reported to recognize Aβ fibrils, oligomers, and monomers in vitro, with the highest affinity for fibrils and lowest affinity for monomers.22 Studies in transgenic mice found that G-mab can effectively bind and reduce brain Aβ plaques but does not affect plasma Aβ.22,44 Phagocytosis of Aβ plaques induced by G-mab was observed in AD brain slices.22,23 Clinical studies showed a dose-dependent reduction in brain Aβ, but no benefit in cognition has been reported yet.23,45–47 Though vasogenic edema developed in several patients,23 G-mab was reported having an acceptable safety profile at a higher dose of 1200 mg.46 It has been shown that G-mab can reduce Aβ plaques in patients with prodromal to moderate AD.46 Epitope mapping revealed that G-mab can recognize both N-terminal (residues 1–12) and the middle segment (residues 23–28) of Aβ, but only structures of G-mab Fab binding with Aβ1–11 or Aβ3–11 monomers have been determined by X-ray diffraction method (PDB ID 5CSZ).22 In this complex, Aβ1–11 monomer contacts extensively with CDRs (H1, H2, H3, and L1) and with F4Aβ deeply buried in a hydrophobic pocket.
C-mab, a humanized monoclonal antibody, is able to recognize Aβ monomer, oligomer, and fibril, with higher affinity for aggregated forms.48 C-mab can bind to Aβ plaques in the brain sections of both AD patients and transgenic mice.17 Compared to other antibodies, C-mab was not outstanding in lowing Aβ level but caused minimum inflammatory reactions in transgenic mice.18 The high safety performance was also confirmed by clinical studies.21 Lower oligomer level and higher monomer level were reported by Yang et al. in a 69-week clinical study,49 but no improvement of cognition has been observed as of now. Crystal structure of CreneFab (C-mab with different constant domain) in complex with Aβ13–24 monomer was resolved by Ultsch et al. in 2016 (PDB ID 5KNA).48 Aβ13–24 monomer, with an extended conformation, interacted with H1, H2, and L3. Solution NMR analysis revealed that CreneFab binds with Aβ1–42 monomer at the same epitope.48
The G-mab and C-mab have different preferred Aβ binding locations in aqueous solution. The G-mab can interact with both N-terminal and central regions of Aβ peptide, while the C-mab binds to the central motif of Aβ peptide. However, both of them can bind to monomeric, oligomeric, and fibril forms of Aβ, with increased affinities. In order to delineate the underlying mechanisms, we systematically simulate and examine the interactions of C-mab and G-mab with Aβ1–40 fibril consisting of 16 chains (PDB ID 2M4J,37 hereinafter referred to Aβ4016mer or Aβ40 fibril). We choose 16 chains to construct Aβ fibril because 16 chains can well represent fibril instead of oligomer and also for the purpose of binding energy comparison with previous study.43 We find that the protonation of His6 in Aβ is necessary for G-mab’s recognition of monomeric Aβ. Our results show that the G-mab–Aβ1–11 complex with neutral histidine is energetically unstable, which may explain the low affinity of G-mab with monomeric Aβ. In addition, on the basis of multiple MD simulations of 9 G-mab-Aβ4016mer complexes and 7 C-mab–Aβ4016mer complexes, we find that (1) G-mab binds to the N-terminus of the fibril mainly by π–π and salt bridge interactions, (2) C-mab binds to the N-terminal region of the fibril through Arg59L2–Ser8Aβ H-bonding and hydrophobic interactions, and (3) C-mab displays a strong interaction with the cross-section of the Aβ40 fibril by hydrophobic interactions with Met35Aβ. Our study demonstrates that antibody–Aβ fibril recognition depends on the amino acid sequences of antibody and the conformations of Aβ fibril.
RESULTS AND DISCUSSION
Protonation of His6 of Aβ1–11 Controls G-mab–Aβ1–11 Interaction.
We first examine the structure of G-mab-Aβ1–11 complex (PDB ID 5CSZ). In the X-ray structure, there is a sulfate anion located near the His6Aβ. The distance between the N atom of the imidazole ring and the S atom of the sulfate anion is only 0.36 nm (Figure 1a), indicating that the His6 could be protonated. The weakly acidic crystallization conditions (25% PEG 3350, 0.1 M Bis-Tris 6.5, and 0.2 M ammonium sulfate)22 can further provide evidence for the protonation of His6. Therefore, we simulate two different G-mab–Aβ1–11 complexes, with His6Aβ in either protonated or neutral state. The average binding energies between Aβ1–11 and G-mab are calculated using the data from 50 to 100 ns. We find that the complex with neutral His6Aβ has a positive binding energy of 12.5 kcal/mol while the one with protonated His6Aβ has a negative binding energy of −24 kcal/mol (Figure 1b). These data demonstrate that G-mab can bind to Aβ1–11 with protonated His6 in low pH environment. The RMSF of G-mab is plotted in Figure 1c (light chain) and Figure 1d (heavy chain). The six CDR regions are highlighted by boxes. As can be seen in Figure 1c,d, both LC and HC regions in the complex with neutral-His6Aβ display obviously higher RMSF values than those in the complex with protonated-His6Aβ, suggesting that the protonated-His6Aβ complex is more stable than the neutral-His6Aβ complex. Interestingly, the residues with higher RMSF values are mainly located in regions outside the CDR domains, which do not interact directly with Aβ1–11, indicating the existence of an allosteric effect.
Figure 1.
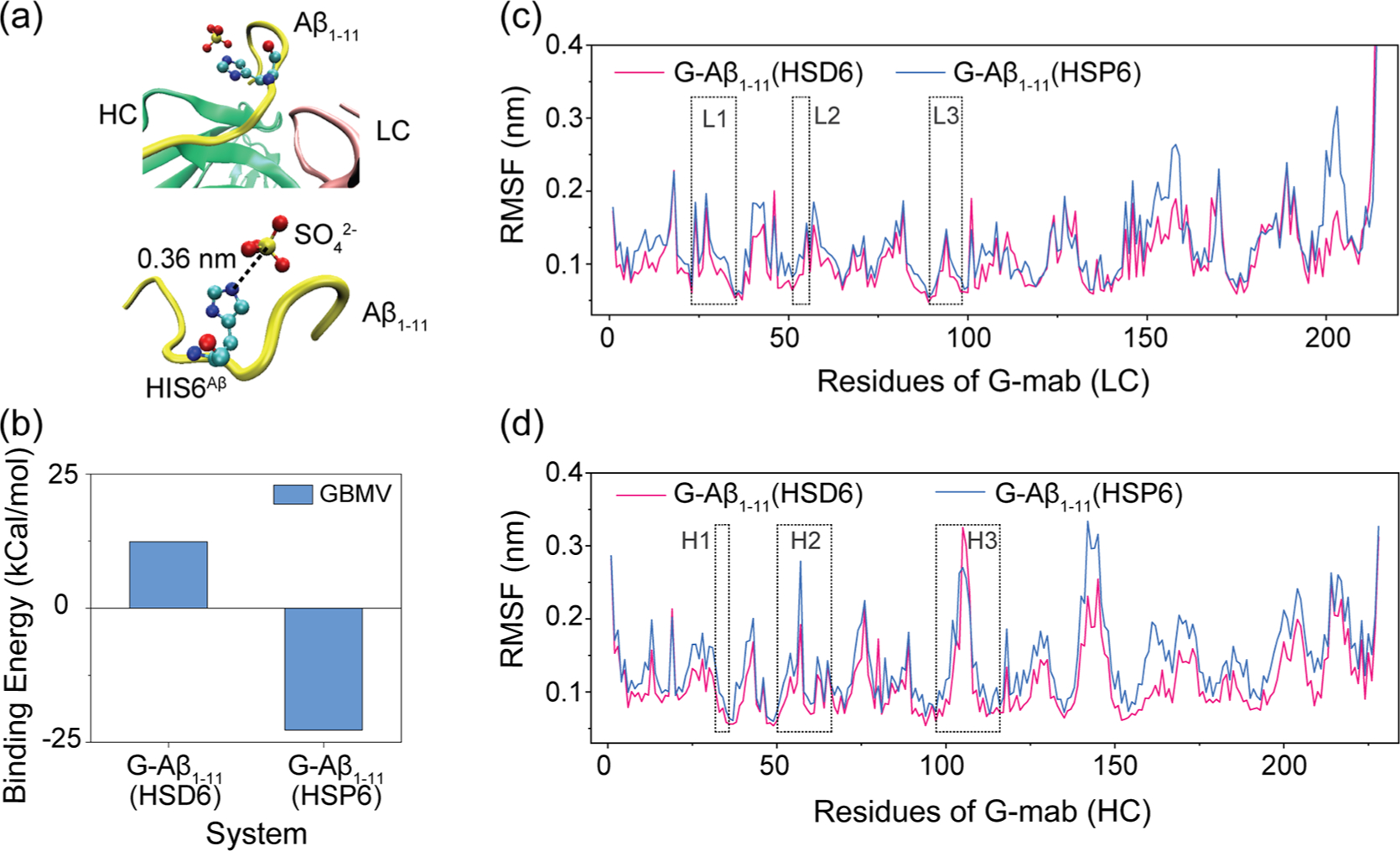
Effects of protonation of His6Aβ on G-mab–Aβ1–11 complex. (a) Part of the G-mab–Aβ1–11 complex in crystal structure (PDB ID 5CSZ). Aβ1–11, light chain, and heavy chain are colored yellow, pink, and green, respectively. His6Aβ and SO42− are shown in VDW representation. A SO42− is located near the nitrogen atoms of His6Aβ side chain (the distance between NE2 and S is 0.36 nm), indicating that His6Aβ is protonated. (b) The binding energy between G-mab and Aβ1–11 with neutral His6 (HSD6) or protonated His6 (HSP6) in our MD simulations. (c, d) Cα root mean square fluctuation (Cα-RMSF) of G-mab’s light chain (c) and heavy chain (d). CDR loops are highlighted in boxes.
It has been found that G-mab could bind to Aβ monomer with a high dissociation rate (kd = 1.2 × 10–2 s−1),22 suggesting rapid exchange of antibody-bound monomeric Aβ. The pH sensitivity and high flexibility of the G-mab–Aβ complex observed in our simulations are consistent with the experimental findings.22 It should be noted that the 1–11 segment used in our simulation cannot replace the full-length peptide in G-mab–Aβ monomer recognition, since there were two epitopes (N-terminal and central region) revealed by experiment but only the complex of G-mab and Aβ1–11 was resolved. However, with the pH change from 7.4 to 6.4, only the charge of the histidine is altered. Since Aβ1–11 is the strongest epitope of G-mab, we can still conclude that the nonprotonation of His6Aβ at neutral pH will decrease the affinity of Aβ1–40 monomer and G-mab.
G-mab and C-mab Bind with Integrated Aβ40 Fibril Rather than Binding Monomer within the Fibril.
We searched the stable structures of G-mab/C-mab binding with Aβ40 fibril by a combination of initial docking and subsequent MD simulations. Since G-mab mostly binds to Aβ40 N-terminal region, a large number of combinations are generated due to the 3-fold symmetry of the Aβ40 fibril conformation. Three clusters of docking poses were selected based on Aβ404 fibril epitope, and we chose 2–3 structures with the lowest energy from each cluster. (Figure S1). In the first group (G-MD1, G-MD2, and G-MD3), G-mab binds to the joint region between two Aβ monomers, where the N-terminal residues of one Aβ monomer interact with the middle region residues of the other Aβ monomer. The second group (G-MD4, and G-MD5) includes the conformers in which G-mab only binds to Aβ N-terminal residues. In the third group (G-MD6, G-MD7, and G-MD8), G-mab binds to the cross-section A40β fibril, rather than the lateral surface. Similarly, 6 structures of C-mab-A40 16mer complex were selected as the initial states (data not shown).
We then performed 20 ns MD simulations starting from these structures and discarded some systems in which the antibody and Aβ were obviously separated from each other. The remaining 10 systems were extended to 100 ns, including two C-mab-Aβ4016mer complexes (C-MD1, C-MD2) and eight G-mab-Aβ40 16mer complexes (G-MD1–8).
In order to examine the possibility of antibody binding to only one monomer within fibril, we used the Chimera software50 to construct two antibody–Aβ4016mer complexes (C-MD0 and G-MD0), in which the epitope fragments (residue 13–22 in C-MD0 and residue 1–11 in G-MD0) in the 16th chain (the edge chain) of Aβ40 fibril were replaced by the Aβ segment in the crystal structure. These two complexes, together with the above ten complexes, were subjected to 100 ns MD simulations.
We first examined if the antibody can bind one monomer within the fibrils and keep the binding interactions in the same ways as in the crystal structure. This question can be answered using the data from the 100 ns simulations of the G-MD0 and C-MD0 systems. The results are shown in Figure 2a; the LC and HC regions of antibodies are colored in pink and green, respectively. The Aβ segments binding to antibodies are highlighted in yellow, and the remaining regions of Aβ40 fibrils are shown in blue. Their binding energies are positive (Figure 2b), indicating that extracting one monomer from Aβ40 fibril by these antibodies is energetically unfavorable.
Figure 2.
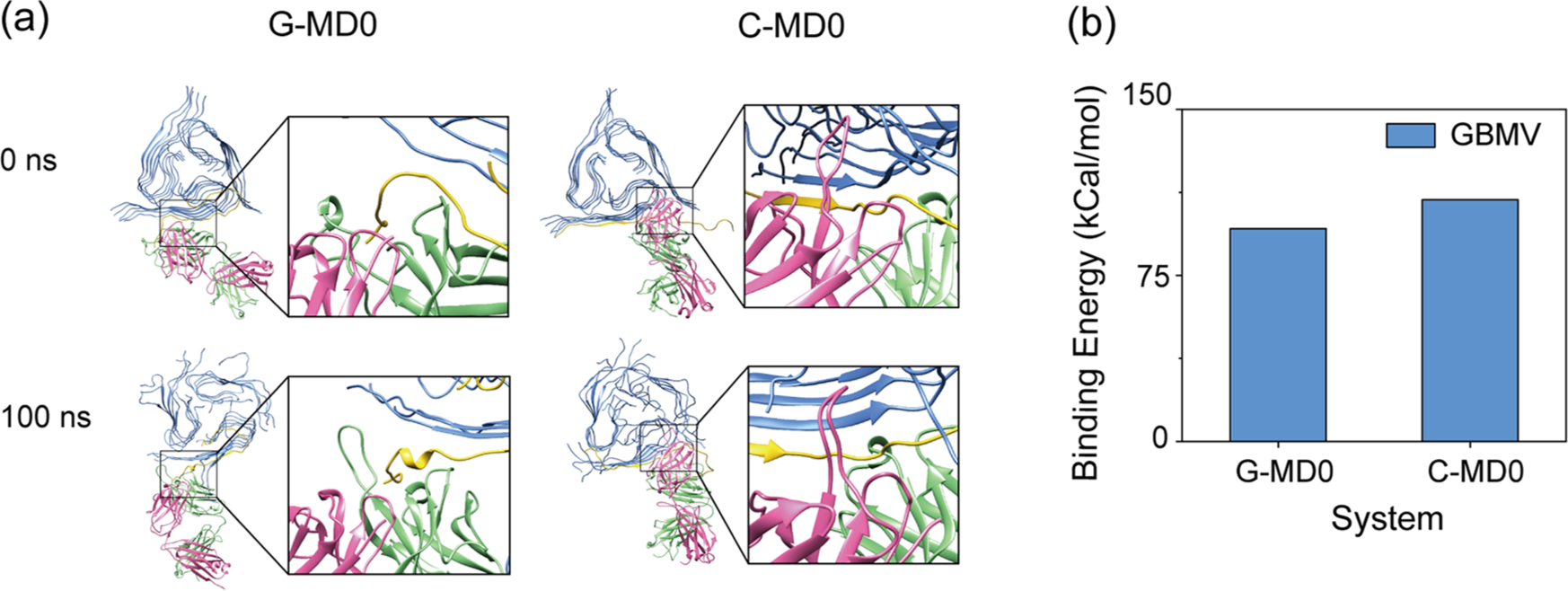
G-mab and C-mab cannot bind to one Aβ monomer within Aβ4016mer. C-MD0 and G-MD0 are two structures with the epitope fragments (residue 13–22 in C-MD0 and residue 1–11 in G-MD0) in the 16th chain (the edge chain) of Aβ40 fibril keeping the same monomeric conformation in the crystal structures. (a) Initial and final structure of G-MD0 and C-MD0, with Aβ, HC, and LC colored in blue, green, and pink, respectively. The segments binding to antibody are highlighted in yellow. (b) Binding energies between G-mab/C-mab and Aβ4016mer of system G-MD0 and C-MD0.
The structures of the ten systems with the antibodies binding with integrated fibril can be seen in Figure S1. We examined the average binding energy between Aβ40 fibril and antibodies using the conformations in the last 50 ns. Systems with positive binding energy are considered to be unstable. As seen in Figure 3a, three systems with negative binding energy are considered to be the stable structures: (1) G-mab binding with the of the N-terminal residues of Aβ40 fibril on the lateral surface (G-MD4), (2) C-mab binding with the N-terminal residues of Aβ40 fibril on the lateral surface (C-MD1), and (3) C-mab interacting with the cross-section of Aβ40 fibril (C-MD2). Snapshots of the three stable structures at 0 and 100 ns are shown in Figure 3b. These results indicate that G-mab and C-mab preferentially bind to integrated fibril, rather than monomer within fibril.
Figure 3.
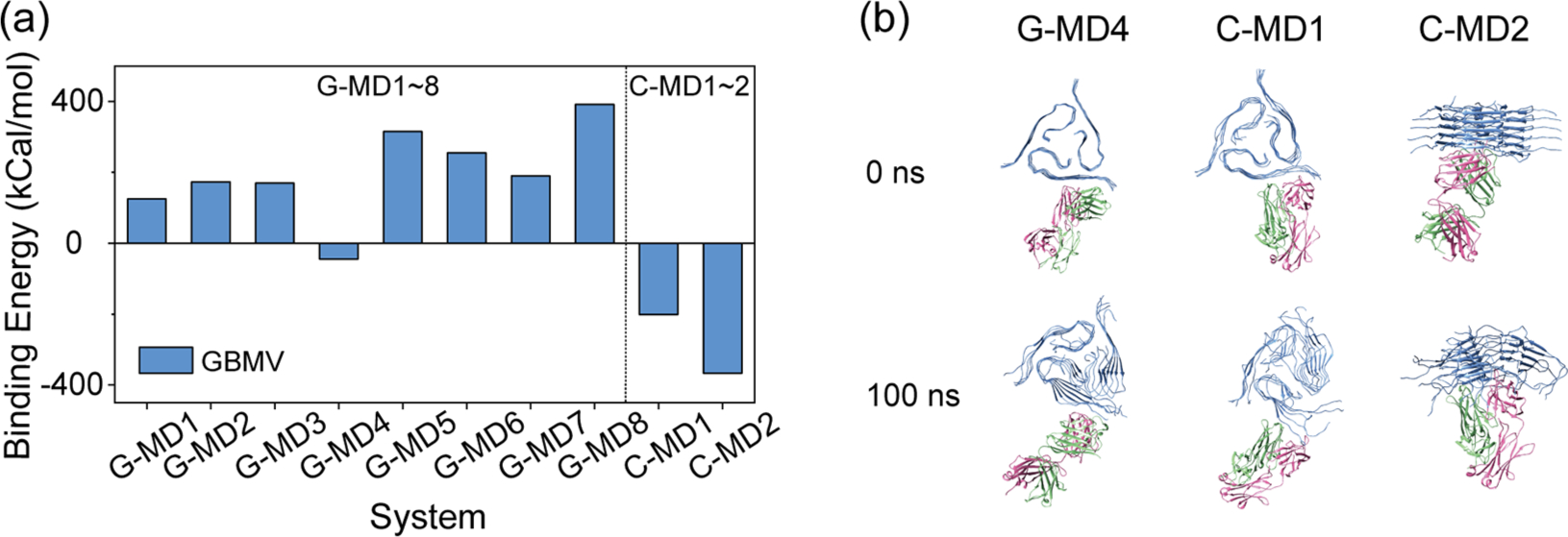
Stabilities of the 10 antibody–Aβ4016mer structures obtained from docking. The initial and final structures of all the systems can be seen in Figure SI. (a) Average binding energy between Aβ40 fibril and antibodies. The binding energy is calculated by 〈Ebind〉 = 〈Ecomplex〉 − 〈Eantibody〉 – 〈Epeptide〉. (b) Initial and final structures of three antibody–Aβ40l6mer complexes with negative binding energy (C-MD1, C-MD2, and G-MD4). Aβ, HC, and LC are colored in blue, green, and pink, respectively.
The Binding of G-mab to the N-Terminal Region of Aβ40 Fibril Is Driven by Aromatic Interactions.
Bohrmann et al.22 proposed a model of G-mab binding to the Aβ fibril surface in their experiment study: G-mab first binds to the disordered N-terminus, followed by its binding to the 19VFFAEDVG26 segment on the fibril surface. However, in the last section, we have shown that binding to the disordered N-terminus is energetically unfavorable. For the Aβ fibril structure in the 2M4J conformation, we find that G-mab binds on the lateral surface of Aβ fibril in the N-terminal region. The contact map between each residue of Aβ and G-mab is plotted in Figure 4a to gain an overall understanding of G-mab-Aβ4016mer interactions. The epitope revealed by X-ray experiment is highlighted in the blue box. As can be seen in Figure 4a, the Aβ interaction region from simulation (residue 1–15) is highly maintained as that in crystal structure (residue 1–13), indicating that G-mab recognizes a similar epitope for both Aβ monomer and Aβ fibril. H2 and H3 regions contribute the most contacts among all the residues of the CDR.
Figure 4.
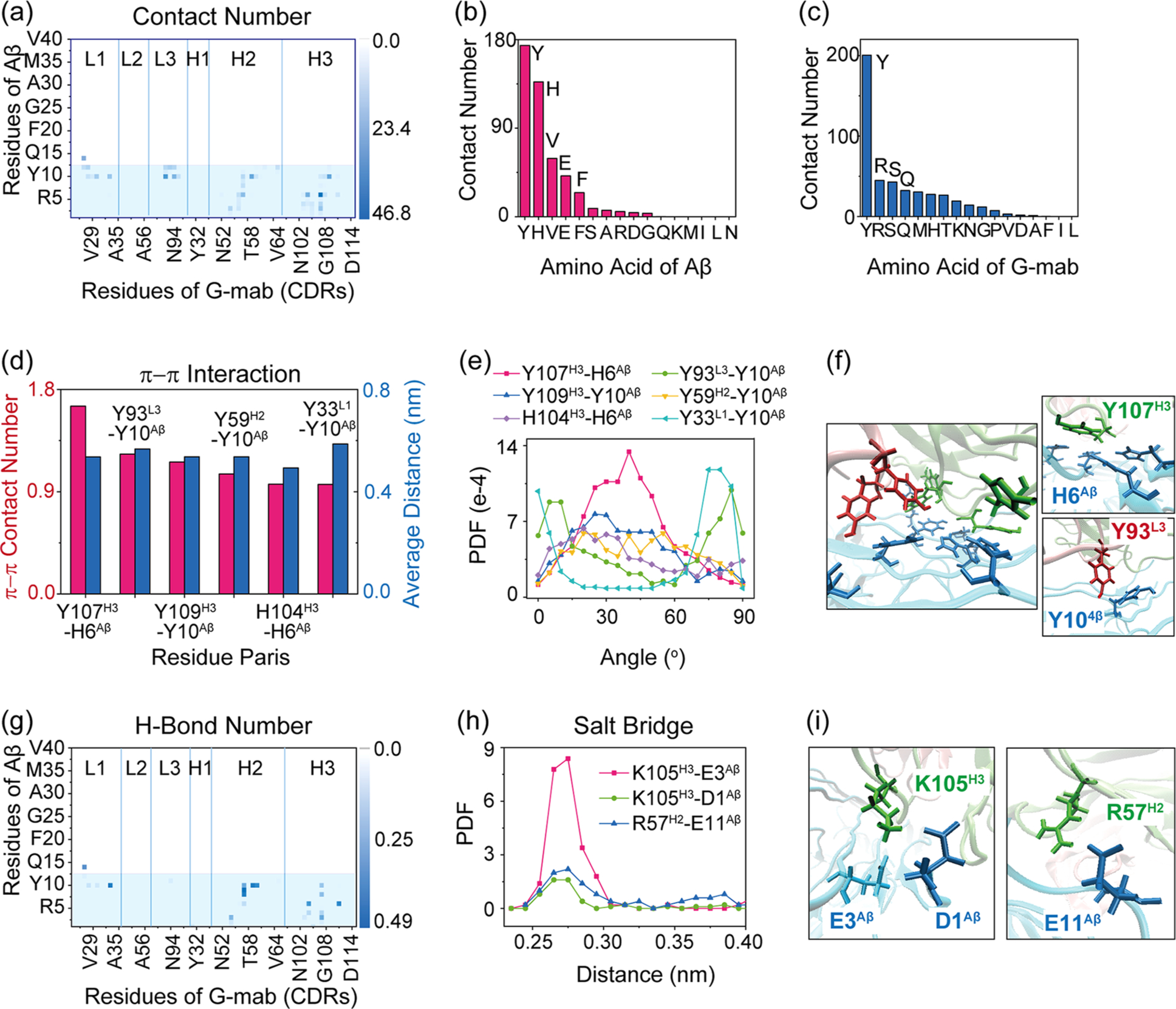
Interaction analysis of G-mab-Aβ4016mer interactions for G-mab binding to the N-terminal region of Aβ40 fibril in simulation G-MD4. (a) Contact probabilities between each residue of G-mab and Aβ40 fibril. The residue pairs with the top six contact numbers are listed in Table 1. (b, c) Total contact number for each residue type of Aβ (b) and antibody (c). (d-f) π–π interactions between G-mab and Aβ40 fibril. (d) π–π stacking number and average distance between two aromatic rings. (e) PDF of the dihedral angle between two aromatic planes. (f) Snapshots of typical aromatic interactions. (g) H-bond number map of G-mab and Aβ40 fibril. (h) PDF of three oppositely charged residue distances. (i) Snapshots showing the Lys105H3-Glu3Aβ, Lys105H3-Asp1Aβ, and Arg57H2-Glu11Aβ salt bridges. Aromatic residues are shown in bond representation.
We examine residue-residue interactions within the binding site by summing up the contact numbers of each kind of residue for Aβ and G-mab (Figure 4b). The residue with the highest contact number in Aβ is Tyr, significantly larger than the second one (His). Similarly, His and Tyr in G-mab contribute the largest and second-largest contact numbers, respectively. Furthermore, most residue pairs with top contact numbers are Tyr–Tyr, Tyr–His, or His–His (Table 1). Both Tyr and His are aromatic residues, indicating that π–π interactions play an important role in G-mab-Aβ4016mer interaction. To further characterize the π–π interactions, we calculate the average π–π stacking numbers and distance for each aromatic residue pair between Aβ and G-mab. As seen in Figure 4d, the six aromatic residue pairs with the top π–π stacking number are Tyr107H3-His6Aβ, Tyr93L3-Tyr10Aβ, Tyr109H3-Tyr10Aβ, Tyr59H2-Tyr10Aβ, His104H3-His6Aβ, and Tyr33L1-Tyr10Aβ, and their angle distributions are plotted in Figure 4e. A snapshot of the interface is presented in Figure 4f to show the importance of π–π stacking intuitively. A large amount of π–π interaction pairs can be seen at the interface. The snapshots of the top two pairs (Tyr107H3-His6Aβ and Tyr93L3-Tyr10Aβ) are placed on the right side of panel Figure 4f. There are also many polar or charged residues located at the N-terminal of Aβ. Thus, H-bonding and electrostatic interactions also play roles in the G-mab-Aβ4016mer interactions. The H-bond number matrix is plotted in Figure 4g. H2 loop of G-mab can form strong H-bonds with segment Ser8–Tyr10Aβ, while H3 loop can form H-bond extensively with Aβ1–11 segment. Three salt bridges exist between Aβ and G-mab: Lys105H3-Glu3Aβ, Lys105H3-Asp1Aβ, and Arg57H2–Glu11Aβ. The PDF of the distance of the three pairs with opposite charges is plotted in Figure 4h, and the snapshots are given in Figure 4i. All three curves exhibit sharp peaks in distance = 0.28 nm, indicating the formation of salt bridges in the three pairs. In short, the binding of G-mab to the lateral of Aβ40 fibril is stabilized by π–π stacking, H-bonding, and salt bridge interactions. It is interesting to note that these interactions are shared for small molecules binding with Aβ fibrils as well, similar binding sites and mechanism were also observed in several computational works.51–55 We also calculate the above interactions in G-MD5 system, which has similar structures with G-MD4 but positive binding energy (Figure S1). Data show that the number of both π–π stacking and salt bridges in G-MD5 are significantly smaller than that in G-MD4 (Figure S2), further underlining the importance of π–π stacking and salt bridges on the binding between G-mab and N-terminal region of Aβ.
Table 1.
Contact Number, H-Bond Number, and π–π Contact Number between Top 6 Contact Pairs in System G-MD4
| residue of G-mab | Y107H3 | Y93L3 | Y109H3 | Y59H2 | Q27L1 | H104H3 |
| residue of Aβ | H6 | Y10 | Y10 | Y10 | H14 | H6 |
| contact number | 46.8 | 31.1 | 27.0 | 25.5 | 25.1 | 23.5 |
| H-bond number | 0 | 0 | 0 | 0.44 | 0.35 | 0.34 |
| π–π contact number | 1.65 | 1.23 | 1.16 | 1.06 | 0.97 |
Interestingly, our simulations showed that Aβ1–11 monomer with neutral histidine (HSD6) cannot form a stable complex with G-mab, while Aβ fibril with HSD6 can be recognized by G-mab in the N-terminal region, indicating that the pH dependence is specific to the binding of G-mab to Aβ1–11. Currently, there is no report about the protonation state of histidine residues in the Aβ fibril. Following the reviewer’s suggestion, we compared the G-mab–Aβ1–11 interaction between system G-Aβ1–11(HSD6) and system G-MD4. The contact maps in Figure S3 show different binding modes of G-mab to Aβ1–40 fibril and Aβ1–11 monomer. In system G-MD4, the interaction concentrates on residues 6–11Aβ and CDRs H2/H3, while in system G–Aβ1–11(HSD6), G-mab mainly recognizes residues 1–5 with L3/H1/H2/H3.
Multiple Binding Modes of C-mab in Aβ40 Fibril Recognition.
The binding characteristics of C-mab in recognizing Aβ40 fibril with the 2M4J conformation through the lateral interface are summarized in Figure 5 and Table 2. Even though the C-mab is an antibody that was reported to mainly recognize the central region of Aβ fibril (residues 12–28),48 we find that it can also bind to Aβ4016mer N-terminal region in the 2M4J conformation (Figure 5a). The main epitope residues are Tyr10, Ser8, and His6 of Aβ (Figure 5b), interacting with paratope residues Ser, Asn, and Arg of C-mab (Figure 5c). The top six residue pairs with the highest contact numbers are listed in Table 2. The first pair is Arg59L2 and Ser8Aβ, maintained by an extremely stable H-bond (the existence probability is 100%), which can also be seen in the H-bond number matrix in Figure 5d. Further calculation reveals that the Arg59L2–Ser8Aβ H-bond is mainly formed between the main-chain of Arg59L2 and the side-chain of Ser8Aβ (~91%). The other five pairs are Asn33L1–Tyr10Aβ, Ser31H1–His14Aβ, Ser61L2–Tyr10Aβ, Asn58L2–Ser8Aβ, and Phe60L2–Tyr10Aβ. Interestingly, despite the existence of polar residues Asn, Ser, and His and aromatic residues His, Tyr, and Phe, almost no hydrogen bond or π–π stacking is formed among the five pairs (Table 2), which illustrates that the main stabilizing force is hydrophobic interaction (for example, between the aromatic ring of Tyr10Aβ and the nonpolar aliphatic groups of Asn33L1). Albeit with very low formation stabilities, π–π stacking interactions are observed in three pairs of aromatic residues: Tyr31L1–Tyr10Aβ, Phe60L2–Tyr10Aβ, and Tyr32H1–His14Aβ (Figure 5e). The PDF of angles in π–π stacking are plotted in Figure 5f. The snapshots of typical π–π stacking are shown in Figure 5g. No salt bridge is observed between Aβ and C-mab. To sum up, the binding of C-mab to the N-terminal region of Aβ40 fibril is primarily by Arg59L2–Ser8Aβ H-bonding and hydrophobic interactions. In system C-MD1, C-mab recognizes the N-terminal region of Aβ40 fibril mainly by L1 and L2 loops (Figure 5a), two CDRs with relatively high content of aromatic and charged residues. However, the π–π and electrostatic interactions in C-MD1 are not as important as those in G-MD4, which can be explained by the sequence difference of C-mab and G-mab. According to Figure 5h, most of the aromatic and charged residues in H2 and H3 of G-mab are centered around the middle of CDRs, while those in L1 and L2 of C-mab are located in the ends of loops, which greatly reduce the freedom of C-mab aromatic and charged residues that interact with Aβ40 fibril. The similar N-terminal binding position of Aβ40 fibril but totally different mechanisms between C-MD1 and G-MD4 indicate the importance of sequence specificity for antibody in recognizing Aβ fibril.
Figure 5.
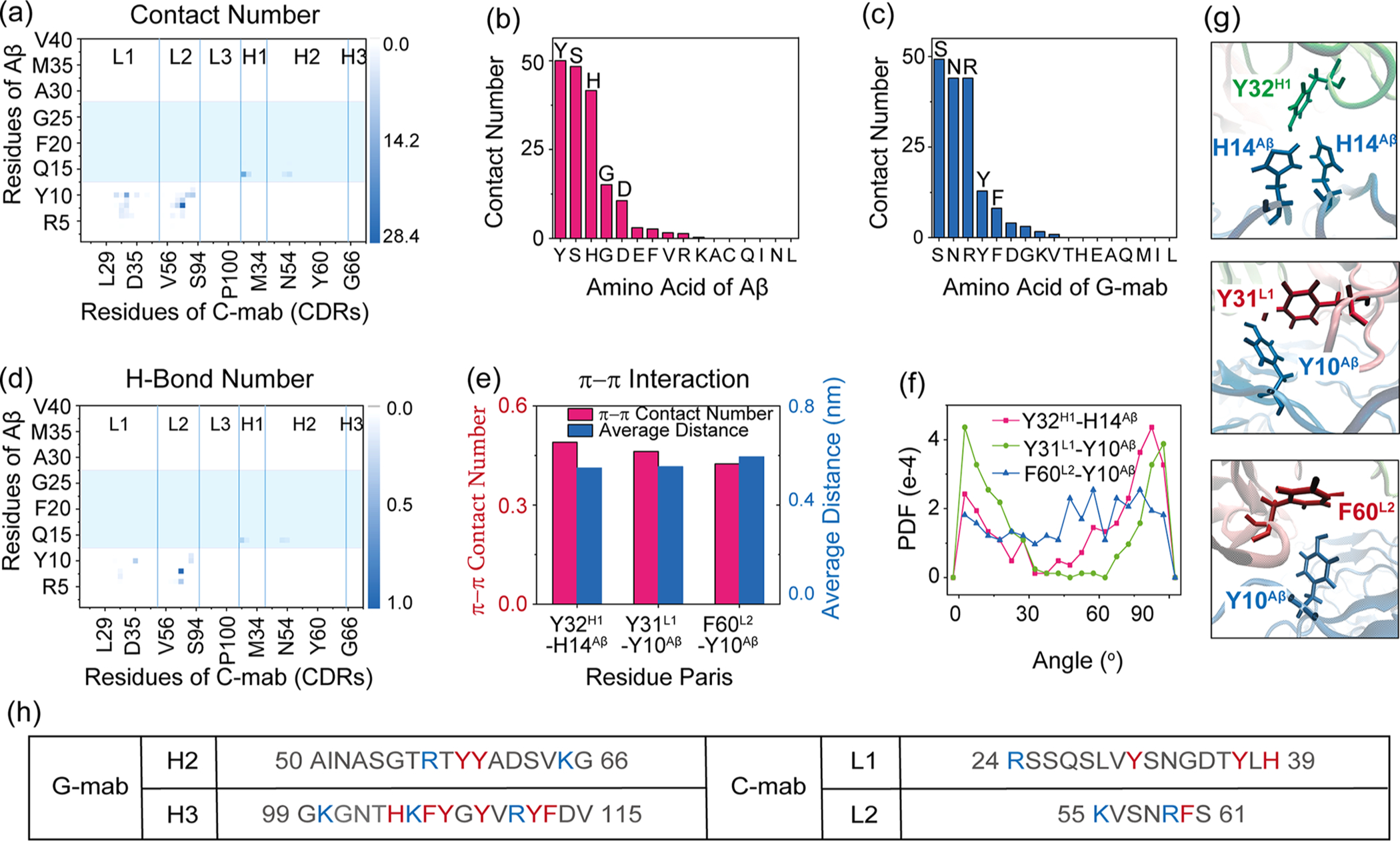
C-mab-Aβ4016mer interactions in system C-MD1. (a) Contact probabilities between each residue of C-mab and Aβ40 fibril. (b, c) Total contact number for each residue type of Aβ with antibody (b) and each residue type of antibody with Aβ (c). (d) Map of H-bond number between C-mab and Aβ4016mer. (e–g) π–π interactions between C-mab and Aβ4016mer: (e) π–π stacking number and average distance between two aromatic rings; (f) PDF of dihedral angle between two aromatic planes; (g) snapshots showing aromatic interactions. (h) Comparison of the sequences of the two CDRs in C-mab and G-mab that contribute most to antibody–Aβ4016mer contact. Residues with positive charge are colored in blue, while aromatic residues are colored in red.
Table 2.
Contact Number, H-Bond Number, and π–π Contact Number between Top 6 Contact Pairs in System C-MD1
| residue of C-mab | R59L2 | N33L1 | S31H1 | S61L2 | N58L2 | F60L2 |
| residue of Aβ | S8 | Y10 | H14 | Y10 | S8 | Y10 |
| contact number | 28.4 | 16.1 | 15.2 | 9.0 | 7.9 | 7.6 |
| H-bond number | 1.0 | 0 | 0 | 0.2 | 0 | 0 |
| π–π contact number | 0 |
Previously, we studied the C-mab binding to the central region of Aβ11–42 fibril (16mer) with the S-shaped 2MXU conformer.43 It is interesting to see that C-mab used different paratope residues to interact with Aβ fibril with different conformations, with similar binding energies (−195.8 kcal/mol for C-mab and Aβ11–4216mer and −201.2 kcal/mol for C-mab and Aβ1–4016mer). In the C-mab/Aβ11–42 binding pattern, the C-mab mainly uses its heavy chain, while in the C-mab/Aβ1–4016mer with the 2M4J conformation, the C-mab mainly uses the light chain. We find that only S31H1 is involved in both binding modes.
In this study, we also examine the possibility of an antibody to bind with Aβ40 fibril on its cross-section. While we do not find an energetically favorable binding mode for G-mab, the system C-MD2 is found to be a stable C-mab–Aβ4016mer complex of C-mab and Aβ. The binding energy for the C-mab binding on the cross-section of Aβ is very low (−366.7 kcal vs −201.2 kcal/mol for C-MD1), indicating strong binding interactions.
We calculated the contact numbers between each residue of C-mab and Aβ (Figure 6a). Data show that residues in all of six CDRs participate in contact with the Aβ40 fibril, while the involved residues of Aβ are mainly distributed in the C-terminal region (29GAIIGLMVGGVV40). Total contact number for each residue of Aβ with C-mab in Figure 6b shows that Met35 contributes most to C-mab–Aβ4016mer interactions, followed by Val40 and Gly33. Remarkably, Met35 plays an important role in stabilizing the Aβ fibril (PDB ID 2M4J).37 Previous studies also reported that the oxidation or mutation of Met35 can greatly reduce the formation rate and neurotoxicity of Aβ aggregates.56,57 The top six contact pairs in Table 3 indicate that M35Aβ mainly contacts aromatic residues Tyr31L1 and Tyr32H1, and the representative snapshots are presented in Figure 6c. The hydrophobic interactions between Tyr residues in C-mab and Met35Aβ help CDR loops stretch into and firmly bind to the cross section of the Aβ40 fibril, which may disaggregate the fibril.
Figure 6.
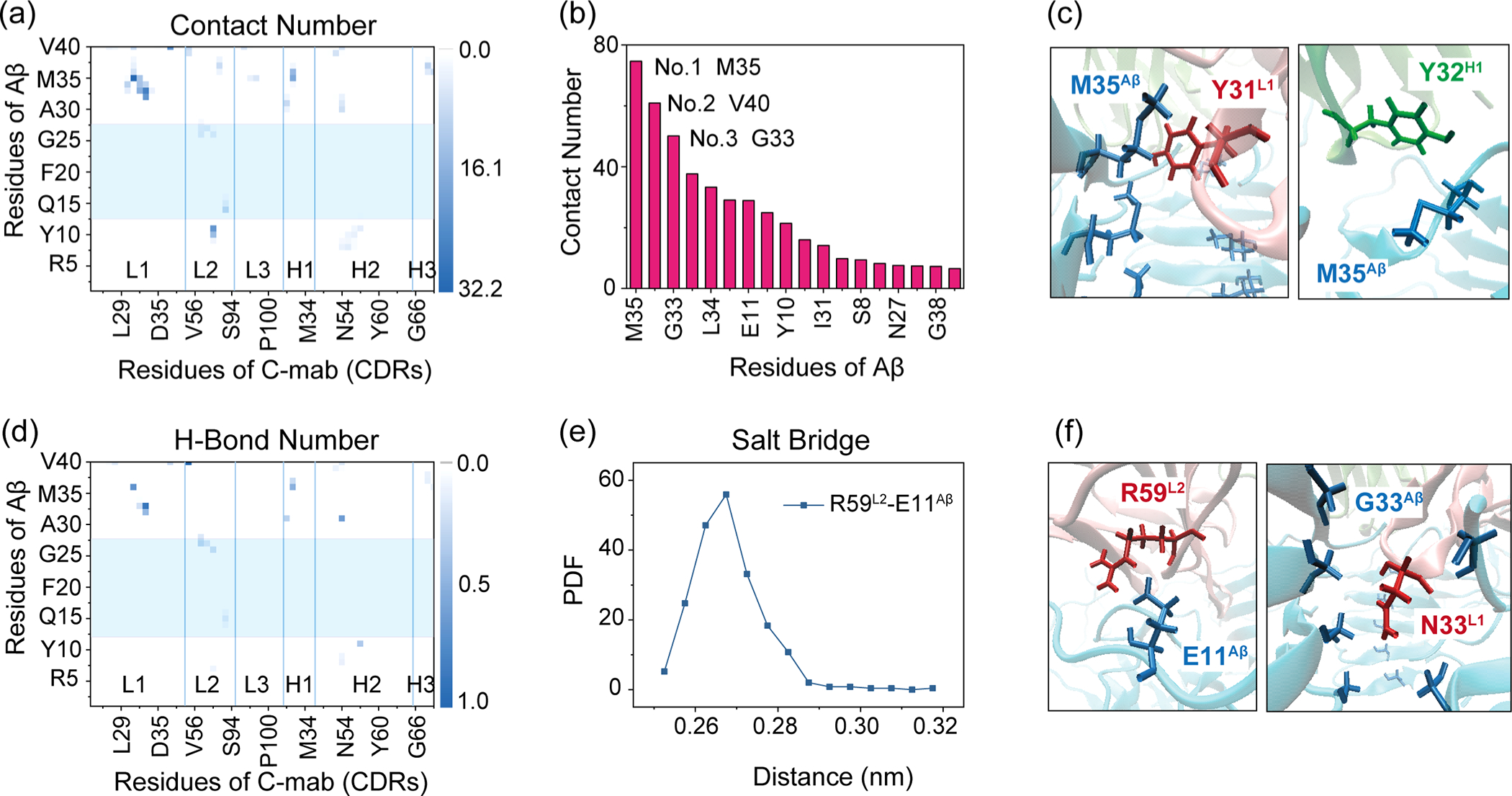
C-mab-Aβ4016mer interactions in C-MD2. (a) Contact probabilities between each residue of G-mab and Aβ4016mer. (b) Total contact number for each residue of Aβ with C-mab. (c) Snapshots of hydrophobic interactions between M35Aβ and C-mab. (d) H-bond number map of G-mab and Aβ4016mer. Paratope residues are highlighted in blue box. (e) PDF of Arg59L2–Glu11Aβ distance. (f) Snapshots of Arg59L2–Glu11Aβ salt bridge and Asn33L1–Gly33Aβ H-bond.
Table 3.
Average Contact Number and H-Bond Number for Top 6 Contact Pairs in System C-MD2
| residue of C-mab | Y31L1 | N33L1 | Y37L1 | R59L2 | N33L1 | Y32H1 |
| residue of Aβ | M35 | G33 | V40 | E11 | I32 | M35 |
| contact number | 32.1 | 26.4 | 22.0 | 21.8 | 21.6 | 17.8 |
| H-bond number | 0 | 0.91 | 0.26 | 0 | 0.47 | 0 |
| salt bridge | 1.00 |
Polar interactions are also essential in C-mab recognizing the Aβ40 fibril. As shown in Table 3, Asn33L1–Gly33Aβ form strong hydrogen-bonding interactions mainly between main-chains (~84.2%). H-bonds for Asn33L1–Ile32Aβ and Asn33L1–Gly33Aβ are relatively weak. More H-bond pairs can be seen in Figure 6d. Salt bridges are observed for the Arg59L2 and Glu11Aβ pair (Figure 6e). Snapshots showing the Arg59L2–Glu11Aβ salt bridge and Asn33L1–Gly33Aβ H-bond are given in Figure 6f. In a word, C-mab in C-MD2 can firmly bind with the cross section of Aβ40 fibril by hydrophobic interactions between Tyr residues of C-mab (Tyr31L1 and Tyr32H1) and Met35Aβ, Arg59L2–Glu11Aβ salt bridge, and Asn33L1–Gly33Aβ H-bonds. In comparison with the binding of C-mab on the lateral surface of the Aβ40 fibril, binding on the cross section of Aβ40 fibril, though with the small binding surface area, would effectively recognize Aβ40 fibril and block the elongation of Aβ40 fibrils.
CONCLUSIONS
The recognition ability of an antibody to the disease-related amyloid conformers may determine its potential effectiveness toward amyloid inhibition in a patients’ brain. In this study, we compare two different antibodies, gantenerumab (G-mab) and crenezumab (C-mab), focusing on the structure of the antibody–Aβ complexes and interaction features with the amyloid conformer 2M4J derived from a patient’s brain. Using all atom MD simulations, we investigate the influence of two protonation states of His6Aβ on G-mab binding with Aβ1–11 monomer and screen the possible binding modes of G-mab/C-mab with Aβ4016mer. We find that G-mab recognizes Aβ1–11 monomer fragment only in slightly lower pH than that of the physiological environment, when His6 of Aβ1–11 is protonated. Since His6 is most likely to be neutral under physiological conditions, our results may explain the low affinity of G-mab and Aβ monomer in vitro and in mouse experiments.22,44
Through extensive search of possible binding modes of antibodies with the Aβ4016mer in the 2M4J conformation, we identify three stable structures of antibodies binding to Aβ40 fibril (one G-mab–Aβ4016mer complex (G-MD4) and two C-mab–Aβ4016mer complexes (C-MD1 and C-MD2)). G-MD4 and C-MD1 share a common feature of antibody binding on the lateral part of the Aβ40 fibril N-terminal region but with totally different molecular mechanisms. The binding of G-mab to Aβ N-terminus is mainly through many of π–π interactions and three pairs of salt bridges (Lys105H3–Glu3Aβ, Lys105H3–Asp1Aβ, and Arg57H2–Glu11Aβ), while the binding of C-mab to Aβ N-terminus is mainly through Arg59L2–Ser8Aβ H-bond and hydrophobic interactions. Their interaction difference results from distinct CDR sequences between the G-mab and the C-mab, reflecting the sequence specificity of antibodies. In the C-MD2 system, the C-mab binds on the cross section of the Aβ40 fibril, with CDR loops stretching into and interacting extensively with the hydrophobic C-terminal region of Aβ, especially with Met35Aβ. Arg59L2–Glu11Aβ salt bridge and Asn33L1–Gly33Aβ H-bond also contribute greatly to C-mab–Aβ4016mer interactions. It is noteworthy that both G-mab and C-mab are able to recognize the central region of Aβ as reported by experiments. However, stable structures of antibodies binding at the central region are not available in our simulations. According to the experiments, residues His13, His14, Lys16, Phe19, Phe20, Glu22, and Asp23 in Aβ contribute significantly to C-mab–Aβ interactions in crystal structures.48 Despite the lack of information about atomic structure, we can infer from system G-MD4 that the aromatic residues Phe19 and Phe20 and charged residues Glu22 and Asp23 will play an important role in G-mab recognition of the Aβ central region. However, the side chain of residues His13, Phe19, Phe20, and Asp23 are pointing toward the interior of the Aβ40 fibril with structure 2M4J, making it unfavorable for antibodies binding with the central region of Aβ. To investigate if our simulation results can be extended to other fibril structures, we compared the exposed residues of four full-length Aβ fibril models (2M4J,37 6SHS,58 5OQV,36 2NAO59). It can be seen in Figure S4 that some residues in the N-terminal region (residues 1–6) in fibril 2M4J are exposed and available to antibodies, while fibril 2NAO has a longer exposed N-terminal region (residues 1–14) than 2M4J. Models 6SHS and 5OQV exhibit very similar solvation-exposure mode in the N-terminal region (residues 1–14): half of the residues are exposed to solvent. According to the different solvent-exposure, the order of antibody–N-terminal region binding affinities can be estimated as follows: 2NAO > 2M4J > 6SHS = 5OQV. In addition, our simulations reveal that C-mab can bind to the cross-section of fibril 2M4J through hydrophobic interaction mainly with M35Aβ (C-MD2). Both 5OQV and 2NAO have a packed C-terminus with the side chain of M35Aβ pointing toward the inner surface, indicating that they may adopt a similar binding mode with C-MD2. It is noteworthy that the side chains of M35Aβ point outward in 6SHS, which might result in a new complex structure in which C-mab binds to the lateral part of the fibril at the C-terminus. It is known that amyloid antibodies are amyloid conformation sensitive, and we are going to examine these differences and antibody/amyloid conformation selection in future studies.
Overall, both C-mab and G-mab bind with integrated Aβ40 fibril rather than binding to monomers within the fibril. The binding of G-mab at the N-terminal region of the Aβ40 fibril is driven by aromatic interactions. C-mab may have multiple binding modes in Aβ fibril recognition. The interaction energy of C-mab with the N-terminal region of the Aβ1–40 fibril is comparable to that of C-mab binding to the central region of Aβ17–42 fibril in S-shaped three β-sheet conformation. These results clearly demonstrate conformation-dependent antibody-Aβ recognition.
METHODS
System Preparation.
The structure of G-mab is taken from the crystal structure 5CSZ22 with the Aβ1–11 fragment removed. The missing sequences are modeled by SWISS-MODEL.60 The structure of C-mab is modeled using the Web server PIGSPro61 with a template of 5KNA, as the CreneFab in that crystal structure is a mutant version of the C-mab with 9 mutated residues.48 The CDR loops are defined according to the Kabat62 definition and listed in Table 4. The structure of Aβ40 fibril (16mer) is constructed according to the ssNMR structure (PDB ID 2M4J37).
Table 4.
Summary of the Properties of the Two Antibodies C-mab and G-maba
| crenezumab (C-mab) | gantenerumab (G-mab) | |
|---|---|---|
| epitope | Aβ12–28 | Aβ1–11 |
| Aβ form | monomers, oligomers, and fibrils | monomers, oligomers, and fibrils |
| affinity | nM level | nM level |
| template | IGg4 | IGg1 |
| CDR-L1 | 24RSSQSLVYSNGDTYLH39 | 24RASQSVSSSYLA35 |
| CDR-L2 | 55KVSNRFS61 | 51GASSRAT57 |
| CDR-L3 | 94SQSTHVPWT102 | 90LQIYNMPIT98 |
| CDR-H1 | 31SYGMS35 | 31SYAMS35 |
| CDR-H2 | 50SINSNGGSTYYPDSVKG66 | 50AINASGTRTYYADSVKG66 |
| CDR-H3 | 99GDY101 | 99GKGNTHKFYGYVRYFDV115 |
L1–L3, 1st–3rd CDR regions of LC; H1–H3, 1st–3rd CDR regions of HC.
Using the web server HADDOCK,63 we perform antibody–Aβ4016mer complex molecular docking to search for their potential binding modes. The active residues during docking processes are those in the CDR loops of antibodies and the residues 1–6, 7–15, and 15–24 of Aβ40 fibril.
MD Simulations.
The four disulfide bonds in antibodies are kept according to the IgG subtypes. Both the N-termini and C-termini in antibodies/Aβ are charged as NH3+ and COO− groups. We first solvate the complex structures using TIP3P water molecules64 with a rectangular box, followed by adding Na+/Cl− with a concentration of 0.15 M to mimic a neutral condition.
Each solvated system is gradually relaxed by performing energy minimizations with 10000 steps using conjugate gradient method,65 in which proteins are first restrained and then set flexible to optimize the protein–water interactions. After all the systems achieve equilibrium, we perform a 100 ns MD simulation for each system using the NAMD software66 with CHARMM36 force field.67 All simulations are carried out in the NPT ensemble at a temperature of 310 K and a pressure of 1 bar. The MD trajectories are saved every 2 ps for analysis. A summary of all simulation systems is listed in Table 5.
Table 5.
Summary of Simulation Details for Each System
| systema | total atoms | time (ns) | template PDB |
|---|---|---|---|
| G-mab + Aβ4016mer | |||
| G-MD1 | 210094 | 100 | 5CSZ/2M4J |
| G-MD2 | 189947 | 100 | 5CSZ/2M4J |
| G-MD3 | 211482 | 100 | 5CSZ/2M4J |
| G-MD4 | 207984 | 100 | 5CSZ/2M4J |
| G-MD5 | 206329 | 100 | 5CSZ/2M4J |
| G-MD6 | 223006 | 100 | 5CSZ/2M4J |
| G-MD7 | 227175 | 100 | 5CSZ/2M4J |
| G-MD8 | 217074 | 100 | 5CSZ/2M4J |
| G-MD0 | 225218 | 100 | 5CSZ/2M4J |
| C-mab + Aβ4016mer | |||
| C-MD1 | 225599 | 100 | 5KNA/2M4J |
| C-MD2 | 215840 | 100 | 5KNA/2M4J |
| C-MD0 | 233698 | 100 | 5KNA/2M4J |
| G-mab + Aβ1–11 Monomer | |||
| G-Aβ1–11 (HSD6) | 90750 | 100 | 5CSZ |
| G-Aβ1–11 (HSP6) | 90746 | 100 | 5CSZ |
| Separate Components | |||
| Aβ1–11 (HSD6) | 18135 | 100 | 5CSZ |
| Aβ1–11 (HSP6) | 10070 | 100 | 5CSZ |
| Aβ4016mer | 124075 | 100 | 2M4J |
| C-mab | 91058 | 100 | 5KNA |
| G-mab | 90918 | 100 | 5CSZ |
HSD, nonprotonated histidine; HSP, protonated histidine.
To check the convergence and repeatability of our simulations, we perform another 100 ns simulation for each of the three stable antibody–Aβ fibril systems (C-MD1, C-MD2, and G-MD4). Convergence check is given in Supporting Information (Figures S5 and S6) and shows that our simulations are reasonably converged.
Analysis.
All the analyses are based on the last 50 ns trajectories using our in-house-developed codes and tools in the NAMD/GROMACS software package.66,68 Here we employ the widely used MM-GBMV (molecular mechanism-generalized born with molecular volume) method69 to calculate the free energy. The binding energy between antibody and Aβ is calculated using 〈Ebind〉 = 〈Ecomplex〉 − 〈Eantibody〉 − 〈EAβ〉, where 〈Ecomplex〉, 〈Eantibody〉, and 〈EAβ〉 all consist of the gas phase energy and the solvation free energy. The conformational entropy is not accounted in MM-GBMV method as done in previous works,43,70–73 because the contribution of entropy to total free energy is much smaller than the other items.74–76 Overall, the binding energy calculated by MM-GBMV without entropy provide a reasonable estimate.43,70 The dielectric constant of water is set to 80. Simulations of isolated C-mab, G-mab, Aβ40 fibril, and Aβ1–11 monomer are performed for the calculation of GBMV energy.
We define a contact as the distance between two heavy atoms being within a distance cutoff. The contact cutoff for two carbon atoms is 0.54 nm; otherwise it is 0.46 nm. Two conditions are needed to define a hydrogen bond (H-bond): (i) the N–O distance is less than 0.35 nm and (ii) the angle of N–H···O is larger than 150°. Positively charged residues and negatively charged residues can form salt bridges when the minimal distance between the N atom of NH3+ group and the COO− group is less than 0.4 nm. For the case of Arg, we include all three side-chain N atoms. Two aromatic rings are considered to form a π–π stacking if their central distance is less than 0.65 nm.77–79 The structure representations are drawn using the VMD software.80
Supplementary Material
ACKNOWLEDGMENTS
This work has been supported by the National Key Research and Development Program of China (Grant No. 2016YFA0501702) and the NSF of China (Grant No. 11674065). It has also been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. All simulations reported in this work had were performed using the high-performance computational facilities at the NIH biowulf supercomputer.
ABBREVIATIONS
- MD
molecular dynamics
- CDR
complementarity determining region
- LC
light chain
- HC
heavy chain
- C-mab
crenezumab
- G-mab
gantenerumab
- Aβ
amyloid-β
probability distribution function
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The supporting material contains the The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00364.
Initial and final structures of Aβ4016mer binding with G-mab and C-mab, comparison of two similar systems (G-MD4 and G-MD5), contact maps of G-mab (CDRs) and Aβ1–11 segment in system G-MD4 and G–Aβ1–11HSD, comparison of side-chain exposure in 4 Aβ fibril models (2M4J, 6SHS, 5OQV, and 2NAO), and testing of the simulation convergence and repeatability (PDF)
Contributor Information
Yujie Chen, Department of Physics, State Key Laboratory of Surface Physics, and Key Laboratory for Computational Physical Sciences (MOE), Multiscale Research Institute of Complex Systems, Fudan University, Shanghai 200438, P. R. China.
Guanghong Wei, Department of Physics, State Key Laboratory of Surface Physics, and Key Laboratory for Computational Physical Sciences (MOE), Multiscale Research Institute of Complex Systems, Fudan University, Shanghai 200438, P. R. China.
Jun Zhao, Basic Science Program, Leidos Biomedical Research, Inc., Computational Structural Biology Section, Frederick National Laboratory for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, United States.
Ruth Nussinov, Basic Science Program, Leidos Biomedical Research, Inc., Computational Structural Biology Section, Frederick National Laboratory for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, United States; Sackler Institute of Molecular Medicine, Department of Human Genetics and Molecular Medicine, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel.
Buyong Ma, Basic Science Program, Leidos Biomedical Research, Inc., Computational Structural Biology Section, Frederick National Laboratory for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, United States; Engineering Research Center of Cell & Therapeutic Antibody (MOE), School of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China.
REFERENCES
- (1).Holtzman DM, Morris JC, and Goate AM (2011) Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hardy J, and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- (3).Selkoe DJ, and Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO. EMBO Mol. Med. 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guillozet AL, Weintraub S, Mash DC, and Mesulam MM (2003) Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729. [DOI] [PubMed] [Google Scholar]
- (5).Stefani M, and Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699. [DOI] [PubMed] [Google Scholar]
- (6).Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- (7).Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, and Ashe KH (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. [DOI] [PubMed] [Google Scholar]
- (8).Li S, and Selkoe DJ (2020) A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 154, 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jokar S, Khazaei S, Behnammanesh H, Shamloo A, Erfani M, Beiki D, and Bavi O (2019) Recent advances in the design and applications of amyloid-β peptide aggregation inhibitors for Alzheimer’s disease therapy. Biophys. Rev. 11, 901–925. [DOI] [PubMed] [Google Scholar]
- (10).Cummings J, Lee G, Ritter A, Sabbagh M, and Zhong K (2019) Alzheimer’s disease drug development pipeline: 2019. Alzheimers. Dement. 5, 272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xi W-H, and Wei G-H (2016) Amyloid-β peptide aggregation and the influence of carbon nanoparticles. Chin. Phys. B 25, 018704. [Google Scholar]
- (12).Takahashi T, and Mihara H (2008) Peptide and Protein Mimetics Inhibiting Amyloid β-Peptide Aggregation. Acc. Chem. Res. 41, 1309–1318. [DOI] [PubMed] [Google Scholar]
- (13).Saunders JC, Young LM, Mahood RA, Jackson MP, Revill CH, Foster RJ, Smith DA, Ashcroft AE, Brockwell DJ, and Radford SE (2016) An in vivo platform for identifying inhibitors of protein aggregation. Nat. Chem. Biol. 12, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dhouafli Z, Cuanalo-Contreras K, Hayouni EA, Mays CE, Soto C, and Moreno-Gonzalez I (2018) Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 75, 3521–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Panza F, Lozupone M, Logroscino G, and Imbimbo BP (2019) A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15, 73–88. [DOI] [PubMed] [Google Scholar]
- (16).Moreth J, Mavoungou C, and Schindowski K (2013) Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun. Ageing 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bouter Y, Noguerola JSL, Tucholla P, Crespi GAN, Parker MW, Wiltfang J, Miles LA, and Bayer TA (2015) Abeta targets of the biosimilar antibodies of Bapineuzumab, Crenezumab, Solanezumab in comparison to an antibody against N-truncated Abeta in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol. 130, 713–729. [DOI] [PubMed] [Google Scholar]
- (18).Fuller JP, Stavenhagen JB, Christensen S, Kartberg F, Glennie MJ, and Teeling JL (2015) Comparing the efficacy and neuroinflammatory potential of three anti-abeta antibodies. Acta Neuropathol. 130, 699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, and Donoghue S (2005) Evaluation of the safety and immunogenicity of synthetic Aβ42 (AN1792) in patients with AD. Neurology 64, 94–101. [DOI] [PubMed] [Google Scholar]
- (20).Wiessner C, Wiederhold K-H, Tissot AC, Frey P, Danner S, Jacobson LH, Jennings GT, Luond R, Ortmann R, Reichwald J, et al. (2011) The Second-Generation Active Aβ Immunotherapy CAD106 Reduces Amyloid Accumulation in APP Transgenic Mice While Minimizing Potential Side Effects. J. Neurosci. 31, 9323–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, and Watts RJ (2012) An Effector-Reduced Anti-β-Amyloid (Aβ) Antibody with Unique Aβ Binding Properties Promotes Neuroprotection and Glial Engulfment of Aβ. J. Neurosci. 32, 9677–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, et al. (2012) Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J. Alzheimer’s Dis. 28, 49. [DOI] [PubMed] [Google Scholar]
- (23).Ostrowitzki S (2012) Mechanism of Amyloid Removal in Patients With Alzheimer Disease Treated With Gantenerumab. Arch. Neurol. 69, 198. [DOI] [PubMed] [Google Scholar]
- (24).Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, and Nicoll JA (2008) Long-term effects of Aβ42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223. [DOI] [PubMed] [Google Scholar]
- (25).Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, Caputo A, Winblad B, and Graf A (2015) Long-term treatment with active Abeta immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, and Mohs R (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. [DOI] [PubMed] [Google Scholar]
- (27).Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Rabe C, Brunstein F, Quartino A, Honigberg LA, Fuji RN, Clayton D, Mortensen D, Ho C, and Paul R (2018) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90, e1889–e1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Salloway S, Honigberg LA, Cho W, Ward M, Friesenhahn M, Brunstein F, Quartino A, Clayton D, Mortensen D, Bittner T, Ho C, Rabe C, Schauer SP, Wildsmith KR, Fuji RN, Suliman S, Reiman EM, Chen KW, and Paul R (2018) Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimer’s Res. Ther. 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gilman S, Koller M, Black RS, Jenkins L, Orgogozo JM, et al. (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. [DOI] [PubMed] [Google Scholar]
- (30).Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, Walz T, Pepinsky RB, Bussière T, Hamann S, Cameron TO, and Weinreb PH (2018) Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 8, 6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Selkoe DJ (2019) Alzheimer disease and aducanumab: adjusting our approach. Nat. Rev. Neurol. 15, 365–366. [DOI] [PubMed] [Google Scholar]
- (32).Howard R, and Liu KY (2020) Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 16, 63–64. [DOI] [PubMed] [Google Scholar]
- (33).Tolar M, Abushakra S, and Sabbagh M (2020) The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dementia, 1–8. [DOI] [PubMed] [Google Scholar]
- (34).Miller Y, Ma B, and Nussinov R (2010) Polymorphism in Alzheimer Aβ Amyloid Organization Reflects Conformational Selection in a Rugged Energy Landscape. Chem. Rev. 110, 4820–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, and Picone D (2002) Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- (36).Gremer L, Scholzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, Willbold D, and Schroder GF (2017) Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, and Tycko R (2013) Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schütz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Güntert P, Böckmann A, Glockshuber R, and Meier BH (2015) Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem., Int. Ed. 54, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Warmack RA, Boyer DR, Zee C-T, Richards LS, Sawaya MR, Cascio D, Gonen T, Eisenberg DS, and Clarke SG (2019) Structure of amyloid-β (20–34) with Alzheimer’s-associated isomerization at Asp23 reveals a distinct protofilament interface. Nat. Commun. 10, 3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, and Ishii Y (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).De S, Wirthensohn DC, Flagmeier P, Hughes C, Aprile FA, Ruggeri FS, Whiten DR, Emin D, Xia Z, Varela JA, Sormanni P, Kundel F, Knowles TPJ, Dobson CM, Bryant C, Vendruscolo M, and Klenerman D (2019) Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 10, 1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hodgson J (2019) Thousands of amyloids may foil Alzheimer’s drugs. Nat. Biotechnol. 37, 114–115. [DOI] [PubMed] [Google Scholar]
- (43).Zhao J, Nussinov R, and Ma B (2017) Mechanisms of recognition of Aβ monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 292, 18325–18343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jacobsen H, Ozmen L, Caruso A, Narquizian R, Hilpert H, Jacobsen B, Terwel D, Tanghe A, and Bohrmann B (2014) Combined Treatment with a BACE Inhibitor and Anti-Aβ Antibody Gantenerumab Enhances Amyloid Reduction in APPLondon Mice. J. Neurosci. 34, 11621–11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nikolcheva T, Lasser R, Ostrowitzki S, Scheltens P, Boada M, Dubois B, Dorflinger E, Volz D, Eichenlaub U, Rabe C, Bittner T, Schmitz M, Edgar C, Garibaldi G, Fontoura P, and Santarelli L (2016) CSF and Amyloid PET Biomarker Ddta from Scarlet RoAD - a Global Phase 3 Study of Gantenerumab in Patients with Prodromal AD. Neurobiol. Aging 39, S28–S29. [Google Scholar]
- (46).Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S, Wang G, Bateman R, Kerchner GA, Baudler M, Fontoura P, and Doody R (2019) Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimer’s Res. Ther. 11, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, et al. (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, Pfeifer A, Pihlgren M, Bainbridge TW, Reichelt M, et al. (2016) Structure of Crenezumab Complex with Aβ Shows Loss of β-Hairpin. Sci. Rep. 6, 39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yang T, Dang Y, Ostaszewski B, Mengel D, Selkoe DJ, et al. (2019) Target Engagement in an Alzheimer Trial: Crenezumab Lowers Aβ Oligomers in CSF. Ann. Neurol. 86, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (51).Parikh ND, and Klimov DK (2015) Molecular Mechanisms of Alzheimer’s Biomarker FDDNP Binding to Aβ Amyloid Fibril. J. Phys. Chem. B 119, 11568–11580. [DOI] [PubMed] [Google Scholar]
- (52).Kai T, Zhang L, Wang X, Jing A, Zhao B, Yu X, Zheng J, and Zhou F (2015) Tabersonine Inhibits Amyloid Fibril Formation and Cytotoxicity of Aβ(1–42). ACS Chem. Neurosci. 6, 879–888. [DOI] [PubMed] [Google Scholar]
- (53).Ngo ST, Fang ST, Huang SH, Chou CL, Huy PDQ, Li MS, and Chen YC (2016) Anti-arrhythmic medication Propafenone is potential drug for Alzheimer’s disease by inhibiting aggregation of Aβ: in silico and in vitro studies. J. Chem. Inf. Model. 56, 1344–1356. [DOI] [PubMed] [Google Scholar]
- (54).Kalhor HR, and Jabbari MP (2017) Inhibition Mechanisms of a Pyridazine-Based Amyloid Inhibitor: As a β-Sheet Destabilizer and a Helix Bridge Maker. J. Phys. Chem. B 121, 7633–7645. [DOI] [PubMed] [Google Scholar]
- (55).Zou Y, Qian Z, Chen Y, Qian H, Wei G, and Zhang Q (2019) Norepinephrine Inhibits Alzheimer’s Amyloid-β Peptide Aggregation and Destabilizes Amyloid-β Protofibrils: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 10, 1585–1594. [DOI] [PubMed] [Google Scholar]
- (56).Hou L, Kang I, Marchant R, and Zagorski M (2002) Methionine 35 Oxidation Reduces Fibril Assembly of the Amyloid-beta (1–42) Peptide of Alzheimer’s Disease. J. Biol. Chem. 277, 40173–40176. [DOI] [PubMed] [Google Scholar]
- (57).Butterfield DA, and Boyd-Kimball D (2005) The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta, Proteins Proteomics 1703, 149–156. [DOI] [PubMed] [Google Scholar]
- (58).Kollmer M, Close W, Funk L, Rasmussen J, et al. (2019) Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 10, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, and Riek R (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, and Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lepore R, Olimpieri PP, Messih MA, and Tramontano A (2017) PIGSPro: prediction of immunoGlobulin structures v2. Nucleic Acids Res. 45, W17–W23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kabat EA, and Wu TT (1991) Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J. Immunol. 147, 1709–1719. [PubMed] [Google Scholar]
- (63).de Vries SJ, van Dijk M, and Bonvin AMJJ (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897. [DOI] [PubMed] [Google Scholar]
- (64).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- (65).Dai Y-H, Liao L-Z, and Li D (2004) On Restart Procedures for the Conjugate Gradient Method. Numer. Algorithms 35, 249–260. [Google Scholar]
- (66).Kalé L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, and Schulten K (1999) NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 151, 283–312. [Google Scholar]
- (67).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, and Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (68).Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJC (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701. [DOI] [PubMed] [Google Scholar]
- (69).Lee MS, Feig M, Salsbury FR Jr., and Brooks CL 3rd. (2003) New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 24, 1348–1356. [DOI] [PubMed] [Google Scholar]
- (70).Wang Q, Yu X, Patal K, Hu R, Chuang S, Zhang G, and Zheng J (2013) Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 4, 1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhang T, Zhang J, Derreumaux P, and Mu Y (2013) Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ1–42 Dimer. J. Phys. Chem. B 117, 3993–4002. [DOI] [PubMed] [Google Scholar]
- (72).Berhanu WM, and Hansmann UHE (2013) The stability of cylindrin β-barrel amyloid oligomer models-A molecular dynamics study. Proteins: Struct., Funct., Genet. 81, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Zhou X, Xi W, Luo Y, Cao S, and Wei G (2014) Interactions of a Water-Soluble Fullerene Derivative with Amyloid-β Protofibrils: Dynamics, Binding Mechanism, and the Resulting Salt-Bridge Disruption. J. Phys. Chem. B 118, 6733–6741. [DOI] [PubMed] [Google Scholar]
- (74).Park J, Kahng B, and Hwang W (2009) Thermodynamic Selection of Steric Zipper Patterns in the Amyloid Cross-β Spine. PLoS Comput. Biol. 5, e1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, and Cheatham TE 3rd. (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
- (76).Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, and Eisenberg D (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Burley SK, and Petsko GA (1985) Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229, 23–28. [DOI] [PubMed] [Google Scholar]
- (78).Burley SK, and Petsko GA (1986) Amino-aromatic interactions in proteins. FEBS Lett. 203, 139–143. [DOI] [PubMed] [Google Scholar]
- (79).Gazit E (2002) A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83. [DOI] [PubMed] [Google Scholar]
- (80).Humphrey W, Dalke A, and Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.