
Figure 1.
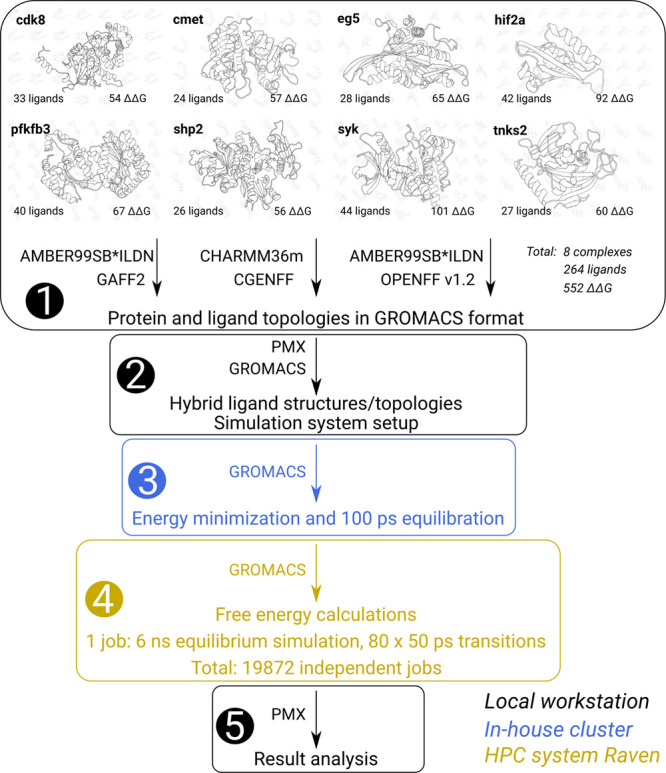
Workflow for the simulation procedure. (1) We start with the protein–ligand complexes prepared and made public by Schindler et al.7 The protein topologies are prepared in AMBER99SB*ILDN and CHARMM36m force fields, while for the ligands we used GAFF 2.11, CGENFF 3.0.1, and OpenFF 1.2.0 force fields. (2) Hybrid ligand structures and topologies for alchemical calculations are created with the pmx software, and further, the systems are assembled and prepared for the simulations with GROMACS. (3) Energy minimization and a brief 100 ps equilibration was performed on an in-house cluster. For the further automation of the workflow, this procedure could be merged with the following step and run in an HPC facility. (4) The main step where the simulations were performed on the Raven Supercomputer. We were able to make use of the trivial parallelization of the calculations by dividing the whole set into individual jobs as detailed in the text. (5) After finalizing the simulations on the HPC machine, the data were retrieved and analyzed locally.