
Figure 1.
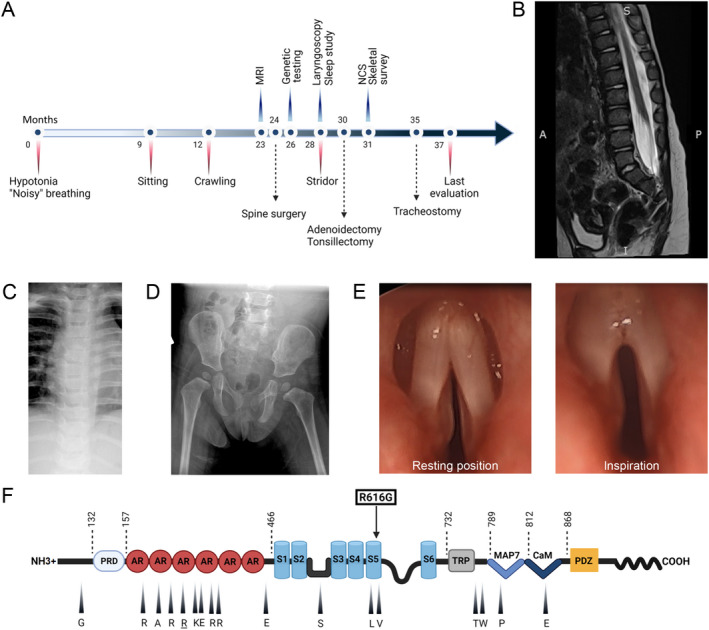
(A) Time line of clinical course showing most significant clinical events, diagnostic studies, and therapeutic interventions. (B) Lumbosacral spine MRI, T2 weighted sequence, shows evidence of tethered cord with thickening of the filum terminale, but without evidence of intrathecal mass. (C) Plain film of the chest shows no evidence of skeletal dysplasia. (D) Plain film of the pelvis and hips shows no abnormalities. (E) Direct laryngoscopy demonstrates vocal cords fixed in a paramedian position and no abduction during inspiration. (F) Representation of TRPV4 protein domains and mutations with corresponding clinical phenotypes. The ligand binding site and the pore region are located between S2‐S3 and S5‐S6, respectively. Amino acids that are mutated in mixed phenotype TRPV4 channelopathies are indicated with an arrowhead and corresponding symbol (WT residue shown). Arginine 269 (underlined) is the most commonly reported mutation site in the literature. The novel R616G missense mutation (squared) affects a residue in transmembrane S5 helix that has been previously associated with skeletal dysplasia. Abbreviations: NCS, nerve conduction studies; PRD, proline‐rich domain; AR, ankyrin repeat; S1 to 6, transmembrane domains; TRP, transient receptor potential; MAP7, microtubule associated protein 7 binding site; CaM, calmodulin binding site. [Colour figure can be viewed at wileyonlinelibrary.com]