

Abstract
Studies of the phenotype and population distribution of rare genetic forms of parkinsonism are required, now that gene-targeting approaches for Parkinson disease have reached the clinical trial stage. We evaluated the frequencies of PRKN, PINK1, and DJ-1 mutations in a cohort of 1,587 cases. Mutations were found in 14.1% of patients; 27.6% were familial and 8% were isolated. PRKN was the gene most frequently mutated in Caucasians, whereas PINK1 mutations predominated in Arab-Berber individuals. Patients with PRKN mutations had an earlier age at onset, and less asymmetry, levodopa-induced motor complications, dysautonomia, and dementia than those without mutations.
Our understanding of the genetic basis of Parkinson disease (PD) has improved with the identification of several disease-causing genes.1 Trials targeting these genes are underway, and the development of cohorts ready for precision clinical trials that target genetic forms of PD are now required.2,3
PRKN, PINK1, and DJ-1 mutations are the most frequent cause of early onset (EO) autosomal recessive (AR) typical PD. We investigated the frequency and nature of pathogenic variants of these 3 genes in a cohort of 1,587 PD probands, comparing clinical characteristics between patients with PRKN mutations (PRKN-PD), and those without pathogenic variants of known PD genes (PD-NM).
Patients and Methods
Patient Selection
Patients were enrolled between 1990 and 2018, through the French Parkinson Disease Genetics Study Group network and North African and Turkish collaborations. PD was diagnosed according to the clinical diagnostic criteria of the UK Parkinson Disease Society Brain Bank.4 Cases with mutations responsible for recessive atypical parkinsonism or those carrying the common LRRK2 Gly2019Ser variant were not included. We selected a cohort of 1,587 probands from 497 AR PD families (at least 2 affected siblings and isolated cases born to consanguineous parents), and 1,090 isolated cases, all screened for PRKN.
Standardized neurological examinations were performed by movement disorder experts, and 28 variables were used to obtain comparable data.
Most probands were Caucasian (n = 1,324, 83.4%; 927 French, 134 Turkish), Arab-Berber (n = 213, 13.4%), or of other ethnicities (n = 50, 3.2%). We included 1,587 PD probands and 52 mutation-carrying relatives in the genotype/phenotype correlation analysis. Informed consent and approval from institutional review boards were obtained for sample collection.
Procedures
Probands were screened by denaturing high-performance liquid chromatography and/or direct sequencing,5–8 next generation sequencing with a targeted gene panel, or whole-exome sequencing.9,10
PRKN was screened in 1,587 probands, PINK1 and DJ-1 in 1,223. Sanger sequencing was performed to confirm variants and cosegregation analyses, where possible. Exon rearrangements were detected by semi-quantitative polymerase chain reaction (PCR) for PRKN5 or with the SALSA multiplex ligation-dependent probe amplification (MLPA, MRC Holland, Amsterdam, the Netherlands; http://www.mlpa.com) P051/P052 Parkinson kits, according to the manufacturer’s instructions. Patients with an age at onset (AAO) ≤ 40 years and lymphoblastoid cells available for real-time (RT)-PCR analysis (n = 15)5 or unaffected relatives for cosegregation analysis (n = 30) were investigated for possible PRKN rearrangements undetectable by MLPA.
Statistical Analysis
The PD-NM and PRKN-PD groups were compared with Welch t tests for continuous variables and Fisher exact tests for categorical variables.
We used generalized linear models (GLMs) to compare clinical features between PRKN-PD with biallelic or double heterozygous mutations and PD-NM, adjusting for sex, AAO, disease duration, and dopaminergic medication. We used GLMs with identity links and normal distributions for continuous clinical features, and GLMs with logit links and Bernoulli distributions for binary clinical features. Interactions between AAO and disease duration were also included. Disease duration and dopaminergic medication were not included in models for clinical features at onset. Effects were assessed with Fisher type II tests, and effect size was estimated with Cohen f2. Benjamini–Hochberg correction for multiple testing was performed. GLMs were generated for 15 of 28 clinical features for at least 20% of the patients in each group with available data.
Results
Demographic and Clinical Data
In our cohort, men (n = 951, 60%) and EO cases (mean AAO = 40.2, standard deviation [SD] = 12.1 years) were overrepresented, particularly among isolated cases (p < 0.0001; Table 1).
TABLE 1.
Demographic Data for Our Study Population
| Whole PD Group | n | AR PD, Including Isolated Cases with Consanguinity | n | Isolated Ceases | n | AR PD vs Isolated Ceases, p Value | |
|---|---|---|---|---|---|---|---|
| % men (n) | 60.0 (951) | 1,587 | 57.1 (284) | 497 | 61.2 (667) | 1,090 | 0.14 |
| Ethnic background | 1,587 | 497 | 1,090 | <0.0001a | |||
| % Caucasian (n) | 83.4 (1,324) | 75.5 (375) | 87.1 (949) | ||||
| % Arab-Berbers (n) | 13.4 (213) | 22.1 (110) | 9.4 (103) | ||||
| % others/mixed (n) | 3.2 (50) | 2.4 (12) | 3.5 (38) | ||||
| % consanguinity (n) | 12.9 (202) | 1,564 | 40.9 (202) | 494 | 0 | 1,070 | NA |
| Mean age at onset, y (SD), range | 40.2 (12.1), 2–81 | 1,543 | 43.8 (15.0), 3–81 | 485 | 38.6 (10.1), 2–74 | 1,058 | <0.0001a |
| Mean age at examination, y (SD), range | 49.7 (13.2), 9–87 | 1,575 | 54.2 (14.5), 16–87 | 495 | 47.7 (12.0), 9–79 | 1,080 | <0.0001a |
| Mean disease duration, y (SD), range | 9.8 (8.6), 0–63 | 1,530 | 10.9 (9.6), 0–63 | 482 | 9.2 (8.0), 0–48 | 1,048 | 0.0005a |
Frequencies were compared with Fisher exact tests for qualitative traits, and means were compared with t tests for continuous variables.
p < 0.05.
AR = autosomal recessive; NA = not appropriate; PD = Parkinson disease; SD = standard deviation.
Distribution and Nature of Recessive PD-Associated Gene Mutations
Biallelic or double heterozygous mutations of known AR PD-causing genes were present in 224 of the 1,587 probands (14.1%), 27.6% (137/497) of familial and 8% (87/1,090) of isolated cases. The most frequendy mutated genes were PRKN (199/1,587, 12.5%), then PINK1 (23/1,223, 1.9%) and DJ-1 (2/1,223, 0.16%). We identified 56 patients with single heterozygous variants in the 3 genes in whom AAO was significantly later than cases with biallelic or double heterozygous mutations (36.9 [SD = 10.6] vs 31.3 [SD = 11.1], p = 0.0009); they were removed from genotype/phenotype correlation analyses.
The 199 cases carried either homozygous (n = 92) or compound heterozygous (n = 59) PRKN mutations, confirmed by segregation analysis of all available unaffected relatives, or double heterozygous (n = 48) PRKN mutations for which phasing was still unknown. These carriers displayed 77 different variants, including 19 absent from public databases (www.mdsgene.org; Fig 1A).11 RT-PCR identified a single French family with PRKN Ex3del/Ex3dup compound heterozygous rearrangements not detectable by MLPA but elucidated by cosegregation analysis.
FIGURE 1:
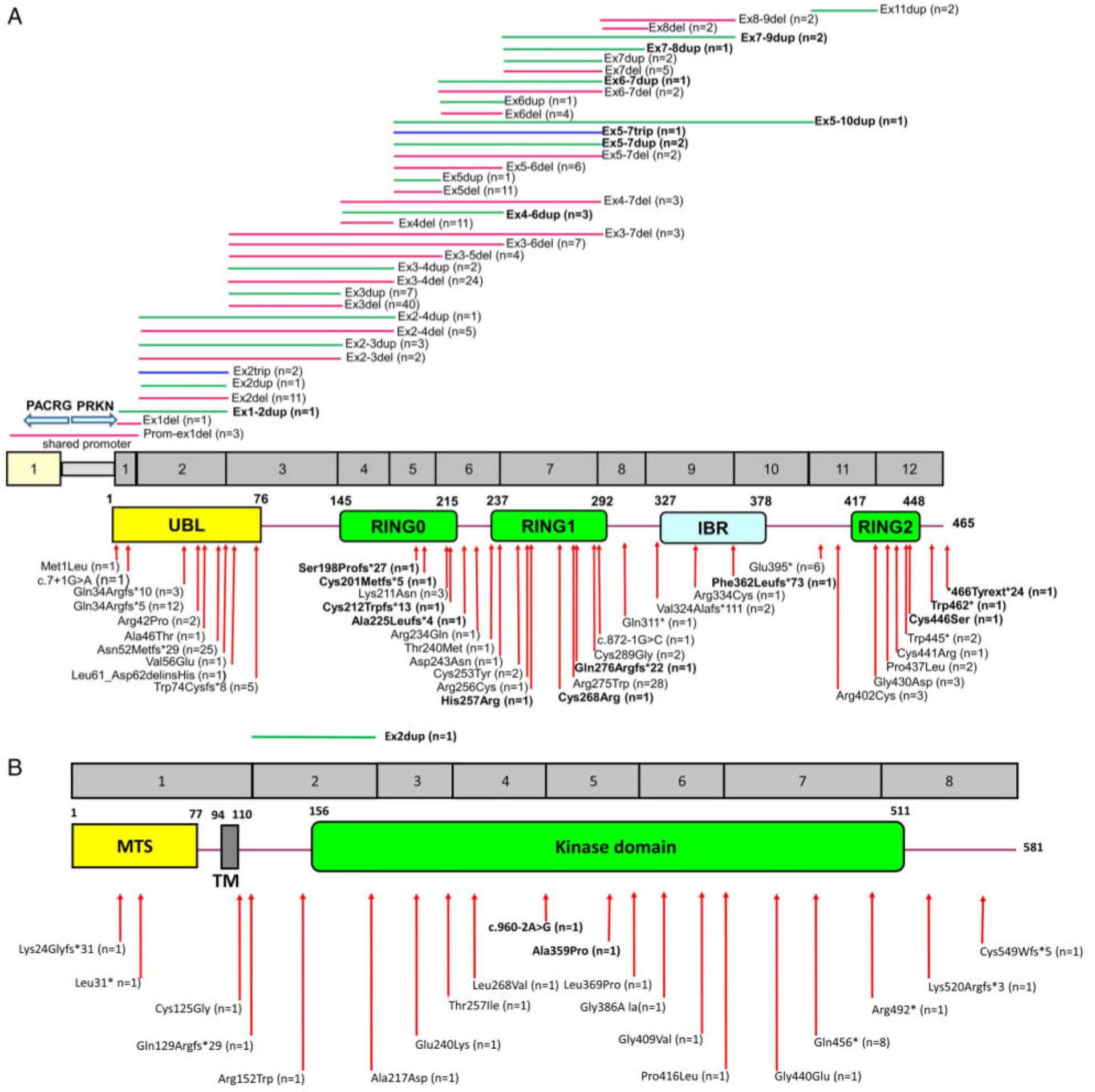
Schematic representation of the (A) PRKN and (B) PINK1 genes and respective proteins and associated disease-linked mutations. Exonic deletions (in red), duplications (in green), or triplications (in blue) are shown in the upper panel, and point mutations (missense, frameshift, nonsense, and splice) are shown in the lower panel. Newly identified mutations are shown in bold. Numbers in brackets indicate the number of mutation carriers. PRKN cDNA numbering: NM_004562.2; PINK1 cDNA numbering: NM_032409.2. IBR, in-between RING; MTS, mitochondrial targeting sequence; RING, really interesting new gene; TM, transmembrane helix; UBL, ubiquitin-like.
Most of the 23 probands with pathogenic PINK1 variants were Arab-Berbers (n = 12, 52.2%); 8 carried the homozygous Gln456* mutation. Most variants were homozygous (n = 18, 78.3%). We identified 21 different pathogenic variants, including 3 not in databases (Fig 1B).
Distribution of PRKN and PINK1 Mutation Carriers by AAO, Pattern of Presentation, and Ethnicity
The proportion of probands with PRKN mutations decreased with increasing AAO: 42.2% for AAO ≤ 20 years, 29% for 21 to 30 years, 13% for 31 to 40 years, and 4.4% for 41 to 60 years; Fig 2A. This decrease was more marked in isolated than familial cases (Fig 2B). PINK1 mutations were less frequent than PRKN mutations (1.9% vs 12.5%) but more evenly distributed among familial cases for AAOs up to 60 years. Neither PRKN nor PINK1 variants were found in patients with onset after 60 years (Fig 2A–D). PRKN mutations were more frequent in Caucasians (179/1324, 13.5%) than in Arab-Berbers (16/213, 7.5%; Fig 2E). Conversely, PINK1 mutations were more common in Arab-Berbers (12/188, 6.4%) than Caucasians (9/1,005, 0.9%; Fig 2E).
FIGURE 2:
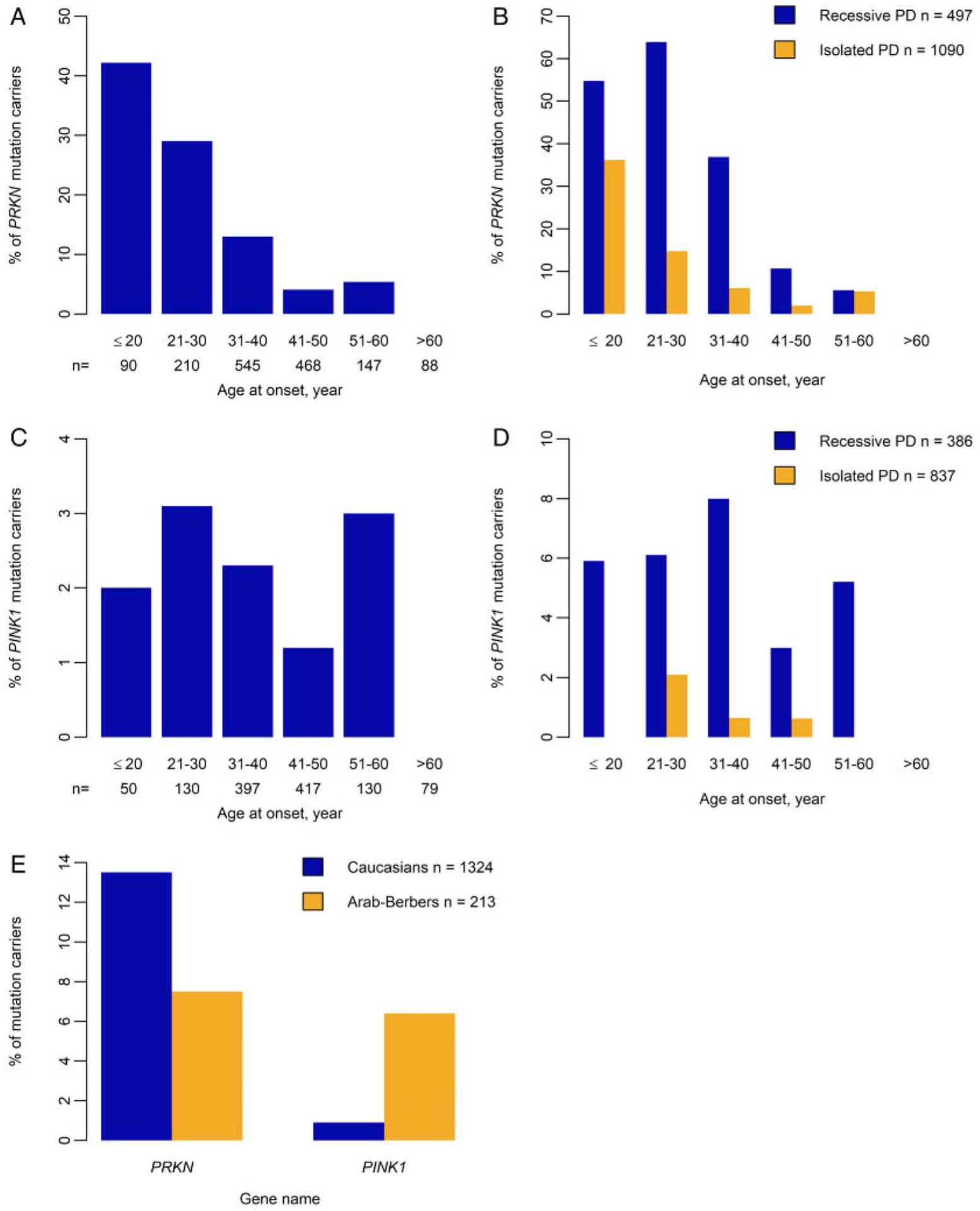
Distribution of PRKN and PINK1 mutation carriers by age at onset, pattern of disease presentation, and ethnicity. (A) Proportion of probands by age at onset for PRKN (199 carriers among 1,587 probands). (B) Proportion of PRKN mutation carriers from 497 cases with autosomal recessive Parkinson disease (PD; in blue) versus 1,090 isolated cases (in orange) by age at onset and pattern of presentation of PD. (C) Proportion of probands by age at onset for PINK1 (23 carriers among 1,223 probands). (D) Proportion of PINK1 mutation carriers from 386 cases with autosomal recessive PD (in blue) versus 837 isolated cases (in orange) by age at onset and pattern of presentation of PD. (E) Proportion of PRKN and PINK1 mutation carriers, according to their ethnicity: Caucasians (n = 1,324, in blue) or Arab-Berbers (n = 213, in orange).
Comparison of PRKN-PD with PD-NM: Genotype-Phenotype Correlations
We compared the clinical features between the 228 of 241 PRKN-PD and 1,181 of 1,307 PD-NM subjects without missing data (Table 2; Supplementary Table 1). The proportion of men (padj = 0.016) and AAO (padj < 0.0001) were greater in the PD-NM than the PRKN-PD group. Dopaminergic treatment was similar between groups (padj = 0.69), but L-dopa responsiveness was higher in PRKN-PD than PD-NM (padj = 0.045).
TABLE 2.
Demographic and Clinical Characteristics of Patients with Parkinson’s Disease with Pathogenic PRKN Variants (PRKN-PD) and of Patients without Pathogenic Variants (PD-NM)
| PD-NM and PRKN-PD Unadjusted Comparisons | PD-NM and PRKN-PD Adjusted Comparisons | |||||
|---|---|---|---|---|---|---|
| Characteristics | PD-NM n=1181 | PRKN-PD n=228 | p Valueb | Coefficient or odds ratio (OR) (SE) | Cohen’s f2 | p Valueb |
| Baseline | ||||||
| Sex (% male) | 727/1,181 (61.6%) | 118/228 (51.8%) | 0.016c | |||
| Mean age at examination (SD), y | 50.5 (13.2) | 45.4 (12.9) | <0.0001c | |||
| Mean disease duration (SD), y | 8.8 (7.8) | 14.1 (10.4) | <0.0001c | |||
| Mean age at onset (SD), y | 41.6 (12.0) | 31.2 (10.7) | <0.0001c | |||
| L-DOPA-treated | 791/1,181 (67%) | 148/228 (64.9%) | 0.69 | |||
| Levodopa responsivenessa | 659/738 (89.3%) | 142/149 (95.3%) | 0.045c | 1.9 (0.82) | 0.007 | 0.15 |
| Motor symptoms and signs | ||||||
| Dystonia at onset | 164/990 (16.6%) | 37/201 (18.4%) | 0.69 | 0.84 (0.18) | 0.001 | 0.46 |
| Akinesia at onset | 590/946 (61.3%) | 99/206 (48.1%) | 0.0010c | 0.51 (0.09) | 0.015 | 0.0003c |
| Tremor at onset | 570/960 (59.4%) | 142/205 (69.3%) | 0.021c | 1.7 (0.29) | 0.007 | 0.0076c |
| Micrographia | 308/927 (33.2%) | 42/204 (20.6%) | 0.0013c | 0.58 (0.11) | 0.007 | 0.010c |
| Asymmetry | 1,001/1,035 (96.7%) | 181/198 (91.4%) | 0.0049c | 0.26 (0.09) | 0.010 | 0.0005c |
| Bradykinesia | 1,033/1,069 (96.6%) | 201/208 (96.6%) | 1.0000 | 1.4 (0.63) | 0.002 | 0.46 |
| Rigidity | 1,008/1,066 (94.6%) | 194/204 (95.1%) | 0.90 | 1.1 (0.42) | <0.001 | 0.77 |
| Tremor | 796/1,056 (75.4%) | 168/205 (82%) | 0.057 | 1.5 (0.32) | 0.002 | 0.072 |
| Mean UPDRS III “ON” state (/108) (SD) | 19.6 (13.8) | 15.9 (11.9) | 0.0049c | −3.3 (1.2) | 0.008 | 0.014c |
| Mean Hoehn & Yahr “ON” state (/5) (SD) | 2 (0.91) | 2.00 (0.93) | 0.74 | −0.14 (0.09) | 0.003 | 0.16 |
| Dyskinesia | 457/667 (68.5%) | 97/178 (54.5%) | 0.0025c | 0.44 (0.09) | 0.020 | 0.0005c |
| Motor fluctuations | 485/663 (73.2%) | 82/177 (46.3%) | <0.0001c | 0.32 (0.07) | 0.041 | <0.0001c |
| Non-motor symptoms and signs | ||||||
| Dysautonomia | 254/470 (54.0%) | 36/192 (18.8%) | <0.0001c | 0.19 (0.05) | 0.095 | <0.0001c |
| Dementia | 67/667 (10.0%) | 6/153 (3.9%) | 0.037c | 0.34 (0.16) | 0.009 | 0.014c |
Data are expressed as mean (standard deviation) for continuous variables, and as counts (percentages) for categorical variables. We used t-tests to compare the two groups for continuous variables and Fisher’s exact tests for binary variables. Coefficients for continuous clinical features and odds ratios (ORs) for binary clinical features and standard error (SE), Cohen’s f2 and p-values were calculated from GLMs with mutation status, sex, age at onset, disease duration, L-DOPA group and age at onset vs disease duration for all 15 variables except for onset variables for which only mutation status, sex and age-at-onset were added. Linear models were used for continuous variables; GLMs with logit links and Bernouilli distributions were used for binary variables; UPDRS III, the motor subsection of the Unified Parkinson’s Disease Rating Scale.
Levodopa responsiveness was defined as a >30% improvement in subjective perceived motor symptoms.
p corrected for multiple testing by the Benjamini-Hochberg procedure.
p < 0.05.
After adjustment for covariables, PRKN-PD had a higher initial frequency of tremor (padj = 0.0076), but lower frequencies of akinesia (padj = 0.0003), micrographia (padj = 0.010), and asymmetry (padj = 0.0005) than PD-NM patients (Table 2). Dystonia at onset and cardinal symptom (bradykinesia, rest tremor, and rigidity) frequencies were similar in both groups. Motor severity after adjustment for disease duration was lower in PRKN-PD than in PD-NM (Unified Parkinson’s Disease Rating Scale score, padj = 0.014), and PRKN-PD patients developed fewer L-dopa–induced motor complications (dyskinesia: padj = 0.0005; motor fluctuations: padj < 0.0001). Nonmotor symptoms, including dysautonomia (padj < 0.0001) and dementia (padj = 0.014), were less frequent in PRKN-PD patients.
Mutational and Phenotypic Characteristics of PINK1 and DJ-1 Mutation Carriers
The PINK1-associated phenotype of the 33 carriers resembled that of PRKN, but with a slightly later AAO (mean = 34.6 [SD = 12.2] years vs 31.3 [SD = 10.9] years; p = 0.20), a lower frequency of tremor (16.7% vs 68.5%; p = 0.0005), and a higher rate of dystonia (90% vs 18.2%; p < 0.0001) at onset (Supplementary Table 2). Nonmotor symptoms, such as dysautonomia (72.2% vs 19.6%; p < 0.0001) and dementia (25% vs. 3.8%; p = 0.0007), were more frequent in patients with PINK1 variants, who were also more likely to display L-dopa–induced dyskinesia (93.3% vs 54.1 %; p = 0.0024) and motor fluctuations (66.7% vs 46.2%; p = 0.06).
The 2 pathogenic DJ-1 variant carriers (1 Caucasian, 1 North African) each carried a previously unknown homozygous Glu94* and a compound heterozygous variant affecting the same highly conserved amino acid (Thr154Ile/Thr154Ala). They developed PD, with the 4 cardinal signs, at the ages of 29 and 28 years. Dystonia at onset, dyskinesia, and orthostatic hypotension were noted in 1 patient, without cognitive signs.
Discussion
We established the spectrum and relative frequencies of mutations in a large cohort of AR PD cases, elucidating the genotype–clinical phenotype relationship. Homozygous/compound or double heterozygous pathogenic variants of PRKN, PINK1, and DJ-1 account for 14.1% of our PD patients, with PRKN the most frequently mutated. This study included a large number of genotyped and extensively phenotyped patients (276 mutation carriers) compared with a group of cases not carrying mutations of known AR PD–causing genes. However, the clinical data were cross-sectional, the numbers of patients with mutations of genes other than PRKN were small, and our populations were biased toward EO cases. In addition, 24% (48/199) of our PD patients with 2 PRKN mutations lacked information on their mutational phasing. However, given that we found no mutations in cis in a cosegregation analysis of 59 index cases, we are confident that the vast majority of patients with 2 mutations carry them in trans. Nevertheless, our findings may have major implications for patient selection for genetic testing based on AAO, pattern of disease presentation, and ethnicity. The frequency of pathogenic variants of AR PD–associated genes (1) decreased with increasing AAO, to zero for an AAO > 60 years; (2) in cases with a positive family history or consanguinity was more than triple that in isolated cases; and (3) was much higher for PRKN than PINK1 in Caucasians, but similar for these 2 genes in Arab-Berbers. However, pathogenic PRKN variants were more frequent in our EO PD cases (<50 years, 190/1,273, 14.9%) than in 4 other cohorts (10.1% in a Taiwanese cohort12 and 2.8% in a Norwegian cohort,13 both with EO defined as <45 years; 5.9% in a UK series,14 and 2.8% in a larger multicenter sample,15 both with EO defined as <50 years). A meta-analysis of >5,800 PD patients found PRKN variants in 8.6% of PD cases with an AAO < 50 years.15 We provide more precise data for PRKN and PINK1 genes, according to AAO.
GLM analyses revealed that AAO was lower, disease progression slower, and the response to L-dopa stronger in patients with PRKN mutations than in those without pathogenic variants, as previously reported.12,15–17 However, these mutations were not associated with higher rates of dystonia at onset, a trait more strongly associated with EO (see Supplementary Table 1) than with genetic status. Patients with PRKN mutations also had a distinctive non-motor symptom profile, with lower frequencies of dementia and dysautonomia, consistent with previous reports.18 After adjustment for disease duration and dopaminergic medication, these patients had fewer treatment-induced complications, such as dyskinesia and motor fluctuations, than PD-NM patients. This very pure and slowly progressive phenotype makes patients with PRKN variants very good candidates for deep-brain stimulation.19,20
The causal gene(s) remained unidentified for a number of families with AR PD (~72.5%), suggesting that pathogenic variants of known genes may have been missed, or the involvement of unknown genes.
These findings will help to guide routine genetic testing and to establish cohorts of patients for clinical trials targeting the gene defects or their physiopathological consequences.
Supplementary Material
Acknowledgment
This work was supported by the Fondation de France, France-Parkinson Association, la Fédération pour la Recherche sur le Cerveau, the French program “Investissements d’avenir” (ANR-10-IAIHU-06), and the Intramural Research Program of the National Institute on Aging, NIH, Department of Health and Human Services (project number ZO1 AG000949).
We thank the patients and their families; and the DNA and Cell Bank of the ICM Brain Institute for sample preparation.
Footnotes
Additional supporting information can be found in the online version of this article.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson’s disease. Rev Neurol 2018;174:628–643. [DOI] [PubMed] [Google Scholar]
- 2.Strafella C, Caputo V, Galota MR, et al. Application of precision medicine in neurodegenerative diseases. Front Neurol 2018;9:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vollstedt EJ, Kasten M, Klein C. MJFF Global Genetic Parkinson’s Disease Study Group. Using global team science to identify genetic Parkinson’s disease worldwide. Ann Neurol 2019;86:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesage S, Magali P, Lohmann E, et al. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat 2007;28:27–32. [DOI] [PubMed] [Google Scholar]
- 6.Ibáñez P, Lesage S, Lohmann E, et al. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 2006;129:686–694. [DOI] [PubMed] [Google Scholar]
- 7.Leutenegger AL, Salih MA, Ibáñez P, et al. Juvenile-onset parkinsonism as a result of the first mutation in the adenosine triphosphate orientation domain of PINK1. Arch Neurol 2006;63:1257–1261. [DOI] [PubMed] [Google Scholar]
- 8.Lohmann E, Dursun B, Lesage S, et al. Genetic bases and phenotypes of autosomal recessive Parkinson disease in a Turkish population. Eur J Neurol 2012;19:769–775. [DOI] [PubMed] [Google Scholar]
- 9.Bouhouche A, Tesson C, Regragui W, et al. Mutation analysis of consanguineous Moroccan patients with Parkinson’s disease combining microarray and gene panel. Front Neurol 2017;8:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesage S, Drouet V, Majounie E, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet 2016;98:500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasten M, Hartmann C, Hampf J, et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 2018;33:730–741. [DOI] [PubMed] [Google Scholar]
- 12.Lin CH, Chen PL, Tai CH, et al. A clinical and genetic study of early-onset and familial parkinsonism in Taiwan: an integrated approach combining gene dosage analysis and next-generation sequencing. Mov Disord 2019;34:506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gustavsson EK, Trinh J, McKenzie M, et al. Genetic identification in early onset parkinsonism among Norwegian patients. Mov Disord Clin Pract 2017;4:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilarski LL, Pearson JP, Newsway V, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov Disord 2012;27:1522–1529. [DOI] [PubMed] [Google Scholar]
- 15.Marder KS, Tang MX, Mejia-Santana H, et al. Predictors of parkin pathogenic variants in early-onset Parkinson disease: the Consortium on Risk for Early-Onset Parkinson Disease study. Arch Neurol 2010; 67:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohmann E, Periquet M, Bonifati V, et al. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 2003;54: 176–185. [DOI] [PubMed] [Google Scholar]
- 17.Khan NL, Graham E, Critchley P, et al. Parkin disease: a phenotypic study of a large case series. Brain 2003;126:1279–1292. [DOI] [PubMed] [Google Scholar]
- 18.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA Neurol 2014;71:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal GD, Hall D, Ouyang B, et al. Genetic and clinical predictors of deep brain stimulation in young-onset Parkinson’s disease. Mov Disord Clin Pract 2016;3:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizzone MG, Martone T, Balestrino R, Lopiano L. Genetic background and outcome of deep brain stimulation in Parkinson’s disease. Parkinsonism Relat Disord 2018;64:8–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.