

Summary
Proteins assemble into a variety of dynamic and functional structures. Their structural transitions are often challenging to distinguish inside cells, particularly with a high spatiotemporal resolution. Here, we present a fluorescence resonance energy transfer (FRET)-based method for continuous and high-throughput monitoring of protein self-assemblies to reveal well-resolved transient intermediate states. Intermolecular FRET with both the donor and acceptor proteins at the same target protein provides high sensitivity while retaining the advantage of straightforward ratiometric imaging. We apply this method to monitor self-assembly of three proteins. We show that the mutant Huntingtin exon1 (mHttex1) first forms less-ordered assemblies, which develop into fibril-like aggregates, and demonstrate that the chaperone protein DNAJB6b increases the critical saturation concentration of mHttex1. We also monitor the structural changes in fused in sarcoma (FUS) condensates. This method adds to the toolbox for protein self-assembly structure and kinetics determination, and implementation with native or non-native proteins can inform studies involving protein condensation or aggregation.
Keywords: FRET probe, aggregation, condensate, self-assembly, protein oligomers, Huntingtin exon 1, DNAJB6b, intermolecular FRET, FUS protein
Graphical abstract
Highlights
-
•
Method to monitor transient structural intermediates in protein assemblies
-
•
Allows for monitoring at both ensemble and single-foci levels
-
•
Reveals transition of mHttex1 from a less-ordered to a fibril-type state
-
•
DNAJB6b increases critical saturation concentration of mHttex1
Motivation
Proteins assemble into a wide range of different supramolecular structures often related to their biological function. However, monitoring and characterizing such assemblies and their structural stability inside living cells is challenging. Here, we develop a method that provides a precise readout of structural changes within protein aggregates, condensates, and oligomers. The method is non-invasive and allows long-term imaging and straightforward combinations with other orthogonal methods.
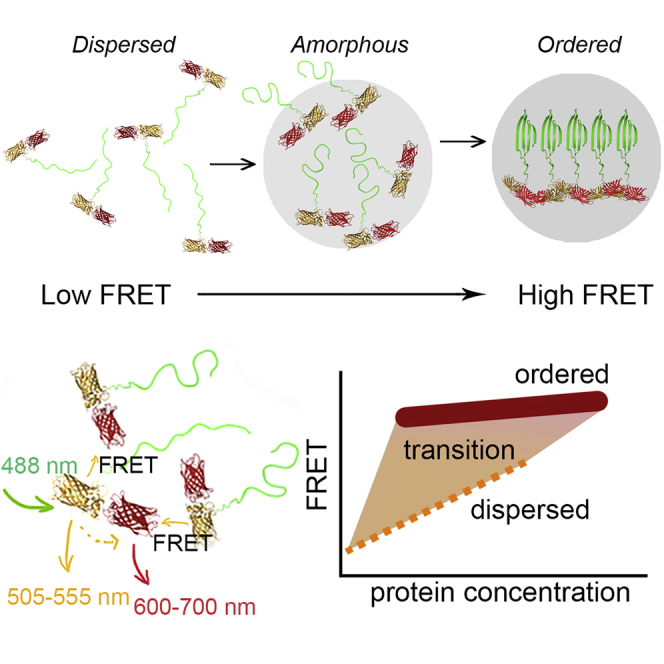
Proteins self-associate into a wide variety of structures such as aggregates, condensates, and oligomers. Here, Wan et al. present a FRET-based method for monitoring structural transitions of these assemblies, both at the ensemble and single-foci levels. They apply the method to study the structural changes of mutant Huntingtin, the chaperone DNAJB6b, and the FUS protein.
Introduction
Proteins assemble into different structures that range from quaternary structures, such as the cytoskeleton and multi-subunit enzymes, to dynamic protein condensates and misfolding-induced aggregates. The structure and dynamics of condensates and aggregates depend on the local biochemistry. For example, aggregate-forming proteins associated with Parkinson's, Alzheimer's, and Huntington's diseases have structures and aggregation pathways resulting from chaperone activity, posttranslational modifications, the inclusion of other biomolecules, and the physicochemical properties of the cell (Iadanza et al., 2018; Lackie et al., 2017; Owen et al., 2019; Schaffert and Carter, 2020). Thus, aggregate structures vary considerably, and better monitoring of the structures may improve the ability to identify the best drug targets. Condensate structures also vary: the protein fused in sarcoma (FUS) forms dynamic liquid-like assemblies but also aberrant gel-like states that mislocalize in the cytoplasm, which are sometimes associated with amyotrophic lateral sclerosis (Murakami et al., 2015; Patel et al., 2015). To better understand the biological functions of condensates and related diseases, it is essential to know the structural transitions they undergo.
Proteins can form various types of assemblies. Irreversible misassembly is classified as aggregation and can result in either ill-defined amorphous structures or more structured fibrils involving beta-sheet protein folds. Proteins can also assemble, in a reversible manner, as biomolecular condensates. Condensates are large assemblies that can act as compartments and are often associated with liquid- to gel-like physicochemical properties. Condensates may structurally evolve into (pathogenic) aggregates, indicating the need to monitor this process continuously to assess their biological impact.
The structural transitions of protein assemblies are challenging to determine inside cells. In buffer, reconstitution of purified protein allows detailed study of condensates and aggregates, but results may not be relevant in cells. Chemical or cryofixation of cells allows antibody staining and electron microscopy, providing the microscopic structure of the aggregate and its location in the cell. However, measurements inside living cells are required to understand what structures are present in living cells, how fast they form, what their dynamics are, and whether transient intermediates form. The dynamics of protein assembly can be observed by tagging the corresponding proteins with a fluorescent protein or by adding small-molecule fluorescent dyes such as Thioflavin T (Xue et al., 2017), which usually only indicate amyloid-type structures. The target protein fused with a fluorescent protein allows fluorescent microscopy with a high spatiotemporal resolution, observation of dynamic behavior, and co-localization with other fluorescently tagged molecules.
Förster resonance energy transfer is an ideal method to probe events at the 2–10 nm scale, which is in a size range of a protein. The fluorescence resonance energy transfer (FRET) efficiency between a donor and an acceptor fluorescent protein increases with an inverse distance to the power of six when the fluorophores are moving closer toward each other. The orientation between the fluorophores attenuates the FRET efficiency further. Thus, FRET has found extensive use in determining protein-protein interactions and observing protein aggregation, such as the mutant Huntingtin exon 1 fragment (mHttex1) containing an extended polyglutamine domain (Ast et al., 2018; Lo et al., 2020; Pollitt et al., 2003). To this end, a fluorescent donor is fused to mHttex1 and the acceptor to the second copy of mHttex1. Co-transfection of both plasmids allows the observation of an increase in FRET efficiency as both copies co-aggregate with a short intermolecular distance in between. Unequal expression of donor and acceptor proteins can be corrected by using correction factors (Xia and Liu, 2001). Alternative methods have been developed recently, such as partial photo converting a GFP into an RFP to generate FRET, named DAmFRET (Khan et al., 2018), or to determine the influence of the aggregate on fluorescent properties of GFP (Kaminski Schierle et al., 2011; Laine et al., 2019; Peskett et al., 2018). These methods provided a great number of insights into the mechanism of protein self-assembly, demonstrating the potential of the approach. These methods do not provide an equal number of donors and acceptors in every cell, and continuous quantitative ratiometric imaging is impossible. To complement the existing methods, the donor and acceptor could be fused to the same protein. However, fusing mHttex1 between a FRET pair impedes mHttex1 aggregation and depends on changes in both inter- and intramolecular FRET (Bünig et al., 2017; Caron et al., 2013). Hence, although FRET is a beneficial tool to observe protein assembly, various hurdles remain before it can reach its full potential in cells.
Here, we demonstrate straightforward and precise monitoring of condensate and aggregate properties with a FRET-based method that relies on conjugating the target protein to a fusion that contains both the donor and acceptor. We validate the application of the method by observing different states in single mHttex1 aggregates, the role and the oligomeric state of DNAJB6b during mHttex1 aggregation, and the less-ordered structure in FUS condensates with the high spatiotemporal resolution that confocal fluorescence microscopy allows and in a high-throughput manner by fluorescence-assisted cytometry sorting (FACS).
Results
Sensor design and approach
We aimed to develop a platform for efficient and precise intermolecular FRET measurements by fusing both the donor and acceptor to the same protein. We adopted a FRET pair previously used for monitoring protein clustering in cell membranes to prevent intramolecular FRET changes during assembly (Ma et al., 2017). Ma et al. removed the native linker domains of the mVenus and mCherry proteins (VC) so that the FRET pair adopts a rigid conformation and stabilizes intramolecular FRET to a low level. Hence, we argued that fusing this pair to proteins that form condensates and aggregates would reveal insights into short-lived intermediate states and their kinetics by following the intermolecular FRET efficiency in time. Intermolecular FRET increases with protein concentration, which can be measured by direct acceptor (mCherry) excitation. This dependence of FRET versus concentration should relate to a specific structure of the assembly: while freely moving fluorophores interact randomly, a preferred binding site provides a dominant orientation and distance between the fluorophores. For example, at the same protein concentration, a binding mode where the fluorophores are close together would provide a higher FRET than randomly mixed proteins. Hence, this approach will be sensitive to changes in the relative distance between the VC domains and their respective orientation. Thus, the method allows distinction between ordered states based on the FRET efficiency.
Monitoring mHttex1 self-assembly in human cells
To test the method, we chose to follow the assembly and aggregation of the well-studied mHttex1 with an extended polyglutamine stretch of Q71, the native N-terminal peptide, and the native proline-rich domain. We fused the VC domain to the C terminus to generate Q71-VC (Figure 1A) (Krobitsch and Lindquist, 2000). Fusions of GFP at the C terminus of mHttex1 have been characterized extensively. We expected the FRET efficiency to increase as the distance between donors and acceptors decreased but to be modulated by the orientations sampled between the fluorophores; in the case of Q71-VC, the VC could take up various orientations, also within a fibril. As observed by confocal fluorescence microscopy, transfection and expression of Q71-VC in HEK293t cells led to the corresponding aggregates (Figure 1B). Subsequent cell lysis and SDS-PA gel electrophoresis showed that the constructs were expressed intact and generated the typical SDS-insoluble aggregates (Figure S1C). To confirm the FRET mechanism and the expected static properties of the aggregate, we determined the fluorescence recovery after photobleaching (FRAP) of the acceptor (Figures S1A and S1B). Indeed, bleaching the acceptor gave a substantial increase in donor emission, confirming a high FRET efficiency. Moreover, fluorescence recovery did not occur as expected from mHttex1 aggregates. Hence, the fusion of VC to mHttex1 does not impede its aggregation behavior and gives a high FRET efficiency.
Figure 1.

Tagging mHttex1 with the VC domain allows detailed observation of the aggregation progression in HEK293t
(A) Design of the probe and cartoon of the sensing mechanism. The mVenus-mCherry FRET pair (VC) is fused to the C terminus of mHttex1. Closer association between mHttex1 peptides increases the intermolecular FRET between the VC tags (N17, native N-terminal peptide; polyQ, polyglutamine; polyP, proline-rich domain). Next to intermolecular distance changes, the VC pairs could take up various relative orientations (also in the fibrillar state), influencing FRET.
(B) Fluorescence intensities and ratiometric images of Q71-VC in time measured by confocal microscopy 24 h after transfection. Excitation: 488 nm; emission: 505–555 and 600–700 nm.
(A and C) Quantifications of single-cell Q71-VC intensity in foci (A) and cytoplasm (C) (top) and the corresponding FRET ratios (bottom) over time. This cell displays a 20-min-stable low-FRET state.
(D) Higher resolution images of the Q71-VC aggregate showing the fiber-like sparkles at its periphery. Top: the merge of mVenus, FRET, and bright-field channels. Bottom: the ratiometric image of FRET/mVenus channels.
(E) FRET/mVenus ratios of Q25, Q39, Q54, and Q71 labeled with VC plotted versus the mCherry intensity 24 h after transfection. The data for the foci, the cytoplasm, and the VC control are compared. The VC control is a single experiment performed in parallel. Each data point is from a different cell.
(F) Fluorescence and FRET/donor ratiometric images of mHttex1 with different polyglutamine lengths. Scale bars are 10 μm.
(G) FRET ratios of mature aggregates compared for the different Q lengths. Data correspond to the aggregates of Figure S1D 48 h after transfection to obtain more mature aggregates. Q39-Q71 p = 0.0004 (Tukey's test).
We quantified the FRET readout by dividing the FRET channel fluorescence (excitation at 488 nm, emission 600–700 nm) by the donor channel (excitation at 488 nm, emission 505–555 nm) (Figure 1C). Before aggregation, the FRET/donor ratio of Q71-VC was ∼0.4, equaling the control VC without Q71. Upon foci formation, the FRET/donor ratio was first similar to the solution state but immediately increased, arriving at 1.3–1.4 after about an hour. These data strongly suggest that the initial foci are structurally less ordered, akin to the solution state, after which the order increases to arrive at the final foci. This means that there are two events on-pathway to high-FRET aggregates: first the formation of low-FRET less-ordered foci, and second, the transition to the high-FRET ordered state.
To further confirm the identity of the final aggregate, a higher resolution confocal fluorescence image (Figure 1D) shows a star-like appearance that is very similar to GFP-fused mHttex1 proteins, as observed by cryo transmission electron microscopy (cryo-TEM) (Riguet et al., 2021), thus suggesting that the Q71-VC eventually forms assemblies consisting of fibrils. While the aggregate quickly depletes the cytoplasm of Q71-VC, the free Q71-VC that is not yet incorporated retains the low FRET/donor ratio of the VC control (Figures 1B, 1E, and S1D) with a small dependence of the FRET/donor ratio on the probe concentration due to intermolecular FRET. These data show that when Q71-VC is not in aggregates, it behaves as the VC, which contains monomeric variants mVenus and mCherry; hence, Q71-VC is indistinguishable from a monomeric state when dispersed.
To verify our method with the well-known increased aggregation propensity with Q length, we replaced the 71 glutamines of Q71-VC with 25 (Q25-VC), 39 (Q39-VC), and 54 (Q54-VC) glutamines. The control VC was run in parallel with the Q-length experiments. Longer glutamine stretches are known to increase aggregate formation, while the native 25 glutamine stretch does not aggregate inside cells. Indeed, we do not observe aggregates for Q25-VC under our conditions (Figures 1F, S1D, and S1E), which have FRET/donor ratios with an equal dependence on the concentration as the control VC (Figure 1E). The ratios of the final foci of Q39, Q54, and Q71 decrease somewhat with 1.6 ± 0.18, 1.53 ± 0.18, and 1.48 ± 0.15 (mean ± SD; n = 60) as the Q lengths increase for Q39, Q54, and Q71, respectively (Figure 1G). The shorter Q lengths apparently reduce the average distance between the mHttex1 C termini. The differences are small in comparison to the changes observed during aggregation. The number of aggregates also depends on the Q length: Q39-VC forms aggregates in only 5% of the cells and Q71-VC in 48% after 24 h of incubation. We further find that the concentration at the onset of foci formation decreases with increasing Q length in the order of Q39 > Q54 > Q71, with 9.2 ± 1.9, 7.4 ± 1.7, and 3.5 ± 1.7 (SD; n = 10), respectively. Hence, even though longer Q stretches are more prone to aggregate, the eventual aggregates have a similar structure.
The method allowed us to obtain detailed kinetic information at the single-focus level (Figure 2A), and we analyzed the kinetics of the transition from disordered assemblies to the fibril-like state for the different Q lengths. In most cases, the FRET/donor increase of the foci has a smooth and somewhat sigmoidal appearance; the overall structure of the aggregate matures continuously without long-term stalling at an intermediate state. However, in a few cases, the FRET/donor ratio stalls at the initial low-FRET level for up to 20–60 min before increasing (Figure S2). Fitting the data from the subsequent steep increase to an exponential decay allows the comparison of the kinetics of the transition in the foci and the determination its apparent half-life, t1/2 (Figure 2B). We find that Q71-VC transits to the fibril-type aggregates with t1/2 = 73 ± 30 min (±SD; n = 10) and Q54-VC with t1/2 = 121 ± 43 min (±SD, n = 10). Q39 has the longest half-time with 141 ± 62 (±SD; n = 10). Hence, the Q length increases the rate of the first step that involves forming stable assemblies, as well as the rate of the second stage that involves the transition to fibril-type assemblies.
Figure 2.

mHttex1 single foci ratio, size, and intensity progression with different Q lengths
(A) Progression of single-foci FRET ratios of Q39, Q54, and Q71-VC in time. Each graph is 10 foci. The moment foci emerged was set as t = 0; the data before t = 0 are the FRET/mVenus ratio in the cytoplasm.
(B) Dependence of aggregation rate on the Q length of (A). Statistics Q71-Q54 p = 0.07, Q71-Q39 p = 0.009 (Tukey's test).
(C) Normalized FRET/Venus versus normalized area (left graph) or normalized mCherry intensity (right graph) for all data of (A).
We next determined the relative aggregate size (surface area) and mHttex1 density (mCherry intensity) (Figure 2C) during the structural transition of the mHttex1 foci. If the transition of the focus follows the same mechanism, we would observe a consistent relation between the surface area, density, and assembly structure. Indeed, although the aggregate surface area and mCherry intensity are highly heterogeneous in time between cells and Q lengths (Figures S1G and S1H), fitting the FRET ratio versus the normalized parameters rendered a similar relation for all conditions. Thus, the degree order in the foci follows its relative size and density, independent of Q length or cell-to-cell variation.
Together, the application of the probe in human cell lines allows the observation of single-foci structural progression from a less-ordered to a fibril-type state. Both aggregate occurrence and aggregate progression follow the typical kinetic dependence on Q length, while the progression occurs structurally in a similar manner.
Spatiotemporally resolved mHttex1 transition in yeast
The VC probing method can directly compare mHttex1 aggregation intermediates and kinetics between yeast and human cells: yeast is a model organism to study mHttex1, although yeast aggregates form solid inclusions instead of macroscopic fibrils (Gruber et al., 2018). To this end, we loaded Q72-VC- and control-VC-expressing yeast cells from an exponentially growing culture in a microfluidics chip to follow aggregation under controlled environmental conditions (temperature, nutrients, pH) in individual cells (Crane et al., 2014). Expression was induced with 0.2% galactose on-chip. To resolve the different aggregation states, we measured individual cells every 20 min for 15 h by widefield fluorescence microscopy (Figures 3A and 3E). The control VC displayed stable ratios throughout the experiments with low dependence on the concentration (Figure S3A). For Q72-VC, on the other hand, the first foci appeared after 338 ± 131 min (±SD). The size of the foci increased over time as expression continued and resulted in large aggregates or inclusions.
Figure 3.

Long-term imaging of individual yeast cells reveals distinct phases of mHttex1 aggregate progression
(A) Ratiometric images of a representative yeast cell expressing 72Q-VC. The cell outline is indicated with a white dotted line. The early low-FRET assemblies are visible in the Venus channel (not shown) but invisible in the ratiometric image, as their FRET ratio is similar to the cytosol. The white numbers correspond to the markings in (B). Scale bar is 5 μm.
(B) FRET/mVenus ratios plotted against time for the cell displayed in (A).
(A and C) The cytoplasm (C) is shown in pink and the aggregate (A) in dark red. When additional small foci appear at later stages, a cytoplasmic region without aggregates is difficult to distinguish, and FRET/mVenus ratios are shown in orange (A and C). When the FRET/mVenus ratio in the aggregate starts to increase steeply is indicated with a star.
(C) FRET/mVenus ratios from the cytoplasmic, aggregate, and mixed regions plotted versus the mCherry signal. The acceptor's fluorescence intensity is a direct indication of the probe concentration.
(D) Eight single-cell trajectories aligned to the time point (∗) at which the FRET/mVenus ratios transition to higher FRET/donor values. The transition point was set as t = 0, and trajectories ±200 min were plotted. The bold red line represents the average.
(E) Fold change in aggregate FRET ratios plotted in time for each cell and aligned at the transition point indicated by a gray line and a star.
(F) The timing of the first appearance of the focus correlates with the division time plotted as the number of divisions completed by each cell during the experiment (R2 = 0.4713, p = 0.0023).
(G) Cells that harbor foci with lower FRET divide faster (Mann-Whitney test, p = 0.0101) than those that transitioned to higher FRET foci.
When analyzing the single-cell traces in time, we find that the FRET/mVenus ratios in the assemblies first increase slowly, followed by a faster increase (Figures 3B and S3B). The stars in Figures 3B and 3E indicate the transition from the phase of slower increase to the phase of faster increase; the transition occurs at a FRET/mVenus ratio between 1.1 and 1.3. At lower FRET/mVenus ratios, the ratio is concentration dependent, as assessed from direct acceptor excitation (Figure 3C), while the FRET/mVenus becomes concentration independent at higher ratios after the transition. We interpret these changes in FRET/mVenus ratios as showing a structural transition between a less-ordered (low-FRET) early and an ordered (high-FRET) late state of the Q72-VC assembly. The duration of the less-ordered state varies significantly, with lifetimes between 100 and 700 min (Figure 3E). When averaging a subset of the cells that display the transition to a high FRET/donor ratio (8 out of 16) (Figures 3D and S3B), we observe that the aggregation proceeds through the typical aggregation curve with t1/2 = 42 ± 4 min (±SE; n = 8). Thus, while the duration of the less-ordered state varies, the transit to the high-FRET state occurs with similar kinetics once they enter the transition point. The remainder of the cells display assemblies that do not transform into high-FRET assemblies (FRET/donor remains <1.5; Figure S3C) during the duration of the experiment (15 h); they may well do so at later time points. In line with previous observations that yeast aggregates form solid inclusions instead of macroscopic fibrils (Gruber et al., 2018), we see that the FRET/donor ratio in the yeast high-FRET assemblies is unstable, contrasting with the stable fibril-type aggregate readouts in mammalian cells. This suggests that there are additional structural rearrangements in the high-FRET yeast aggregates.
Assessing the ratiometric images (Figure 3A), we see spatial heterogeneity in the FRET/donor ratios inside some of the aggregates along the aggregation trajectory, where assemblies with low FRET/donor ratios already contain an area with the high FRET/donor ratio of the aggregate (e.g., see Figure 3A; 700 min). This likely means that after the first nucleation event that results in less-ordered foci, we resolve the appearance of a second-phase transition within the less-ordered foci. Further, in contrast to mammalian cells, the ratios outside high FRET/donor aggregates also increase steadily (Figures 3B and S3D), suggesting more widespread aggregation. Indeed, as aggregation proceeds, multiple visible aggregates appear in the cytoplasm, especially for cells with very high expression levels, as noted previously for yeast (Gruber et al., 2018).
The division time of a yeast cell is a readout of cell fitness. There is a natural variation in division times of genetically identical yeast cells grown under these well-controlled conditions (Janssens and Veenhoff, 2016). Interestingly, we find that the timing of the appearance of the focus somewhat correlates with the number of divisions a cell completes during the recording, where foci appear later in the faster-dividing cells (Figure 3F). Cell fitness, as measured by the division time, is a complex phenotype, and therefore we do not expect stronger correlations than those observed. Moreover, we find that foci that do not undergo a transition within the duration of the experiment almost exclusively originate from yeast cells that maintain their division frequency under 200 min per division (Figure 3G). These correlations suggest that fitter cells have a higher protein homeostasis capacity, which delays the appearance of the focus as well as the transition to the high-FRET state.
Together, the mHttex1 aggregation monitored with this sensor in yeast cells shows several distinct features: (1) a variable duration of the low-FRET state but a similar half-time of the transition to the high-FRET state between the foci, (2) the transition from the less-ordered to ordered aggregate is spatially and temporally resolved akin a nucleation event, (3) later foci appearance and absence of high-FRET foci in more fit, faster-dividing cells, and (4) in the later stages of aggregation, widespread condensation/aggregation occurs throughout the cytoplasm. Strikingly, while the less-ordered state is more stable in yeast than in HEK293t cells, the transition kinetics from the less-ordered to the fibril-type state is similar.
DNAJB6b delays the onset of mHttex1 aggregation
One of the advantages of the method is that it allows for an easy combination with a second genetic intervention. The chaperone DNAJB6b is an excellent case study because it prevents polyQ aggregation by inhibiting early oligomer formation (Kakkar et al., 2016). We co-transfected and expressed DNAJB6b and Q71-VC in HEK293t cells and compared the resulting confocal fluorescence images with Q71-VC alone (Figure 4A). Co-expression of DNAJB6b strongly reduced the number of Q71-VC aggregates, and only a few aggregates remained. The FRET/donor ratio of Q71-VC aggregates with and without DNAJB6b is similar (Figure S4A), and the foci with lower FRET/donor ratios were observed with the same frequency as those without DNAJB6b, that is, 3% below 0.6 and 9% below 0.8, for both Q71-VC without DNAJB6b (n = 117 foci) and with DNAJB6b (n = 59 foci). These data suggest that the DNAJB6b does not prominently stabilize foci with a less-ordered structures along the aggregation pathway.
Figure 4.

High-throughput measurement of DNAJB6b suppression of mHttex1 aggregation
(A) Fluorescence intensities and ratiometric images of Q71-VC with and without co-expression of DNAJB6b. Scale bars are 10 μm.
(B) FRET versus mVenus channel of Q71-VC, Q25-VC, co-transfection with DNAJB6b, and VC-DNAJB6b in HEK293T cells as obtained by FACS showing clear differences in populations. Biological replicates are shown in Figure S5.
(C) Violin plots of the individual FRET/donor ratios obtained by FACS in (B).
(D) Left: diagram illustrates the percentage of high-FRET cells at different Q71-VC expression levels when cells are co-transfected with different amounts of DNAJB6b plasmid. Right: density maps of FACS data show the distribution of cells in FRET-ratio versus protein-expression levels, demonstrating two separate groups of Q71-VC when Q71-VC is co-transfected without and with DNAJB6b plasmid (DNAJB6 plasmid amount corresponds to white line in the left graph). Cells were fixed with 4% paraformaldehyde (PFA). Cells were imaged 24 h after transfection.
A powerful application of this approach is the possibility of rapid assessment of chaperone-induced inhibition by FACS (Figure 4B). We compared the absence and presence of DNAJB6b on Q71-VC aggregation in many cells. By plotting the FRET channel versus the donor channel as a measure for the FRET/donor ratio, we find a single main population for the Q25-VC with a slope that depends on the FRET efficiency, while the Q71-VC data show a distinct additional population corresponding to the higher FRET/donor ratio of the aggregate. We analyzed the FACS data by visualizing the FRET/donor-ratio densities in violin plots (Figure 4C). We find that the main population with lower FRET/donor ratios is present in all experiments while the Q71-VC shows a well-populated segment with a FRET/donor ratio >2, which presumably contains the fibril-like aggregate state. Confirming our microscopy experiments, co-expression of the DNAJB6b reduces high-FRET, fibril-like Q71-VC aggregate formation (Figures 4C and S5). Hence, the method allows FRET measurements by FACS.
To obtain more mechanistic insights into the functioning of the chaperone, we subsequently titrated DNAJB6b to the Q71-VC-expressing cells, measured the cells with FACS, and plotted the frequency of high-FRET cells versus the mHttex1 expression level, as determined from direct acceptor excitation, as a phase diagram (Figures 4D, S4B, and S4C). The populations of low- and high-FRET cells are clearly separated in FACS, while microscopy shows a clear continuous transition at the single-foci level. The FACS data further show that DNAJB6b increases the mHttex1 concentration needed to obtain the fibril-type aggregates. We applied a simple kinetic model, classical nucleation theory, which was previously used to obtain insight into phase separation of mHttex1 and other proteins (Posey et al., 2021; Crick et al., 2013). In this analysis, the degree of supersaturation of mHttex1 and the nucleation energy barrier determine the nucleation rate. The degree of supersaturation is the mHttex1 concentration c with respect to its saturation concentration cs and is defined as S = ln(c/cs). In our system, as shown by microscopy experiments, there would be two nucleation events: first from a dispersed state to the disordered assemblies and second from the disordered assemblies to the ordered state. In our human cell experiments, the second state always immediately follows the first, indicating that the first step is rate determining, and our analysis provides information on the first step. We assume that the cells with a transition at the lowest Q71-VC concentration in FACS are closest to the actual Q71-VC saturation concentration, hence, where c→cs. We take the Q71-VC concentration at which 50% of the cells have converted to the high-FRET state to determine the degree of supersaturation where most cells are forming aggregates. We thus find that under our expression conditions, most aggregates form at a supersaturation of S = 0.48 ± 0.05 (n = 3 biological replicates; ±SD).
Using the same approach in the presence of DNAJB6b, we find that the saturation concentration cs of Q71-VC increases up to 5.6 ± 1.8-fold (n = 3 biological replicates; ±SD). However, the degree of supersaturation S to form most aggregates remains strikingly similar with S = 0.46 ± 0.08 (n = 3 biological replicates; ±SD). The simplest explanation is that the presence of DNAJB6b increases Q71-VC solubility but does not influence the nucleation mechanism significantly. Given these observations, which are supported by literature (Kakkar et al., 2016; Mansson et al., 2014b), we hypothesize that DNAJB6b transiently binds Q71-VC (or small soluble clusters thereof) to increase Q71-VC solubility. At higher Q71-VC concentrations beyond the buffering capacity of DNAJB6b, irreversible aggregation captures DNAJB6b into the aggregates (see below).
Probing the oligomerization properties of DNAJB6b
DNAJB6b has been shown to form oligomers in buffer, which may affect its functioning (Mansson et al., 2014b; Soderberg et al., 2018; Karamanos et al., 2020). To observe whether the chaperone oligomerizes in the cell and if oligomerization is needed to inhibit mHttex1 aggregation, we tagged the DNAJB6b with the VC to generate VC-DNAJB6b (Figure 5A). The VC was fused to the N terminus of DNAJB6b to prevent inhibition of the active serine/threonine-rich domain located near the C terminus of DNAJB6b (Kakkar et al., 2016). Transfection and expression lead to a single VC-tagged protein, as assessed by SDS-PA gel electrophoresis (Figure S4D). Monitoring the VC-DNAJB6b in cells by confocal microscopy gave FRET/donor ratios equal to the VC control, with a similar dependence on the probe concentration (Figures 5B and 5C). Dispersed VC-DNAJB6b is thus indistinguishable from the monomeric VC. Further, the FRET/donor ratios of VC-DNAJB6b were homogeneous throughout the cytoplasm, suggesting an absence of any local oligomerization or self-assembly under the conditions used. We did notice, however, an additional population with higher FRET/donor ratios in FACS (Figure 4C). This population occurred at the highest expression levels, and both the FRET ratio and fraction were highly dependent on the experiment. Nonetheless, the method allows the observation of VC-DNAJB6b oligomerization, but this does not occur under more controlled conditions in microscopy.
Figure 5.

Tagging chaperone DNAJB6b allows evaluation of oligomerization during co-assembly with Httex1
(A) The VC-DNAJB6b design and schematic of VC-DNAJB6b inclusions in mHttex1 aggregates.
(B) VC-DNAJB6b co-transfected with and without Q79. Ratiometric images show an increase in FRET in the few foci. Foci and condensates are not observed without Q79. Scale bars are 10 μm.
(C) VC-DNAJB6b FRET/donor ratio versus the protein concentration with (right panel) and without (left panel) Q79. Every data point is a cell. VC control is from the same experiment run in parallel.
(D) Images of representative corona and homogeneous VC-DNAJB6b and mHttex1 Q71 complexes.
(E) The intensities and FRET ratio profiles of red lines of (D) showing no dependence of the FRET ratio on the intensity. Cells were imaged 48 h after transfection.
We next studied the VC-DNAJB6b oligomerization behavior in the presence of unlabeled Q71 (Figures 5B and 5C). The Q71 did not influence the FRET efficiency of dispersed VC-DNAJB6b, which remained similar to the VC. Foci appeared with a similar frequency as Q71-VC with unlabeled DNAJB6b: labeling Q71 with GFP showed that VC-DNAJB6b and Q71 co-localize in these foci (Figure S4E), confirming previous observations (Gillis et al., 2013). This co-assembly is not due to the VC domain, as the control VC does not localize with Q71 (Figure S4E). VC-DNAJB6b that co-localizes in aggregates displays, however, a higher FRET/donor of ∼0.6 than in the cytoplasm, where it has a ratio of ∼0.4. The increase in FRET could be due to the capture of DNAJB6b oligomers in the Q71 aggregates, as previously determined for DNAJB6b incorporation in amyloid-β (Osterlund et al., 2020). Additional quenching by the Q71 fibrils could further influence the ratio. The VC-DNAJB6b is locked in the aggregates, as the fluorescence did not recover after photobleaching (Figure S4F). Photobleaching the acceptor showed an increase in donor intensity, indicative of the presence of FRET. The DNAJB6b distribution somewhat displays a corona in most aggregates (∼80%) 48 h after transfection, where fluorescence increased by about 50% at the rims. The FRET/donor was independent of corona formation or localization in the corona (Figures 5D and 5E), suggesting similar environments for the DNAJB6b. Together, VC-DNAJB6b can co-aggregate with Q71 aggregates, increasing the FRET/donor ratio, indicative of a non-random structure but with less order than the mHttex1 aggregates.
FUS condensates display expected condensates with less order
To verify that the method can probe condensate-type structures, we applied the method to the FUS protein often associated with condensate formation. FUS condensates have mutation-dependent dynamics and structure in buffer with varying RNA affinity (Niaki et al., 2020; Patel et al., 2015). We selected wild-type FUS and a common pathogenic mutant. Wild-type FUS is transported into the nucleus, while mutations in the nuclear localization signal (NLS) retain FUS in the cytoplasm, where it forms condensates and sequesters RNA.
To determine the influence of the VC domain on FUS localization, we first determined the VC fusion site. Fusion at the N terminus (VC-FUS) displays native-like behavior by localizing in the nucleus in a dispersed state, while fusion at the C terminus (FUS-VC) (Figures 6A, 6B, S6A, and S6B) hampers localization in the nucleus and forms foci in the cytoplasm. Hence, we selected the VC-FUS for further studies.
Figure 6.

FUS mutants form foci with distinctly different structures
(A) The structure of wild-type FUS (LC, low complexity domain; NES, nuclear export signal; RRM, RNA recognition motif; NLS, nuclear localization signal).
(B) Probe design of VC-FUS and the corresponding fluorescence intensity images of HEK293t cells transfected. The wild-type and the R521C mutation are depicted. Scale bars are 10 μm.
(C) FRET channel plotted versus mVenus channel for the different FUS constructs. The data in the foci, cytoplasm, and nucleus were analyzed separately, and each was fitted to a linear equation for comparison with the same VC control. Each data point is a different cell. A representative biological replicate is displayed in Figure S6.
(D) Comparison of the slopes as in (C) that represent the FRET/donor ratio for VC-FUSR521C locations and two biological replicates. Error bars are the error in the fit. All samples are 48 h after transfection.
The slope of a linear fit of the FRET versus the donor channel provides the FRET/donor ratio of the assemblies or the dispersed states while comparing the same concentration regimes (Figures 6C, 6D, and S6C) (Liu et al., 2017). The FRET/donor of the dispersed VC-FUS overlaps with that of the control VC, and both proteins likely have similar monomeric and diffusive properties.
We next assessed the structure of the condensate-forming pathogenic R521C mutant that is associated with amyotrophic lateral sclerosis (ALS) (Belzil et al., 2009; Blair et al., 2010; Yamamoto-Watanabe et al., 2010). This mutant binds RNA more tightly in cells than does the wild type, leading to static complexes in a buffer (Niaki et al., 2020), and weakens the binding with karyopherin β2 (Zhang and Chook, 2012), a transportin that carries various RNA-binding proteins to the nucleus. Measurement of the VC-FUSR521C by confocal fluorescence microscopy showed that the mutation induced cytoplasmic foci (Figure 6B) with a dispersed fraction in the nucleus and cytoplasm. Complete bleaching of the foci by FRAP led to a rapid recovery of 2.1 ± 1.0 s (SD; n = 2), suggesting a rapid exchange of the components with the cytoplasm. Interestingly, the VC-FUSR521C foci increased the FRET/donor by 0.15 ± 0.9 (±SD; n = 3) compared with its dispersed state in the cytoplasm or nucleus. The increased FRET/donor suggests that the fluorophores are situated more closely or with a favorable orientation within the condensate, hence with more order than random diffusion in the cytoplasm.
To test if the increase would be the same for other FUS condensates, we compared our findings with the FUS-VC foci. Compared with its dispersed state, these foci provide a somewhat lower average FRET/donor ratio increase of 0.06 ± 0.03 (±SD; n = 3). Also, here, full bleaching of the FUS-VC foci led to rapid recovery with a t1/2 of 2.2 ± 0.8 s (SD; n = 3) (Figures S6D–S6G). The FUS-VC foci split and fused during the experiment, and their FRET/donor ratios did not change in time (Figures S6H and S6I). The R521C mutation showed a minute increase in the FRET/donor ratios for the FUS-VCR521C foci of 0.03 ± 0.02 (±SD; n = 3; Figure S6). The FUS-VCR521C construct provided a recovery half-time of 2.1 ± 0.7 s (SD; n = 3) from FRAP by full bleaching, suggesting a similar rapid exchange of components. Thus, foci formed by the FUS constructs have similar partitioning dynamics and low degrees of order, as expected from a condensate, while small structural differences between the foci of the different FUS constructs remain.
Discussion
Ubiquitous protein assembly in cell biology results in diverse and dynamic architectures that are often part of their biological function. Here, we apply a method that allows better monitoring of the corresponding structural transitions with high spatiotemporal resolution. We show that by fusing an mVenus-mCherry domain to the well-studied aggregating protein mHttex1, the chaperone DNAJB6b, and the condensate-forming FUS, we can distinguish between different assembly structures and can measure their interconversion resolved in space and time.
The readout is exceptionally sensitive to the assembly structure at the distance of ∼2–10 nm: The high nonlinear FRET dependence on the distance and the orientation between the fluorophores provides a unique sensitive signature of a particular structure. The relation between the FRET efficiency and structure can deviate because a fraction of ideally localized fluorophores may change the average FRET disproportionally, and competing donor quenching by fibrils may alter the readout (Kaminski Schierle et al., 2011; Laine et al., 2019). Nonetheless, a high FRET efficiency should relate to highly ordered aggregates, while unstructured condensates will have the same concentration dependence of the FRET efficiency as freely diffusing proteins. Any deviation should result from specific interactions or preferred orientations between the probed proteins, providing a fingerprint for a given assembly. With these considerations in mind, we have shown that the probing method samples structures ranging from disordered condensates to fibril-type aggregates and their transitions at the single-focus level.
The mHttex1 aggregation pathway appears to embark from a more disordered assembly, as the first foci invariably have a low FRET/donor ratio, similar to the foci formed by the FUS proteins and close to that of the VC control. Previously, Peskett et al. showed a liquid-like state in mHttex1 assembly (Peskett et al., 2018), which would indeed correspond to our low FRET values. Various aggregating proteins have been shown first to form a liquid-type condensate before aggregation (Mathieu et al., 2020), including mHttex1 (Peskett et al., 2018). After the first disordered foci, a structural transition occurs toward the fibril-type state. The fibrillar nature follows from the SDS insolubility, FRAP experiments, and fluorescence microscopy. The high FRET would originate from the tighter packing of monomers in fibrils, while additional donor-quenching pathways are also possible. In mammalian cells, this transition starts immediately after foci appearance, while the disordered state is a more stable intermediate in yeast cells. The stepwise nature of the aggregation is clear from the spatiotemporally resolved appearance of high-FRET focus from within a low-FRET focus. We can extract that the first nucleation event would reflect the formation of a disordered structure, and the second, the formation of the fibrillar structure. The gradual increase in FRET over the entire assembly after the second nucleation would align with increasing fibril content. A two-step transition has also been seen in buffer upon deletion of the N17 peptide, while the full-length variant provided immediate fibrils (Crick et al., 2013). One may speculate that in cells, the activity of the N17 peptide is modulated to induce a two-step transition, albeit that there are many other competing possibilities in the complex and confined intracellular environment. Nonetheless, it appears that protein-homeostasis fitness strongly delays the transition to the highly ordered state, as we deduce from the yeast division time in long-term (>10h), single-foci measurements.
We see that the chaperone DNAJB6b inhibits the nucleation of Q71-VC foci, as demonstrated by the microscopy experiments and the increase in saturation concentration of mHttex1 due to the presence of DNAJB6b. After that, the aggregation occurs with a similar dependence on the supersaturation as without DNAJB6b, suggesting that the DNAJB6b is simply out buffered. This mechanism follows the proposed mechanism of DNAJB6 in buffer and in cells as well as that proposed for some other chaperones (Mansson et al., 2014a; Posey et al., 2018). The encapsulation of VC-DNAJB6b by the aggregates does not appear to destabilize the aggregates and does not lead to an increase in soluble mHttex1, suggesting that the DNAJB6b only acts at the soluble state of mHttex1. The DNAJB6b is likely dispersed and monomeric when functional against mHttex1 aggregation, although we cannot exclude that an oligomeric form of DNAJB6b retains functionality.
The method based on the VC domain may in the future be expanded to determine changes in the architectures of protein assemblies during cell stress or aging and also where, for example, dimers or trimers with a fixed architecture are dispersed in the cytoplasm. It allows for the comparison of protein self-assembly under controlled conditions in buffer and cells to determine what influences the assembly architecture. It can be expanded to native proteins and allows localization in various compartments by fusion to localization tags. In addition, the method can generate data in an ensemble and high-throughput fashion by FACS or screening in multiwell arrays by confocal microscopy. The extent of structural information can be multiplied by using time-resolved anisotropy measurements, fluorescence lifetime imaging, or fluorescence correlation spectroscopy, all of which could, in principle, be combined with the method based on the VC moiety. The simplicity of ratiometric imaging combined with proper controls allows straightforward access to great detail in the structural order in space and time and the simultaneous monitoring of biological function with orthogonal assays. These assets enable distinguishing structures and intermediate kinetics, benefiting the study of the biological consequences of the assembly. Together, this highly sensitive method opens up additional routes to investigate the properties of condensates and aggregates in living cells.
Limitations of the study
Fluorescent protein fusions influence the aggregation behavior of proteins (Riguet et al., 2021) (Laine et al., 2019; Peskett et al., 2018; Xu et al., 2020). However, the VC-tagged proteins mostly recapitulate known mutation-dependent transitions and assembly behavior: (1) the glutamine-length dependence of the VC-Q series follows the established glutamine dependence, (2) intermediate phases in mHttex1 aggregation also have been shown to occur in a buffer without fluorescent protein fusions (Posey et al., 2018), (3) the VC does not dominate VC-DNAJB6b binding to Q71 because the VC control does not bind Q71, (4) the VC-DNAJB6b retains its activity by preventing Q71 aggregation, and (5) the tag influences FUS-condensate formation depending on the terminus tagged, while the 521C mutation always induces expected assemblies. Moreover, we use the monomeric versions of the fluorescent proteins to reduce any interactions between the fluorescent proteins themselves. Nonetheless, the interactions of the fluorescent proteins may influence protein behavior that should be assessed for each study.
Previous comparable fluorescent-protein-based methods, such as the DAmFRET method based on photoconverting half of the donor into an acceptor, have shown important mechanistic insights after high-throughput screening using FACS (Nuckolls et al., 2020; Posey et al., 2021; Kandola et al., 2021). In principle, the same data and mechanistic inferences are also available from the VC method presented here. The VC tag is larger than a single fluorescent protein in the other methods, which may influence its behavior. However, the VC tag has a major asset in that it allows precise imaging with high spatiotemporal resolution, continuously over long periods of time at the single-focus level that reveal kinetics of transient intermediates, in combination with second genetic interventions. Hence, the combination of such methods provides an extensive toolbox to determine the interactions of fluorescent-protein-tagged self-assembling proteins.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Albumine bovine serum | Acros organics | 268131000; CAS: 9048-46-8 |
| Yeast synthetic drop-out medium supplements without uracil | Sigma-Aldrich | Cat# Y1501 |
| Difco™ Yeast Nitrogen Base w/o Amino Acids | BD | Cat# 291940 |
| Select agar | Invitrogen | Cat# 30391049 |
| D(+)-Glucose anhydrous | Fischer scientific | Cat# G/0500/65; CAS:50-99-7 |
| D(+)-Raffinose pentahydrate 99+% | Acros organics | Cat# 195671000; CAS: 17629-30-0 |
| D(+)-Galactose, 99+% | Acros organics | Cat# 150610010; CAS: 59-23-4 |
| Lithium acetate dehydrate 98% | Sigma-Aldrich | CAS: 6108-17-4 |
| Deoxyribonucleic acid, low molecular weight, from salmon sperm (ssDNA) | Sigma-Aldrich | Cat# 31149; CAS: 100403-24-5 |
| Polyethylene glycol 35000 | Sigma-Aldrich | Cat# 8188921000; CAS:25322-68-3 |
| SYLGARD 184 Silicone elastomer kit (PDMS) | Dow Corning | N/A |
| Lipofectamine ™ 2000 Transfection Reagent | ThermoFischer | 11668019 |
| Paraformaldehyde (PFA) | Sigma-Aldrich | Cat# 158127 |
| Deposited data | ||
| Analyzed data | This paper | https://doi.org/10.5281/zenodo.6044649 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae: Strain background: BY4741 | This paper | SMY32 |
| S. cerevisiae: Strain background: BY4741 | This paper | SMY33 |
| Recombinant DNA | ||
| Plasmid: pYES2-pGAL1-Venus-Cherry | ThermoFischer Scientific: GeneArt | N/A |
| Plasmid: pYES2-pGAL1-72Q-Venus-Cherry | ThermoFischer Scientific: GeneArt | N/A |
| Plasmid: pMSC | Ma et al., 2017 | Addgene Plasmid |
| #90411 | ||
| Plasmid: pEGFP-Q71 | Harm H. Kampinga’s group | N/A |
| Plasmid: pEGFP-Q25 | Harm H. Kampinga’s group | N/A |
| Plasmid: pDNAJB6b | Harm H. Kampinga’s group | N/A |
| Plasmid: pFUSWT | Harm H. Kampinga’s group | N/A |
| mHTTex Q54 | ThermoFischer Scientific: GeneArt | N/A |
| mHTTex Q54 | ThermoFischer Scientific: GeneArt | N/A |
| Plasmid: pQ71-VC | This paper | N/A |
| Plasmid: pQ54-VC | This paper | N/A |
| Plasmid: pQ39-VC | This paper | N/A |
| Plasmid: pQ25-VC | This paper | N/A |
| Plasmid: pVC | This paper | N/A |
| Plasmid: pQ79 | This paper | N/A |
| Plasmid: pVC-DNAJB6b | This paper | N/A |
| Plasmid: pVC-FUSWT | This paper | N/A |
| Plasmid: pVC-FUSR521C | This paper | N/A |
| Plasmid: pFUSWT-VC | This paper | N/A |
| Plasmid: pFUSR521C-VC | This paper | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Prism GraphPad, version 9.1.2 for Windows | GraphPad Software | www.graphpad.com |
| FlowJo V7.6.1/V.10.7.2 | BD Life Sciences | https://www.flowjo.com/ |
| R studio (R version 4.0.4) | RStudio | https://www.rstudio.com/ |
| Origin 2018 | OriginLab Corporation | https://www.originlab.com/ |
| Other | ||
| Matrix for ALCATRAS microfluidic device | Matt Kaeberlain | (Crane et al., 2014) |
| Fine Bore Polythene Tubing | Smiths medical | Cat#: 800/100/120 |
| Luer-Lok™ Syringe 10mL | BD Plastipak | Cat#: 305959 |
| 25G needle | BD Microlance | Cat#: 300600 |
| Syringe without needle 1mL | Terumo | Cat#: SS+01T1 |
| Glass bottom culture dishes 50 mm uncoated | MatTek | Cat#: P50G-1.5-14-F |
| Leica TCS SP8 confocal microscope | Leica | N/A |
| 8-well Lab-Tek™ Chambered Coverglass | ThermoFisher | Cat#: 155411 |
| DMEM, high glucose, HEPES, no phenol red | Gibco | Cat#: 21063 |
| Opti-MEM™ I Reduced Serum Medium | Gibco | Cat#: 31985 |
| BECKMAN COULTER MoFlo Astrios Cell Sorter | Beckman Coulter | N/A |
| CellTris™ Disposal Filters 100 μm | Sysmex | Cat#: 04-0042-2318 |
| PS Cytometry tubes, 5 mL, 75X12 mm | Sarstedt | Cat#: 55.1579 |
| ChemiDoc MP imaging system | BIO-RAD | Cat#: 17001402 |
Resource availability
Lead contact
Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Arnold J. Boersma (boersma@dwi.rwth-aachen.de).
Materials availability
DNA plasmids that are used in this study are made available from the lead contact.
Experimental model and subject details
Mammalian cell culture
HEK293T cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S) at 37 °C with 5% CO2. HEK293T cells are of female origin. Cells have been authenticated by ATCC.
Yeast cell culture conditions
Yeast cells (S. cerevisiae: Strain background BY4741) were pre-cultured overnight at 30°C, 200 rpm in Synthetic Drop-out media, lacking uracil (SD - ura), and supplemented with 2% glucose. Afterward, cells are transferred to SD – ura, supplemented with 2% raffinose, 0.1% glucose for 24 hours before loading into the microfluidic device. The supplementation with glucose is essential to repress any leaky expression from the GAL1 promoter and prevent premature aggregation during the experiment. The 72Q or control probe expression is induced with 0.2% galactose on-chip. The cells loaded onto the chip are from an exponentially growing culture with OD600 = 0.4-0.6.
Method details
Mammalian plasmids constructs
pMCS (Addgene, 90411) containing Venus-mCherry was bought from Addgene. mHttex1 (Q71/Q25), FUSWT, DNAJB6b were generated from pEGFP-Q71, pEGFP-Q25, pFUS, pDNAJB6b plasmids kindly provided by Harm H. Kampinga. mHttex1 Q54 and Q39 were synthesized by GeneArt (ThermoFisher). Q71/54/39/25 sequences were subcloned into the pMCS plasmid, and Venus-mCherry was subcloned to pFUS and pDNAJB6b by sequence and ligation-independent cloning (SLIC) method to obtain pVC-Q71/54/39/25, pVC-FUSWT, pFUSWT-VC, and pVC-DNAJB6b. FUS mutants pVC-FUSR521C and pFUSR521C-VC were generated from pVC-FUSWT, pFUSWT-VC by site-directed mutagenesis.
Yeast plasmid constructs and strains
pYES2 vectors containing 72Q-mVenus-mCherry or mVenus-mCherry control were synthesized by GeneArt (ThermoFisher). The gene is under the control of the galactose inducible, glucose repressible GAL1 promoter. Yeast cells were transformed according to the LiAc method.
Mammalian cell transfection
For microscopy, cells were seeded into 8-well Lab-Tek™ Chambered Coverglass (ThermoFisher), 1.0×105 cells in 200 μl growth medium (DMEM+10%FBA+1%P/S) for each well. For PAGE gel and FACS experiments, cells were plated into 12-well plates, 4.0×105 cells in 1 mL growth medium for each well. One day after plating, when the confluency reached 70-90%, cells were transfected using Lipofectamine 2000 (ThermoFisher) according to the manufacturer's instruction. For each well of the 8-well Lab-Tek slide, 1 μl Lipofectamine and 250 ng plasmid were diluted with 25 μl Opti-MEM I Reduced Serum Medium (Gibco) separately and incubated for 5 min at room temperature. Then diluted DNA and Lipofectamine were combined (50 μl) and incubated at room temperature for 20 min followed by adding 150 μl DMEM (serum-free, antibiotic-free). Change the cell culture medium in Lab-Tek slide to transfection complexes (200 μl) and after 6 hours the media was changed back to complete growth medium. In polyQ DNAJB6b co-expression experiments, 125 ng of each plasmid was used. 1 μg plasmid and 4 μl Lipofectamine were used for the 12-well plate. Before imaging, the medium was replaced with DMEM (with HEPES, no phenol red, Gibco).
Mammalian cell FRET imaging and analysis
Cells were observed in Leica SP8 confocal microscope and imaged at 37°C. The laser powers were consistent for all measurements, as 3.6% for 488nm and 1% for 561nm. The excitation and emission settings are listed in Table S2. Z-stacks were acquired with 1 μm steps to ascertain correct focus. Imaging was performed with a 63×/1.35 oil-immersion objective. 8-bit images were obtained. Images were processed with ImageJ. First, a background subtraction was applied to all images as indicated by blanc cell samples. Areas were selected inside cells or following the shape of aggregates using manual selection tools in ImageJ combined with visual inspection. In general, aggregates were clearly brighter in the fluorescent channels. All relevant parameters were measured and exported for further analysis. The FRET channel intensity was divided by the donor intensities as indicated in the corresponding graphs. In the case of FUS, nuclear and cytoplasmic FRET/donor could be determined separately because the FUS constructs distributed asymmetrically. All graphs were generated and analyzed by GraphPad Prism. The kinetic curves of the Httex1 mutants were fitted with a one phase exponential association to obtain the half-times. The linear fit to obtain the FUS slopes was set to cross the origin.
FRET imaging in yeast cells
All experiments were performed at 30°C. Images were acquired using a DeltaVision Elite imaging system (Applied Precision (GE), Issaquah, WA, USA) composed of an inverted microscope (IX-71; Olympus) equipped with a UPlanSApo 100x (1.4 NA) oil immersion objective, InsightSSI solid-state illumination, ultimate focus, and a PCO sCMOS camera. Five Z-stacks were acquired with 0.5 μm spacing, every 20 min throughout the experiment. Images were analyzed in ImageJ. The appearance of aggregates was determined by visual inspection and the selection of the aggregate was done manually. Generally, aggregates appeared as bright foci in the cells. Sometimes, more than one aggregate could be detected. The nucleus was indistinguishable from the cytoplasm, and is permeable for the polyQ monomers. The mean background fluorescence was measured in the region of one cell diameter away from the region of interest (ROI) for each channel and subtracted from the cell's average fluorescence. The FRET/Venus was calculated from these background-subtracted intensities. To calculate the ratiometric images, we set the background to zero by subtracting the maximum background fluorescence in each channel, measured in immediate proximity to the ROI. The transition point between the phases of slow and faster increase in the FRET/mVenus ratio (indicated by a star in Figure 3) were determined as the time point where the slope increased 3-fold or more.
FRAP measurements
The FRAP experiments were carried out in the same microscope with 100% bleaching laser power at 561nm. The mHttex1 and DNAJB6b aggregates were bleached for 2s. FUS condensates were bleached for 0.139s for the whole assembly regions. The image acquisitions after the bleaching shared the same settings as in Table S2. The data was exported from Leica software LAS X, and the graphs were generated by GraphPad Prism. To calculate the recovery half time, we first normalized the donor and FRET intensities, and FRET/Venus ratios as below:
Then the curves were fitted with one-phase exponential equations using GraphPad Prism 7.
PAGE analysis
48-well plates were used to prepare cell samples. HEK293T cells were washed with 200 μL 1×PBS 24h or 48h after transfection. Cells were then lysed with 50 μL 1×SDS sample buffer with additional protease inhibitor (Roche cOmplete™ EDTA-free protease inhibitor cocktail), 1 mg/mL DNaseI (Roche), and 2 mM MgCl2. Cell lysates were incubated on ice for 30 min, after which 20 μL of each sample was loaded to a 10% SDS-PAGE or a native PAGE gel. The gels were evaluated under 365nm UV light (VWR Genosmart) to get the fluorescent images. The multi-channel fluorescent images were generated from ChemiDoc MP imaging system (Bio-Rad), Alexa 488 (excitation: Epi-Blue 460−490 nm, emission: 532/28) for Venus, Alexa 546 (excitation: Epi-Green 520-545 nm, emission: 602/50) for mVenus&mCherry.
FACS
12-well plates were used to prepare cell samples. HEK293T cells were washed by 500μl 1×PBS 24h after transfection. 0.25% Trypsin was used to detached the cells from the bottom. Then the cells were resuspended in 1ml 1×PBS after washing and centrifuging. After filtration with 100μm filters, the cell suspensions were measured with the flow cytometer (BECKMAN COULTER MoFlo Astrios Cell Sorter). Cells were not fixed unless specified (Figure 4D). For fixed cells, the cells were resuspended in 4% Paraformaldehyde (PFA) (in PBS) and incubated for 30 min after being washed and centrifuged with PBS. Then cells were centrifuged with 350 rcf and the pellet was resuspended in PBS and stored at 4°C until being measured. All steps were in room temperature unless specified otherwise. The excitation wavelength was 488nm for mVenus and FRET with 576/21 and 664/22 detector filters. The populations were selected as in Figures S4G–S4I. For mCherry, 561 nm laser and 614/20 filter were chosen. The graphs were plotted with FlowJo 7.6.1 or 10.7.2. Raw data was exported. Data points at the edge of detection were removed. Violin maps were generated in R studio. The 2D diagrams were plotted with Origin 2018.
Quantification and statistical analysis
The quantification and statistics methods are described with the figures and in the method details section. In general, all measurements have been performed as three independent biological repeats. P values were calculated using Tukey’s or Mann Whitney’s test using Graphpad Prism.
Acknowledgments
We thank Prof. Andreas Herrmann, Prof. Bert Poolman, Prof. Harm H. Kampinga, and Dr. Steven Bergink for valuable discussions. We thank Prof. Harm H. Kampinga and Dr. Steven Bergink for sharing mHttex1, DNAJB6b, and FUS plasmids. The work was funded by the ERC Consolidator Grant (PArtCell; no. 864528) to A.J.B., the China Scholarship Council grant to Q.W., and the Netherlands Organization of Scientific Research (OCENW.GROOT.2019.068 en NWO-vici [VI.C.192.031]) to L.M.V. Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which is supported by the European Commission and the federal state of North Rhine-Westphalia (no. 300088302).
Author contributions
Q.W. and S.N.M. performed the experiments and analyzed data. All authors contributed to the design and planning of the experiments. L.M.V. supervised the yeast experiments. A.J.B. supervised the project. Q.W., S.N.M., and A.J.B. wrote the paper with input from L.M.V.
Declaration of interests
The authors declare no competing interests.
Published: March 28, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100184.
Supplemental information
Data and code availability
-
•
Analysis data has been made available through Zenodo (https://doi.org/10.5281/zenodo.6044649). Microscopy images reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ast A., Buntru A., Schindler F., Hasenkopf R., Schulz A., Brusendorf L., Klockmeier K., Grelle G., McMahon B., Niederlechner H., et al. mHTT seeding activity: a marker of disease progression and neurotoxicity in models of Huntington's disease. Mol. Cell. 2018;71:675–688.e6. doi: 10.1016/j.molcel.2018.07.032. [DOI] [PubMed] [Google Scholar]
- Belzil V.V., Valdmanis P.N., Dion P.A., Daoud H., Kabashi E., Noreau A., Gauthier J., Team S.D., Hince P., Desjarlais A., et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair I.P., Williams K.L., Warraich S.T., Durnall J.C., Thoeng A.D., Manavis J., Blumbergs P.C., Vucic S., Kiernan M.C., Nicholson G.A. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry. 2010;81:639–645. doi: 10.1136/jnnp.2009.194399. [DOI] [PubMed] [Google Scholar]
- Bünig S.S., Sharma A., Vachharajani S., Newcombe E., Ormsby A., Gao M., Gnutt D., Vöpel T., Hatters D.M., Ebbingaus S. Conformational dynamics and self-association of intrinsically disordered huntingtin exon 1 in cells. Phys. Chem. Chem. Phys. 2017;19:10738–10747. doi: 10.1039/c6cp08167c. [DOI] [PubMed] [Google Scholar]
- Caron N.S., Desmond C.R., Xia J., Truant R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc. Natl. Acad. Sci. U S A. 2013;110:14610–14615. doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane M.M., Clark I.B., Bakker E., Smith S., Swain P.S. A microfluidic system for studying ageing and dynamic single-cell responses in budding yeast. PLoS One. 2014;9:e100042. doi: 10.1371/journal.pone.0100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick S.L., Ruff K.M., Garai K., Frieden C., Pappu R.V. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U S A. 2013;110:20075–20080. doi: 10.1073/pnas.1320626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis J., Schipper-Krom S., Juenemann K., Gruber A., Coolen S., van den Nieuwendijk R., van Veen H., Overkleeft H., Goedhart J., Kampinga H.H., et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem. 2013;288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A., Hornburg D., Antonin M., Krahmer N., Collado J., Schaffer M., Zubaite G., Luchtenborg C., Sachsenheimer T., Brugger B., et al. Molecular and structural architecture of polyQ aggregates in yeast. Proc. Natl. Acad. Sci. U S A. 2018;115:E3446–E3453. doi: 10.1073/pnas.1717978115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadanza M.G., Jackson M.P., Hewitt E.W., Ranson N.A., Radford S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018;19:755–773. doi: 10.1038/s41580-018-0060-8. [DOI] [PubMed] [Google Scholar]
- Janssens G.E., Veenhoff L.M. The natural variation in lifespans of single yeast cells is related to variation in cell size, ribosomal protein, and division time. PLoS One. 2016;11:e0167394. doi: 10.1371/journal.pone.0167394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkar V., Mansson C., De Mattos E.P., Bergink S., van der Zwaag M., van Waarde M., Kloosterhuis N.J., Melki R., van Cruchten R.T.P., Al-Karadaghi S., et al. The S/T-Rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell. 2016;62:272–283. doi: 10.1016/j.molcel.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Kaminski Schierle G.S., Bertoncini C.W., Chan F.T.S., van der Groot A.T., Schwedler S., Skepper J., Schlachter S., van Ham T., Esposito A., Kumita J.R., et al. A FRET sensor for non-invasive imaging of amyloid formation in vivo. Chemphyschem. 2011;12:673–680. doi: 10.1002/cphc.201000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandola T., Venkatesan S., Zhang J., Lerbakken B., Blanck J.F., Wu J., Unruh J., Berry P., Lange J.J., Von Schulze A., et al. The polyglutamine amyloid nucleus in living cells is a monomer with competing dimensions of order. bioRxiv. 2021 doi: 10.1101/2021.08.29.458132. Preprint at. 2021.08.29.458132. [DOI] [Google Scholar]
- Karamanos T.K., Tugarinov V., Clore G.M. An S/T motif controls reversible oligomerization of the Hsp40 chaperone DNAJB6b through subtle reorganization of a beta sheet backbone. Proc. Natl. Acad. Sci. U S A. 2020;117:30441–30450. doi: 10.1073/pnas.2020306117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T., Kandola T.S., Wu J., Venkatesan S., Ketter E., Lange J.J., Rodriguez Gama A., Box A., Unruh J.R., Cook M., et al. Quantifying nucleation in vivo reveals the physical basis of prion-like phase behavior. Mol. Cell. 2018;71:155–168.e7. doi: 10.1016/j.molcel.2018.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krobitsch S., Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie R.E., Maciejewski A., Ostapchenko V.G., Marques-Lopes J., Choy W.Y., Duennwald M.L., Prado V.F., Prado M.A.M. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front. Neurosci. 2017;11:254. doi: 10.3389/fnins.2017.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine R.F., Sinnige T., Ma K.Y., Haack A.J., Poudel C., Gaida P., Curry N., Perni M., Nollen E.A.A., Dobson C.M., et al. Fast fluorescence lifetime imaging reveals the aggregation processes of alpha-synuclein and polyglutamine in aging Caenorhabditis elegans. ACS Chem. Biol. 2019;14:1628–1636. doi: 10.1021/acschembio.9b00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Aberg C., Van Eerden F.J., Marrink S.J., Poolman B., Boersma A.J. Design and properties of genetically encoded probes for sensing macromolecular crowding. Biophys. J. 2017;112:1929–1939. doi: 10.1016/j.bpj.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C.H., Pandey N.K., Lim C.K., Ding Z., Tao M., Thomas D.D., Langen R., Sachs J.N. Discovery of small molecule inhibitors of huntingtin exon 1 aggregation by FRET-based high-throughput screening in living cells. ACS Chem. Neurosci. 2020;11:2286–2295. doi: 10.1021/acschemneuro.0c00226. [DOI] [PubMed] [Google Scholar]
- Ma Y., Pandzic E., Nicovich P.R., Yamamoto Y., Kwiatek J., Pageon S.V., Benda A., Rossy J., Gaus K. An intermolecular FRET sensor detects the dynamics of T cell receptor clustering. Nat. Commun. 2017;8:15100. doi: 10.1038/ncomms15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson C., Arosio P., Hussein R., Kampinga H.H., Hashem R.M., Boelens W.C., Dobson C.M., Knowles T.P., Linse S., Emanuelsson C. Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Abeta42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 2014;289:31066–31076. doi: 10.1074/jbc.M114.595124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson C., Kakkar V., Monsellier E., Sourigues Y., Harmark J., Kampinga H.H., Melki R., Emanuelsson C. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones. 2014;19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu C., Pappu R.V., Taylor J.P. Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science. 2020;370:56–60. doi: 10.1126/science.abb8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Qamar S., Lin J.Q., Schierle G.S., Rees E., Miyashita A., Costa A.R., Dodd R.B., Chan F.T., Michel C.H., et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaki A.G., Sarkar J., Cai X., Rhine K., Vidaurre V., Guy B., Hurst M., Lee J.C., Koh H.R., Guo L., et al. Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-Linked FUS mutations. Mol. Cell. 2020;77:82–94.e4. doi: 10.1016/j.molcel.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuckolls N.L., Mok A.C., Lange J.J., Yi K., Kandola T.S., Hunn A.M., McCroskey S., Snyder J.L., Bravo Nunez M.A., McClain M., et al. The wtf4 meiotic driver utilizes controlled protein aggregation to generate selective cell death. Elife. 2020;9:e55694. doi: 10.7554/eLife.55694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund N., Lundqvist M., Ilag L.L., Graslund A., Emanuelsson C. Amyloid-beta oligomers are captured by the DNAJB6 chaperone: direct detection of interactions that can prevent primary nucleation. J. Biol. Chem. 2020;295:8135–8144. doi: 10.1074/jbc.RA120.013459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen M.C., Gnutt D., Gao M., Warmlander S., Jarvet J., Graslund A., Winter R., Ebbinghaus S., Strodel B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019;48:3946–3996. doi: 10.1039/c8cs00034d. [DOI] [PubMed] [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M., et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Peskett T.R., Rau F., O'Driscoll J., Patani R., Lowe A.R., Saibil H.R. A liquid to solid phase transition underlying pathological huntingtin Exon1 aggregation. Mol. Cell. 2018;70:588–601.e6. doi: 10.1016/j.molcel.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitt S.K., Pallos J., Shao J., Desai U.A., Ma A.A., Thompson L.M., Marsh J.L., Diamond M.I. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron. 2003;40:685–694. doi: 10.1016/s0896-6273(03)00697-4. [DOI] [PubMed] [Google Scholar]
- Posey A.E., Ruff K.M., Harmon T.S., Crick S.L., Li A., Diamond M.I., Pappu R.V. Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J. Biol. Chem. 2018;293:3734–3746. doi: 10.1074/jbc.RA117.000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey A.E., Ruff K.M., Lalmansingh J.M., Kandola T.S., Lange J.J., Halfmann R., Pappu R.V. Mechanistic inferences from analysis of measurements of protein phase transitions in live cells. J. Mol. Biol. 2021;433:166848. doi: 10.1016/j.jmb.2021.166848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riguet N., Mahul-Mellier A.L., Maharjan N., Burtscher J., Croisier M., Knott G., Hastings J., Patin A., Reiterer V., Farhan H., et al. Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 2021;12:6579. doi: 10.1038/s41467-021-26684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffert L.N., Carter W.G. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: a systematic review. Brain Sci. 2020;10:232. doi: 10.3390/brainsci10040232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C., Rasband W., Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg C.A.G., Mansson C., Bernfur K., Rutsdottir G., Harmark J., Rajan S., Al-Karadaghi S., Rasmussen M., Hojrup P., Hebert H., et al. Structural modelling of the DNAJB6 oligomeric chaperone shows a peptide-binding cleft lined with conserved S/T-residues at the dimer interface. Sci. Rep. 2018;8:5199. doi: 10.1038/s41598-018-23035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.C.S., Loricco J.G., Bishop A.C., James N.A., Huynh W.H., McCallum S.A., Roan N.R., Makhatadze G.I. Sequence-independent recognition of the amyloid structural motif by GFP protein family. Proc. Natl. Acad. Sci. U S A. 2020;117:22122–22127. doi: 10.1073/pnas.2001457117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C., Lin T.Y., Chang D., Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017;4:160696. doi: 10.1098/rsos.160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Watanabe Y., Watanabe M., Okamoto K., Fujita Y., Jackson M., Ikeda M., Nakazato Y., Ikeda Y., Matsubara E., Kawarabayashi T., et al. A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: a clinical, pathological and genetic report. J. Neurol. Sci. 2010;296:59–63. doi: 10.1016/j.jns.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Zhang Z.C., Chook Y.M. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the fused in sarcoma protein (FUS) Proc. Natl. Acad. Sci. U S A. 2012;109:12017–12021. doi: 10.1073/pnas.1207247109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Analysis data has been made available through Zenodo (https://doi.org/10.5281/zenodo.6044649). Microscopy images reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.