

Abstract
The process from high-risk human papillomavirus (HR-HPV) infection to cervical cancer is a continuous and long-term process, but the pathogenesis of the whole process is not completely clear. Here, 59 Chinese women were engaged in this study, and divided into five groups: normal healthy group, HR-HPV infections group, low-grade intraepithelial neoplasia (LSIL) group, high-SIL(HSIL) group, and cervical cancer group. With the occurrence of HR-HPV infection and the development of cervical lesions, the diversity of vaginal microbiota species was increased, and the relative abundance of Lactobacillus (L.), the dominant bacteria in maintaining vaginal microecological balance, was decreased gradually. In contrast, the abundance of Actinobacteria in the four disease groups was significantly higher than that in normal group. Furthermore L. iners may be related to the serious progression of cervical cancer. After analyzing the whole process, we found that Gardnerella(G.), Atopobium(A.) and Dialister(D.) have important effects on both persistent HR-HPV infection and the pathogenesis of cervical cancer. In addition, PICRUSt2 and KEGG results showed that the KEGG pathways enriched by the predicted genes of vaginal microbiota in cancer group included metabolic diseases, endocrine system and immune systems when compared with that in normal group. These findings may provide insights into the pathogenesis of cervical cancer, and help to improve the early detection and prevention of cervical precancerous lesions.
Keywords: HR-HPV, LSIL, HSIL, cervical cancer, vaginal microbiota
Introduction
In recent years, female vaginal microbiota has attracted more and more attention from medical researchers. Microbial communities living in the reproductive tract seem to be involved not only in maintaining female reproductive tract health, but also in reproductive tract diseases, reproductive failure and pregnancy complications (Al-Nasiry et al., 2020). Cervical cancer is one of the most common cancers in women worldwide (Ginsburg et al., 2017). Persistent HR-HPV infection has been recognized as closely associated with squamous intraepithelial lesions (SILs) and cervical cancer (Munoz et al., 2003). Factors that alter the vaginal microbiota, have been identified as cofactors in the persistence of HPV infection, such as sexual intercourse, vaginal irrigation, inflammation of the vagina, or sexually transmitted infections (Clarke et al., 2012; Vriend et al., 2015; Łaniewski et al., 2018). Most women can recover from HR-HPV infection without developing cervical cancer due to the elimination of HR-HPV by their immune system (Insinga et al., 2011; Greer et al., 2016). The vaginal microbiota in healthy women of reproductive age is dominated by Lactobacillus (L.), which is known to produce lactic acid to maintain low vaginal pH (< 4.5), and this is the first line of defense against pathogenic agents (Boskey et al., 2001). Additionally, the vaginal microbiota is able to produce specific metabolites and bacteriocins which synergize or inhibit each other (Aroutcheva et al., 2001; McMillan et al., 2011). This suggests that vaginal microenvironment plays an important role in women’s reproductive health.
Recently, next-generation sequencing of barcoded 16S rRNA has broadened our understanding of vaginal microbiota, and helps to better study the relationship between the composition of vaginal bacterial community and disease. Previous studies have proposed that abnormal vaginal microbiota are classified into five main communities dominated by L. crispatus, L. iners, L. gasseri and L. jensenii, while the other community is characterized by a decrease in L. spp. and an increase in bacterial diversity (Brotman et al., 2014). However, among them, the vaginal microbiota dominated by L. iners is more permissive (for example in receiving potential pathogens in the vaginal ecosystem) than others. And it is associated with microbiological disorders, and increases the risk of cervical and other female genital tract diseases (Mitra et al., 2015; Stokholm et al., 2018; McKinnon et al., 2019). In addition, several studies have shown that the microbiota can promote cellular metabolism and immune disorders, and play an important role in the development of cancer (Banerjee et al., 2018; Porter et al., 2018). Although these results do not explicitly state a causal relationship, they suggest a link between microbiota signatures and cancer in different organs. Recently, some studies have shown that vaginal microbiota are associated with HPV infection, as well as CIN and CC (Di Paola et al., 2017; Kyrgiou et al., 2017; McKee et al., 2019). But there are some differences in reported conclusions. Some epidemiological reports indicate that the vaginal microbiota of women vary by race and region (Łaniewski et al., 2018; Noyes et al., 2018). In this study, we explored the characteristics of vaginal micmbiome in normal, HR-HPV-positive, low-grade squamous intraepithelial lesion (LSIL), high-grade squamous intraepithelial lesion (HSIL) and cervical cancer women. And the correlation between vaginal microbiota and different progression stages of cervical cancer were further analyzed.
Subjects and Methods
Study design and sample collection
Premenopausal nonpregnant women from department of Cervical Disease of International Peace Maternity and Child Health Hospital, Shanghai Jiao Tong University School of Medicine were recruited. The exclusion criteria for the study are as follows: 1) Subjects having immunosuppressive diseases; 2) Subjects using antibiotic within the last 7 days; 3) Subjects currently having a sexually transmitted infection or vaginal infection; 4) Subjects using suppositories, vaginal applied medications and douching substances within 48 hours prior to the visit; 5) Subjects having had sexual intercourse less than 48 hours prior to the visit; 6) Subjects having a previous history of cervical treatment.
The total of 59 subjects were divided into five groups according to the HPV infection status and Histology of colposcopy-directed biopsy samples, including normal controls (normal), HR-HPV infection (HPV), low-grade squamous intraepithelial lesion (LSIL), high-grade squamous intraepithelial lesion (HSIL) and cervical cancer (cancer). HPV status was determined by the Linear Array HPV Genotyping Tests (Roche, Indianapolis, IN), and divided into two groups: HPV-positive (including HPV 16/18 genotypes) and HPV-negative controls (normal).
This study was approved by the Ethics Committee of International Peace Maternity and Child Health Hospital (Approval number: GKLW2017-139). Vaginal swabs were collected by clinicians according to the clinical standards, and patient information and experiments were performed according to approval standards. All samples were sent to the laboratory immediately after collection and saved at −80 °C for subsequent DNA extraction.
Bacterial DNA extraction
Total DNA of samples were extracted according to the Fast DNA SPIN extraction kits (MP Biomedicals, Santa Ana, CA) instruction. DNA concentration and purity were detected by NanoDrop2000, and DNA quality was tested using 1% agarose gel electrophoresis. DNA was stored at -20 °C before further analysis.
DNA amplification and sequencing of barcoded 16S RNA gene fragments
PCR amplification of the V3-V4 regions of the bacterial 16S rRNA gene was performed using 338F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) primers. The PCR reagent system contained 2 μL of 2.5 mM dNTPs, 4 μL of 5× Fast Pfu Buffer, 0.4 μL of Fast Pfu Polymerase, 0.8 μL of each primer (5 μM), and 10 ng of template DNA. PCR amplified procedure was as follows: 95 °C for 3 min, followed by 27 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s and a final extension at 72 °C for 10 min (PCR instrument: ABI GeneAmp9700).
PCR products were separated with 2% agarose gel, and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). Quanti Fluor - ST (Promega, USA) was used for quantitative detection. PE 2*300 library was constructed from purified amplified fragments according to Illumina MiSeq platform (Illumina, San Diego, USA) standard operating procedures. Steps for library construction: (1) Connecting “Y” connector; (2) Using magnetic beads to remove self-connecting segments; (3) Enriching library templates by PCR amplification; (4) Producing single stranded DNA fragments by sodium hydroxide denatures. And the Illumina MiSeq PE300 platform (Illumina, San Diego, USA) was used for sequencing.
HPV genotyping
HPV 16/18 genotyping was performed using the Roche Linear Array HPV Genotyping Test (Roche, Basel, Switzerland) according to manufacturer’s guidelines. A clinically validated in vitro polymerase chain reaction (PCR) assay was used to identify HPV-16, -18 and 12 other HR HPV subtypes (31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68).
Sequence analysis
The original sequencing was controlled by Trimmomatic software (Bolger et al., 2014) and spliced with FLASH software (Quan et al., 2019). Sequences with a length of <150 bp and an average Phred score of <20 were filtered out. Sequences that had ambiguous bases were removed, and sequences that had mononucleotide repeats shall be more than 10. Sequences that could be spliced were removed using UPARSE software (version 7.1), according to the similarity of 97% to OTU sequence clustering. Chimeras were removed using UCHIME software. For each sequence, the RDP classifier was used to annotate the species classification, and the comparison was made to the Silva database (SSU123), with a threshold value of 70%.
Statistical analysis
Quantitative insights into microbial ecology (QIIME) software and R packages were used to analyze the sequence information. Bacterial diversity was assessed using the alpha diversity: Shannon and Simpson diversity index. The difference of alpha diversity index between different groups was analyzed using Student’s t-test (p < 0.05) (Huang et al., 2018). The R language vegan package was adopted to perform species segmentation and aggregation, which was represented on the community heat map. In addition, we used unweighted principal component analysis to carry out UniFrac distance measurement by PCA (principal component analysis), and we observed the interaction between bacterial communities in different groups by R packages. Partial Least Squares Discriminant Analysis (PLS-DA) was used to analyze environmental factors affecting microecology. LEfSe (http://huttenhower.sph.harvard.edu/galaxy/) was employed to detect differentially abundant genera between groups for biomarker discovery (Segata et al., 2011). By calculating the Pearson correlation coefficient, the cooccurrence patterns of the 30 most abundant taxa in different groups are discussed. Using data from the Kyoto Encyclopedia of Genes (KEGG) dataset, PICRUSt made predictions about the functional composition of the bacterial community (Langille et al., 2013).
Results
Subjects information
In the current study, 59 eligible women were recruited and classified into five groups: normal controls (normal; n = 10), HR-HPV infection (HPV; n = 13), low-grade squamous intraepithelial lesion (LSIL; n = 15), high-grade squamous intraepithelial lesion (HSIL; n = 10) and cervical cancer (cancer; n = 11) (Table 1 ). In total, 2,866,307 reads were acquired from 59 samples, with an average number of 47,771±12,155 reads per sample. Following removal of rare OTUs and single tons, in this study, a total of 333 taxa were observed in the vaginal microbiota.
Table 1 -. Characteristics of study participants and hrHPV status, P values were calculated using ANOVA for continuous variables.
| n | normal (n = 10) | HPV+ ( n = 13) | LSIL ( n = 15) | HSIL ( n = 10) | Cancer ( n = 11) | P value | |
|---|---|---|---|---|---|---|---|
| age(mean(SD)) | 59 | 38.50(5.68) | 37.08(7.05) | 38.33(7.35) | 40.50(5.95) | 43.45(5.50) | 0.19 |
| HPV status | |||||||
| HPV 16 positive | 7(53.85) | 5(33.33) | 6(60.00) | 4(36.36) | 0.65 | ||
| HPV 18 positive | 7(53.85) | 2(13.33) | 1(10.00) | 2(18.18) | 0.28 | ||
| other high-risk | 3(23.08) | 8(53.33) | 6(60.00) | 6(54.55) | 0.47 | ||
| Single high-risk | 9(69.23) | 9(60.00) | 7(70.00) | 4(36.36) | 0.79 | ||
| Multiple high-risk | 4(30.77) | 2(13.33) | 3(30.00) | 2(18.18) | 0.79 | ||
Bacterial community characterization
In this study, the compositions of vaginal microbiota in the five groups were investigated (Figure 1A and Table S1). The majority of vaginal bacteria belonged to six major phyla: Firmicutes, Bacteroidetes, Fusobacteria, Actinobacteria, Proteobacteria and Tenericutes. Of these, the number of L. spp. belonging to Firmicutes was the most abundant (78.04%) in all five groups. In addition, the species diversity of vaginal microbiota in the four disease groups (HR-HPV, LISL, HISL and cancer groups) was increased compared with that in normal group. Among them, the species diversity of vaginal microbiota in the cancer group was further increased, and the most significant change at the genus level was the large increase in the relative abundance of low-abundance bacteria or even unknown bacteria, indicating that the community structure of vaginal microbiota was disorganized.
Figure 1-. The comparisons of the five groups in vaginal microbiota. (A) Bar chart describing the difference in bacterial diversity between the normal, HPV, LSIL, HSIL and cancer groups. (B) Simpson index of OTU level and P values calculated using Student`s t-test (P ≤ 0.05 was marked as *; 0.001 < P ≤ 0.01 was marked as **; P ≤ 0.001 was marked as ***). (C) Shannon index of OTU level and P values calculated using Student`s t-test (P ≤ 0.05 was marked as *; 0.001 < P ≤ 0.01 was marked as **; P ≤ 0.001 was marked as ***).
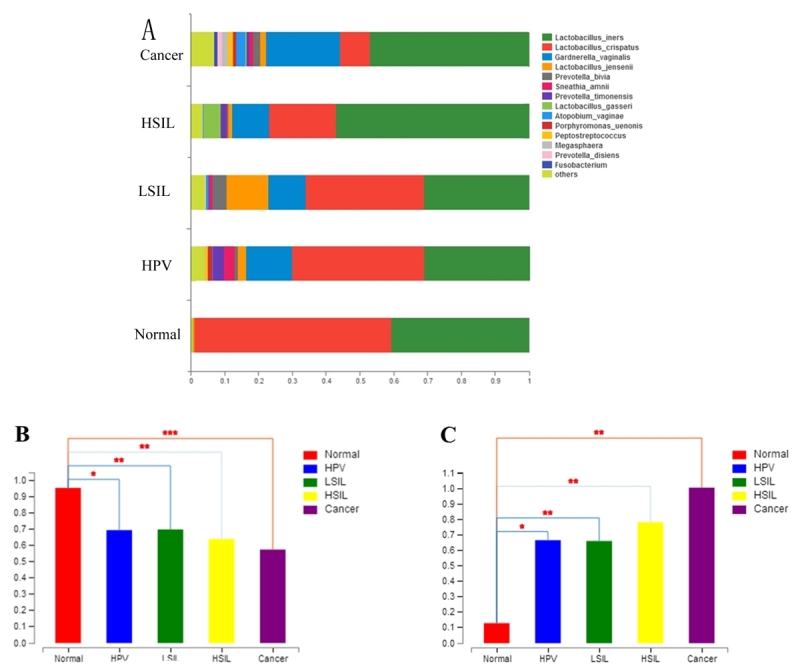
The L. spp. abundance in the HR-HPV infection, LSIL, HSIL and cervical cancer group was significantly decreased compared with the normal group (Figure 1B). In contrast, the abundance of Actinobacteria (mainly G. and A.) in the four disease groups was higher than that in the normal group, and cancer group had the highest abundance (Figure 1C). The increased abundance of Bacteroidetes (mainly Prevotella) and Fusobacteria (mainly Sneathia) was also positively correlated with disease progression, but the increase was not as significant as that of Actinobacteria.
The Simpson diversity index and Shannon diversity index were used to evaluate the α-diversity of the bacterial community. The diversity index calculated by t-test indicated significant differences between the normal control group and disease group; the diversity became more significant with the severity of the disease (HPV-normal p=0.01851; LSIL-normal p=0.003303; HSIL-normal p=0.007463; cancer-normal p= 0.0008941), but there were no statistically significant differences between the disease groups (Figure 1B and Table S2).
Heat map analysis of the sequence data using nearest neighbor linkage at the species level identified four major vaginal microbiota community state types (CSTs) (Figure 2 and Table S3). Among all detected strains, L. spp. was the most abundant microorganism. In the identified L. species, L. iners and L. crispatus were the most abundant (39.7% for L. iners and 32.3% for L. crispatus) in this study. In addition, L. jensenii were identified in 4.3% of subjects, and L. gasseri is was even rarer, with only 0.9%.
Figure 2 -. Heat map analysis, each vertical line represents a sample. Relative abundance is shown in blue and white: blue indicates a high proportion, and white indicates a low proportion. Five groups (normal, HPV, LSIL, HSIL, cancer) were distributed in different CSTs with different colors.
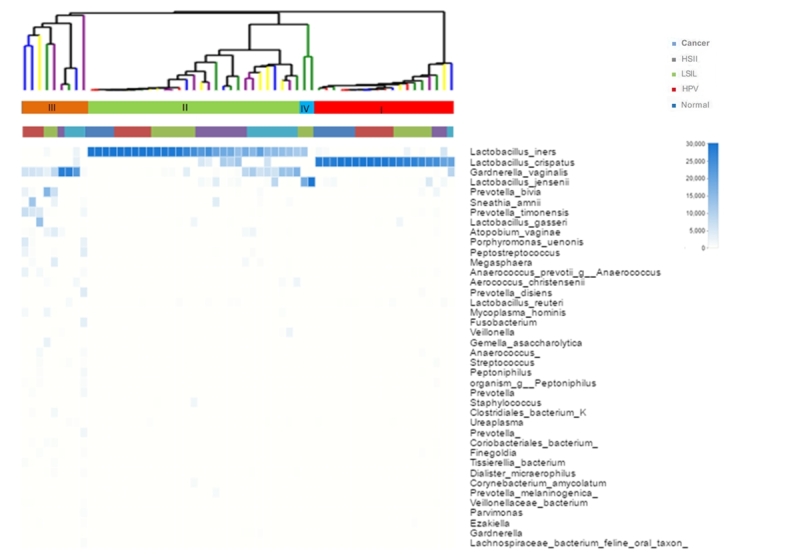
The frequency of CST I (L. crispatus-dominant) decreased with increasing disease severity (normal = 6/10, 60%; HPV =5/13, 38%; LSIL = 4/15, 27%; HSIL = 2/10, 20%; cancer = 1/11, 9%). Conversely, the rate of CST II (L. iners-dominated) was associated with an increase in HSIL and cancer group compared with the normal group (normal = 4/10, 40%; HPV =5/13, 38%; LSIL = 6/15, 40%; HSIL = 7/10, 70%; cancer = 7/11, 64%). None of normal group samples were found in CST III (L.-depleted), and the cancer group accounted for the largest proportion (HPV =3/13, 23%; LSIL = 2/15, 13%; HSIL = 1/10, 10%; cancer = 3/11, 27%). Due to the small sample size, two of the CST IV samples were LSIL, which could not explain the relationship between CST IV and disease.
Principal component analysis (PCA) of species sequence data was used to evaluate the vaginal community structure in the context of disease grade (normal, HPV, LSIL, HSIL and cancer) (Figure 3A). Compared with the results of the heat map analysis, the conclusion was roughly the same; however, due to the low abundance of L. jensenii, the three main clusters were identified, which represented samples dominated by species with higher diversity, and the other two were L. crispatus and L. iners.
Figure 3-. Principal component analysis (PCA) of species sequence data and partial Least Squares Discriminant Analysis (PLS-DA) analysis of the high-dimensional data. (A) Principal component analysis (PCA) showed that the vaginal microbiota identified three major clusters L. crispatus (CST I), L. iners (CST II), and depleted of L. spp. with high diversity (CST III). (B) Partial least square discriminant score plot of vaginal microbiota between the normal, HPV, LSIL, HSIL and cancer groups.

Then, we used Partial Least Squares Discriminant Analysis (PLS-DA) to analysis the high-dimensional data (Figure 3B). The bacterial communities in the normal, HPV, LSIL, HSIL and cancer groups clustered separately, which indicates that the vaginal microbiota composition of the serious progression of cervical cancer and healthy people is significantly different. The normal group and HPV infection group were in the second and third quadrants, respectively. The LSIL and HSIL groups were close to each other, both of which were in the first quadrant and partly intersect, indicating a high correlation between the vaginal microbiota of the two disease stages. The cancer group was located in the fourth quadrant, and it showed the maximum distance from the normal group, indicating a more significant difference between the two groups. In addition, it can be seen from the discrete distribution of sample points in the PLS-DA analysis that the vaginal microbiota composition of different patients varies greatly.
Identification of vaginal microbiota composition markers
The linear discriminant analysis (LDA) effect size (LEfSe) model was used to identify the differences in microbiota composition, which may be associated with increased disease severity. At the genus level, three taxa exhibited significantly higher abundances in the HPV group than in the normal group, including D., Sneathia, and Senegalimassilia. The Acetobacter taxa were remarkably prevalent in the controls (Figure 4A).
Figure 4 -. Linear discriminate analysis effect size (LEfSe) was performed between different disease stages. (A) According to HPV infection, LEfSe was used to detect the difference in relative abundance of microbiota. (B) According to cervical intraepithelial neoplasia status, LEfSe was used to detect difference in microbiota relative abundance. Only taxa with LDA scores of >2 are presented.
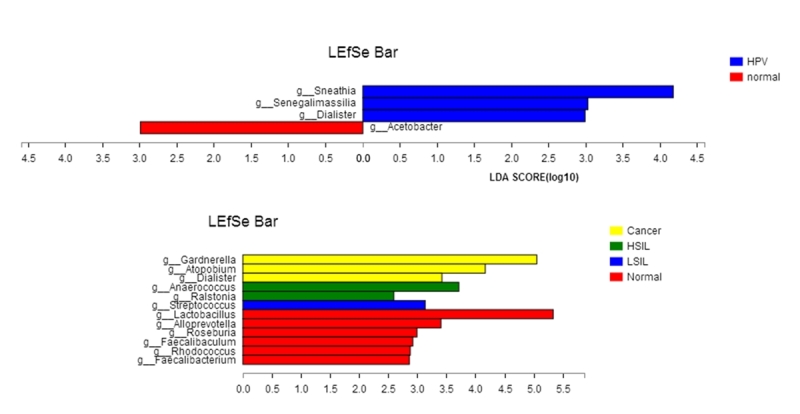
There were significant differences in the community compositions between LSIL, HSIL, and cervical cancer groups at the genus level. There were six significant genera in the normal group: Faecalibaculum, Rhodococcus, Roseburia, L., Faecalibacterium (p<0.01) and Alloprevotella (p<0.04). In the LSIL group, Streptococcus was significantly overrepresented, and in the HSIL group, the highest abundance was Ralstonia (p<0.01) and Anaerococcus (p<0.03). G. (p<0.01), A. and D. (p<0.03) were the predominant genera in the cancer groups. These bacteria with significant differences between groups can be suggested as potential biomarkers for disease diagnosis (LDA> 2, p<0.05) (Figure 4B).
Microbiota functional predictions in cancer and normal groups
PICRUSt was used to infer the bacterial functional metagenomic content of each sample and to classify the inferred genes into the level 2 KEGG pathways. The vaginal microbiota of the normal group was characterized by signal transduction, over representation of membrane transport, excretory system, genetic information translation and processing, all of which may be related to the normal physiological metabolism of bacteria and cells. In contrast, compared with the normal group, the cancer group showed highlighted metabolic diseases, endocrine system, immune system, cellular processes, cell growth and death, cell motility, transport and catabolism as well as “folding, sorting and degradation” (Figure 5).
Figure 5-. PICRUSt infers the cellular functions of bacterial communities in cervical cancer.

Discussion
HR-HPV infection is a recognized risk factor for squamous intraepithelial lesions (SILs) and cervical cancer occurrence. In addition, there are many other factors contributing to cervical cancer development. In this study, we compared the structure and diversity of vaginal microbiota in normal, HR-HPV infection, LSIL, HSIL and cervical cancer groups, and analyzed the differences in bacterial composition and gene function during the development of cervical cancer.
We found that the species diversity and community structure of vaginal microbiota were different among normal, HR-HPV groups, and groups with different grades of cervical lesions. With the occurrence of HR-HPV infection and the development of cervical lesions, the diversity of vaginal microbiota species was increased, and the community structure became more complex and disordered. In addition, the relative abundance of L. spp., the dominant bacteria in maintaining vaginal microecological balance, was decreased gradually with the occurrence of HR-HPV infection and the development of cervical lesions. Among them, the change was most pronounced in the cancer group. In contrast, the abundance of Actinobacteria (mainly G. and A.) in the four disease groups was significantly higher than that in normal group. Among them, the change was also most pronounced in the cancer group. With the development of cervical lesions, the relative abundance of low-abundance bacteria and even unknown bacteria in vaginal microbiota was increased, indicating that the imbalance of vaginal microflora became more and more serious.
The vaginal microbiota in healthy women of reproductive age is dominated by L., which is known to produce lactic acid to maintain low vaginal pH (< 4.5), and this is the first line of defense against pathogenic agents (Boskey et al., 2001). In this study, L. iners and L. crispatus were the most abundant vaginal microbial communities. L. jensenii was the third abundant communitiy in L. spp. While L. gasseri content was only 0.9%. This phenomenon of only a small amount of L. jensenii and even less or no L. gasseri has also been found in Asian women in other studies (Lee et al., 2013; Huang et al., 2018). The characteristic seems to be unique to Asian women, as it differs from the vaginal microbial communities in European and American women that the content of L. gasseri is higher than that of L. jensenii (Brotman et al., 2014). These differences may be related to racial and regional differences, such as genetic and living conditions, innate and adaptive immune systems, ligands on epithelial cell surfaces, as well as the composition and quantity of vaginal secretions (Ravel et al., 2011; Anahtar et al., 2015; Dareng et al., 2016).
This study further found that the distribution of CSTs in normal group was different from the other disease groups. The abundance of L. crispatus was decreased in disease groups compared with normal group. While the frequency of CST III (L.-deficient) was increased during the disease progression. In this study, L. spp. in the HSIL group was slightly higher than that in the LSIL group, but the proportion of L. iners in L. spp. in the HSIL group was significantly higher than that in the LSIL group. The L. spp. content in the cancer group was reduced, while the proportion of L. iners continued to be increased, accounting for 80.44% of the L. spp.. The role of L. iners in the healthy cervix is unclear, since it was detected both in normal and disease groups (Petrova et al., 2017). L. iners is more common than L. crispatus in patients with vaginal dysbiosis caused by HSV-2 and HIV infection (Borgdorff et al., 2014). Some studies have shown that there are two isoforms of lactic acid, D-lactic acid and L-lactic, but they act differently in microbial. D-lactic acid level has been inversely associated with the ability of HIV to transverse cervicovaginal mucus. L. crispatus produces both the two isoforms, whereas L. iners produces only L-lactic acid (Nunn et al., 2015; Norenhag et al., 2020). Most L. spp. can produce H2O2 with antibacterial properties, except L. iners (Aroutcheva et al., 2001; Reimers et al., 2016), indicating that L. iners may be related with disease. Our results are consistent with these findings. The rate of CST II (L. iners-dominated) was increased in HSIL and cancer group compared with that in normal group. L. iners may also related to the serious progression of cervical cancer. In addition, the analysis of PCA and PLS-DA further confirmed the significant difference between the vaginal microbiota in disease group and the normal group. As a result, vaginal bacterial dysbiosis may have existed in the development of cervical cancer.
Based on LEfSe analysis, some potential biomarkers related to different disease stages were found. D., Sneathia and Senegalimassilia were significantly overrepresented in HPV group. Streptococcus were the most representative in LSIL. Whereas Anaerococcus and Ralstonia were significantly enriched in HSIL samples. Three other BV-associated bacteria, G., D. and A., were also observed to be markers of cancer. Previous studies have identified Sneathia spp. as a microbiological marker for HPV infection, cervical neoplasm and increased HIV acquisition (Di Paola et al., 2017; Łaniewski et al., 2018; McClelland et al., 2018). In our study, we further analyzed the vaginal microbiota of HR-HPV infection, SIL and cervical cancer, and found that Sneathia spp. was more abundant in HR-HPV infection women than in patients with cervical cancer. To this extent, we confirmed that cervical disease was correlated with distinct bacterial combinations, which also changed with disease status. In addition, these specific bacteria were present in HR-HPV infection and CIN severity. We can assume that they contribute an important coordinating role in clearing HR-HPV infection or facilitating the transition from normal cytology to cervical squamous intraepithelial lesion status.
Based on the 16S rRNA sequence information, PICRUSt was used to infer the function of the bacterial community, and further analyzed the differences between normal control and cervical cancer groups. The results showed that changes in bacterial abundance and composition could lead to significant changes in gene functional expression, especially in metabolic diseases, endocrine system, and immune system, which might be the factors of the development of cervical lesions.
A major strength of our study is that we have analyzed the vaginal microbiota of Chinese women from HR-HPV infection to precancerous lesions and further to cervical cancer. There are still some limitations, including a relatively modest sample size and uncollected pH data. In summary, our work provides evidences for elucidating the complex relationship between vaginal microbiota composition, HR-HPV infection and cervical intraepithelial neoplasia. We also identified bacterial biomarkers to distinguish HR-HPV infection, LSIL, HSIL and cervical cancer. This may improve our understanding of the role of the bacterial microenvironment in HPV persistence, CIN development and cancer progression. The relationship between vaginal microbiota and functional prediction may provide insights into the pathogenesis of cervical cancer. These specific bacteria will improve women’s health status while helping to improve the early detection and prevention of cervical precancerous lesions. In future studies, we will further study the relationship between vaginal bacteria and the tumor microenvironment.
Acknowledgements
We thank members of our laboratory for helpful discussion.
Supplementary material.
The following online material is available for this article:
References
- Al-Nasiry S, Ambrosino E, Schlaepfer M, Morré SA, Wieten L, Voncken JW, Spinelli M, Mueller M, Kramer BW. The interplay between reproductive tract microbiota and immunological system in human reproduction. 378Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anahtar MN, Byrne EH, Doherty KE, Bowman BA, Yamamoto HS, Soumillon M, Padavattan N, Ismail N, Moodley A, Sabatini ME, et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 2015;42:965–976. doi: 10.1016/j.immuni.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroutcheva A, Gariti D, Simon M, Shott S, Faro J, Simoes JA, Gurguis A, Faro S. Defense factors of vaginal lactobacilli. Am J Obstet Gynecol. 2001;185:375–379. doi: 10.1067/mob.2001.115867. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Tian T, Wei Z, Shih N, Feldman MD, Peck KN, DeMichele AM, Alwine JC, Robertson ES. Distinct microbial signatures associated with different breast cancer types. 951Front Microbiol. 2018;9 doi: 10.3389/fmicb.2018.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Marc L, Bjoern U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgdorff H, Tsivtsivadze E, Verhelst R, Marzorati M, Jurriaans S, Ndayisaba GF, Schuren FH, van de Wijgert JHHM. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014;8:1781–1793. doi: 10.1038/ismej.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskey ER, Cone RA, Whaley KJ, Moench TR. Origins of vaginal acidity: High D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod. 2001;16:1809–1813. doi: 10.1093/humrep/16.9.1809. [DOI] [PubMed] [Google Scholar]
- Brotman RM, Shardell MD, Gajer P, Tracy JK, Zenilman JM, Ravel J, Gravitt PE. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J Infect Dis. 2014;210:1723–1733. doi: 10.1093/infdis/jiu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MA, Rodriguez AC, Gage JC, Herrero R, Hildesheim A, Wacholder S, Burk R, Schiffman M. A large, population-based study of age-related associations between vaginal pH and human papillomavirus infection. 33BMC Infect Dis. 2012;12 doi: 10.1186/1471-2334-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dareng EO, Ma B, Famooto AO, Adebamowo SN, Offiong RA, Olaniyan O, Dakum PS, Wheeler CM, Fadrosh D, Yang H, et al. Prevalent high-risk HPV infection and vaginal microbiota in nigerian women. Epidemiol Infect. 2016;144:123–137. doi: 10.1017/S0950268815000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola M, Sani C, Clemente AM, Iossa A, Perissi E, Castronovo G, Tanturli M, Rivero D, Cozzolino F, Cavalieri D, et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk human papillomavirus infection. Sci Rep. 2017;7:10200. doi: 10.1038/s41598-017-09842-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg O, Bray F, Coleman MP, Vanderpuye V, Eniu A, Kotha SR, Sarker M, Huong TT, Allemani C, Dvaladze A, et al. The global burden of women’s cancers: A grand challenge in global health. Lancet. 2017;389:847–860. doi: 10.1016/S0140-6736(16)31392-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer RL, Dong X, Moraes ACF, Zielke RA, Fernandes GR, Peremyslova E, Vasquez-Perez S, Schoenborn AA, Gomes EP, Pereira AC, et al. Akkermansia muciniphila mediates negative effects of IFNgamma on glucose metabolism. 13329Nat Commun. 2016;7 doi: 10.1038/ncomms13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Li C, Li F, Zhao J, Wan X, Wang K. Cervicovaginal microbiota composition correlates with the acquisition of high-risk human papillomavirus types. Int J Cancer. 2018;143:621–634. doi: 10.1002/ijc.31342. [DOI] [PubMed] [Google Scholar]
- Insinga RP, Perez G, Wheeler CM, Koutsky LA, Garland SM, Leodolter S, Joura EA, Ferris DG, Steben M, Hernandez-Avila M, et al. Incident cervical HPV infections in young women: Transition probabilities for CIN and infection clearance. Cancer Epidemiol Biomarkers Prev. 2011;20:287–296. doi: 10.1158/1055-9965.EPI-10-0791. [DOI] [PubMed] [Google Scholar]
- Kyrgiou M, Mitra A, Moscicki A-B. Does the vaginal microbiota play a role in the development of cervical cancer? Transl Res. 2017;179:168–182. doi: 10.1016/j.trsl.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łaniewski P, Barnes D, Goulder A, Cui H, Roe DJ, Chase DM, Herbst-Kralovetz MM. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. 7593Sci Rep. 2018;8 doi: 10.1038/s41598-018-25879-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Lee S, Lee H, Song Y-M, Lee K, Han MJ, Sung J, Ko G. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One. 2013;8:e63514. doi: 10.1371/journal.pone.0063514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland RS, Lingappa JR, Srinivasan S, Kinuthia J, John-Stewart GC, Jaoko W, Richardson BA, Yuhas K, Fiedler TL, Mandaliya KN, et al. Evaluation of the association between the concentrations of key vaginal bacteria and the increased risk of HIV acquisition in African women from five cohorts: A nested case-control study. Lancet Infect Dis. 2018;18:554–564. doi: 10.1016/S1473-3099(18)30058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee KS, Carter KA, Bassis C, Young V, Reed B, Harper D, Ruffin MT, Bell J. The vaginal microbiota, high-risk human papillomavirus infection, and cervical intraepithelial neoplasia: results from a population-based study. Am J Obstet Gynecol. 2019;221:668–668. doi: 10.21037/gpm-20-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon LR, Achilles SL, Bradshaw CS, Burgener A, Crucitti T, Fredricks DN, Jaspan HB, Kaul R, Kaushic C, Klatt N, et al. The evolving facets of bacterial vaginosis: Implications for HIV transmission. AIDS Res Hum Retroviruses. 2019;35:219–228. doi: 10.1089/aid.2018.0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan A, Dell M, Zellar MP, Cribby S, Martz S, Hong E, Fu J, Abbas A, Dang T, Miller W, et al. Disruption of urogenital biofilms by lactobacilli. Colloids Surf B Biointerfaces. 2011;86:58–64. doi: 10.1016/j.colsurfb.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Mitra A, MacIntyre DA, Lee YS, Smith A, Marchesi JR, Lehne B, Bhatia R, Lyons D, Paraskevaidis E, Li JV, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep. 2015;5:16865. doi: 10.1038/srep16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ, Group International Agency for Research on Cancer Multicenter Cervical Cancer Study. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Norenhag J, Du J, Olovsson M, Verstraelen H, Engstrand L, Brusselaers N. The vaginal microbiota, human papillomavirus and cervical dysplasia: A systematic review and network meta-analysis. BJOG. 2020;127:171–180. doi: 10.1111/1471-0528.15854. [DOI] [PubMed] [Google Scholar]
- Noyes N, Cho K-C, Ravel J, Forney LJ, Abdo Z. Associations between sexual habits, menstrual hygiene practices, demographics and the vaginal microbiome as revealed by Bayesian network analysis. Plos One. 2018;13:e0191625. doi: 10.1371/journal.pone.0191625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn KL, Wang Y-Y, Harit D, Humphrys MS, Ma B, Cone R, Ravel J, Lai SK. Enhanced trapping of HIV-1 by human cervicovaginal mucus is associated with L. crispatus -Dominant Microbiota. Mbio. 2015;6:e01084-15. doi: 10.1128/mBio.01084-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova MI, Reid G, Vaneechoutte M, Lebeer S. Lactobacillus iners: Friend or foe? Trends Microbiol. 2017;25:182–191. doi: 10.1016/j.tim.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Porter CM, Shrestha E, Peiffer LB, Sfanos KS. The microbiome in prostate inflammation and prostate cancer. Prostate Cancer Prostatic Dis. 2018;21:345–354. doi: 10.1038/s41391-018-0041-1. [DOI] [PubMed] [Google Scholar]
- Quan J, Langelier C, Kuchta A, Batson J, Teyssier N, Lyden A, Caldera S, McGeever A, Dimitrov B, King R, et al. FLASH: A next-generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Res. 2019;47:e83. doi: 10.1093/nar/gkz418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Proc Natl Acad Sci. Vol. 108. U S A: 2011. Vaginal microbiome of reproductive-age women; pp. 4680–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers LL, Mehta SD, Stewart ML, Burk RD, Xie X, Ravel J, Cohen MH, Palefsky JM, Weber KM, Xue XJ, et al. The cervicovaginal microbiota and its associations with human papillomavirus detection in HIV-infected and HIV-uninfected women. J Infect Dis. 2016;214:1361–1369. doi: 10.1093/infdis/jiw374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, Schoos AM, Kunøe A, Fink NR, Chawes BL, et al. Maturation of the gut microbiome and risk of asthma in childhood. 141Nat Commun. 2018;9 doi: 10.1038/s41467-017-02573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend HJ, Bogaards JA, van Bergen JE, Brink AA, van den Broek IV, Hoebe CJ, King AJ, van der Sande MA, Wolffs PF, de Melker HE, et al. Incidence and persistence of carcinogenic genital human papillomavirus infections in young women with or without Chlamydia trachomatis co-infection. Cancer Med. 2015;4:1589–1598. doi: 10.1002/cam4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.