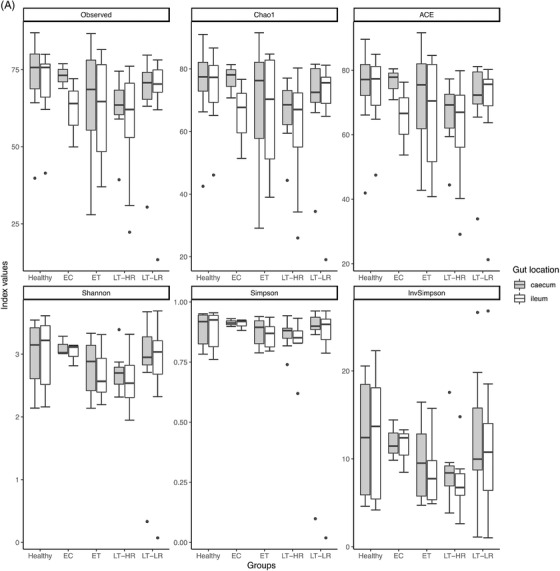
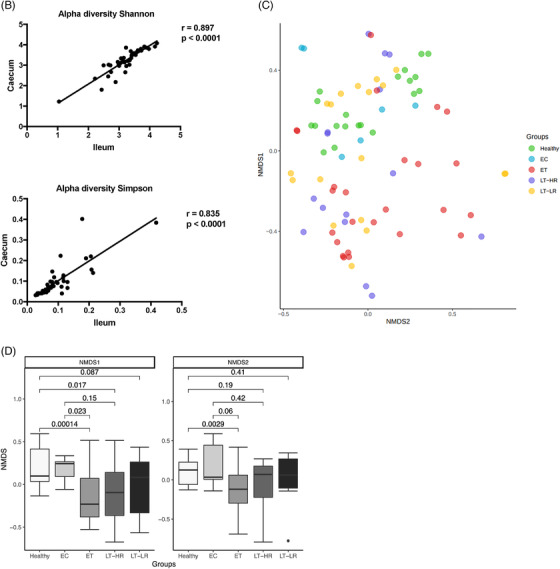
FIGURE 2.
Richness and diversity measures of the microbiota obtained from gut mucosal samples of subjects included in the study groups. Sequencing data of microbial 16S rRNA were processed with Mothur (version 1.43.0), using SILVA (non‐redundant version 138) and Greengenes (version 13_8_99) databases for aligning and taxonomic purposes, respectively. Operational taxonomic units (OTUs) picking was performed at 97% sequence similarity to detect subgenera. Subsequent data analysis was accomplished using the Phyloseq package (R version 3.6.3). Only bacterial taxa present at least in 50% of the analysed samples were considered. (A) Different richness (Chao1 and ACE) and alpha diversity (Shannon and Simpson) indexes were calculated using Phyloseq and Vegan packages from R, and compared among study groups according to the location of sampling area for biopsies, caecum or ileum mucosa (dots represent outliers). (B) Spearman's Rho Coefficient was used to correlate Shannon and Simpson alpha diversity values between caecum and ileum samples of all the groups studied. (C) Non‐metric multidimensional scaling (NMDS) analysis for beta microbiome diversity of ileum and caecum samples from study groups’ subjects, choosing Morisita‐Horn index as it yielded the lowest stress value. Coloured dots represent samples from the different groups according to the legend on the right. (D) NMDS analysis separated by axes, NMDS1, and 2, showing distances among samples of the five study groups (dots represent outliers). Kruskal–Wallis rank sum tests, followed by post hoc Tukey's procedure, were used to test for differences in alpha diversity measures and NMDS axes among study groups. Differences in beta diversity were tested using PERMANOVA and HOMOVA procedures. p < .05 indicates a statistically significant association. EC, elite controllers; ET, early‐treated; LT‐HR, late‐treated high recovery; LT‐LR, late‐treated low recovery; r, Spearman's rho