

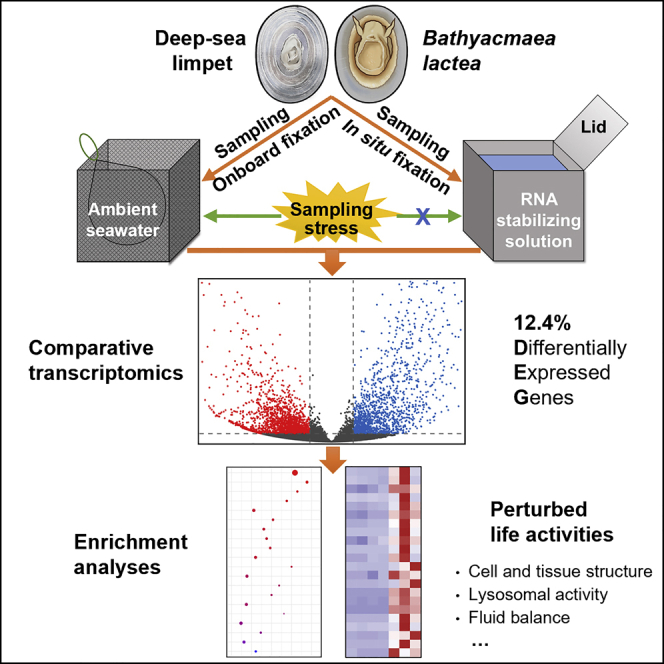
Summary
Precise gene expression reflects the molecular response of deep-sea organisms to their harsh living environments. However, changes in environmental factors during lifting samples from the deep sea to a research vessel can also affect gene expression. By using the transcriptomic approach, we compared the gene expression profiles of the onboard fixed with the in situ fixed samples of the deep-sea limpet Bathyacmaea lactea. Our results revealed that the concomitant stress during conventional deep-sea sampling without RNA in situ fixation greatly influenced the gene expression. Various biological activities, such as cell and tissue structure, lysosomal activity, fluid balance, and unsaturated fatty acid metabolism, were perturbed, suggesting that the sampling stress has exerted systemic impacts on the life of the limpets. These findings clearly illustrate that deep-sea samples without RNA in situ fixation can easily lead to biased results in gene expression analysis, which requires to be appropriately addressed in future studies.
Subject areas: Biotehnology, marine biotechnology, omics, Transcriptomics
Graphical abstract
Highlights
-
•
Deep-sea sampling without RNA in situ fixation influenced the gene expression greatly
-
•
Concomitant sampling stress perturbed various life activities of deep-sea limpets
-
•
Providing a high-quality in situ transcriptome of limpet Bathyacmaea lactea
Biotehnology; Marine biotechnology; Omics; Transcriptomics
Introduction
Approximately 88.3% of the ocean is deeper than 1000 m, commonly known as the deep sea (Weatherall et al., 2015), which is characterized by high hydrostatic pressure, low temperature (except for the hydrothermal vents), deficient food supply, and total darkness, and has therefore been considered hostile to life (Rex and Etter, 2010). However, both microorganisms and macroorganisms have been discovered in the deep sea, which is actually the largest habitat on Earth (Ramirez-Llodra et al., 2010). The deep sea supports various ecosystems with high biodiversity and biomass, such as hydrothermal vents, cold seeps, and whale falls (Kiel, 2016). Deep-sea organisms must have evolved various adaptive strategies to survive and thrive under such harsh environmental conditions. High-quality samples and free access to their genetic information are necessary to obtain a comprehensive and in-depth knowledge of the molecular mechanisms underlying deep-sea adaptation. Nevertheless, deep-sea sampling has always been quite technically challenging because of the great depth and far distance from land.
Owing to the rapid development of deep-sea sampling tools and techniques during the past decades (Clark et al., 2016), obtaining samples from deep-sea ecosystems has become easier. Furthermore, many studies have been conducted to obtain the genomic information of deep-sea organisms and explore the deep-sea adaptation mechanisms through genome, transcriptome, and proteome sequencing and analyses (Lan et al., 2018; Sun et al., 2017; Wang et al., 2019a; Yang et al., 2020). However, most of these studies were based on deep-sea samples obtained by conventional deep-sea sampling method, that is, the samples were fixed on the board of a research vessel after retrieval from the seafloor. For organisms that have adapted well to the deep sea, the harsh local environment has been optimum for them. Sampling these organisms from the seafloor to the surface might cause inevitable stress because of the changes in the ambient environment. Hence, conventionally obtained faunal samples can hardly reflect their natural physiological and biochemical status as they were inhabiting the deep sea. It is worth mentioning that some researchers realized this issue and tried to obtain in situ fixed deep-sea samples and advocated for the utilization of in situ fixed deep-sea samples in their studies (Chen et al., 2021; Gao et al., 2019; Mat et al., 2020; Motoki et al., 2020; Sanders et al., 2013; Sun et al., 2020; Wang et al., 2019b; Watsuji et al., 2014; Wei et al., 2020). Nevertheless, studies that explicitly compare the biological difference between in situ fixed and conventional sampled deep-sea fauna are still lacking.
The cold seep is a typical chemosynthetic ecosystem in the deep sea, where reduced sulfur compounds and methane emanate from the seafloor to the water (Feng et al., 2018; Levin, 2005). In addition to the general deep-sea environmental stressors mentioned earlier, the organisms inhabiting cold seeps also need to cope with extra stressors, such as low oxygen and potentially harmful substances in the seepage fluid (McMullin et al., 2000). The deep-sea patellogastropod limpet Bathyacmaea lactea is one of the dominant macrobenthos in the Haima cold seep in the South China Sea (Liu et al., 2020), and no evidence of harboring endosymbionts in B. lactea has been reported until now. Given its small size and open-shell structure, B. lactea is relatively easier to be in situ fixed during deep-sea sampling than the others because it does not need to be cracked. In this study, two methods were used for sampling this limpet species (Figure 1): the conventional onboard fixation method in which samples are fixed on board by RNA stabilizing solution after retrieval and the in situ fixation method in which the samples are fixed on the seafloor before suffering sampling stress. Further comparative transcriptomic analysis was performed on the limpet samples fixed with different methods to investigate the influences of deep-sea sampling stress on their gene expression at the transcriptional level.
Figure 1.

Pictures showing the two sampling methods applied in this study
(A) Schematic of the two sampling methods.
(B) An on-site photograph of the tuck net filled with the deep-sea limpet Bathyacmaea lactea attached to the mussel shells (indicated with red arrows). Inset: dorsal view of a B. lactea specimen. Scale bar, 0.5 cm.
(C) An on-site photograph of the in situ fixation. Bathyacmaea lactea attached on mussel shells (indicated with red arrows) were fallen off and in situ fixed in the sampling chamber (indicated with a white dotted box) fully filled with in-house RNA stabilizing solution.
Results and discussion
Transcriptome sequencing, assembly, and annotation
A total of 27,674 nonredundant transcripts with predicted open-reading frames were obtained by using 648,458,172 reads from seven individuals of B. lactea for transcriptome assembly, with an N50 value of 3610 bp (Table S1). The Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment results show 98% completeness (single copy: 84.1% and duplicated: 13.9%) for the predicted transcripts. The functional annotation of the transcripts indicated that 23,327 transcripts had hits to the NCBI nonredundant (NR) database; 17,160 to the EggNOG database; 16,462 to Gene Ontology (GO) items; and 8424 to the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Figure S1). To the best of our knowledge, this is the first report on the in situ transcriptome of B. lactea.
Differential gene expression analysis between the in situ and onboard fixed samples
The expression level of each transcript was quantified in seven individuals. The principal component analysis (PCA) result shows a clear separation between the in situ fixed and onboard fixed groups along PC1, explaining the 48.2% variance (Figure 2A). Although variability also exists among replicates of the same group, we consider it acceptable, as these samples grew in a volatile wild environment that might result in individual differences. Moreover, replicates among the onboard fixed group showed more variability than that of in situ fixed group (Figure 2A), possibly because of having suffered the sampling stress.
Figure 2.

Comparative transcriptomic analysis of the in situ and onboard fixed groups
(A) Principal component analysis (PCA) result based on gene expression levels of four in situ and three onboard fixed individuals. Significance of PCA was examined by the PERMANOVA analysis on Bray-Curtis dissimilarities (p value = 0.027).
(B) A volcano plot showing the relationship between false discovery rate (FDR) and fold change (FC). Red spots indicate upregulated transcripts in the onboard fixed group, and blue spots indicate downregulated transcripts in the onboard fixed group.
Differential gene expression analysis was performed between the two groups to investigate the impacts of sampling stress during retrieval from the seafloor on the gene expression of cold seep-adapted limpets. A total of 3,436 (12.4%) differentially expressed genes (DEGs) were identified, with 1,858 upregulated and 1,578 downregulated in the onboard fixed limpets compared with in situ fixed ones (Figure 2B). This result indicated that sampling stress indeed led to dramatic changes in the gene expression of the deep-sea limpets during the ∼80 min sampling process. During the conventional deep-sea sampling process, the samples were exposed to the ambient environment during retrieval from the seafloor to the surface. As a result, the samples suffered from decreased hydrostatic pressure, increased water temperature, increased dissolved oxygen, and decreased salinity (Figure S2). Other environmental parameters, such as hydrogen sulfide, metal ion, and pH, might also have changed. In a recent study conducted under laboratory conditions, few (˂ 0.1%) DEGs were identified in the shallow-water sea cucumber Apostichopus japonicus after the hydrostatic pressure changed in the first hour (Chen et al., 2020), and in another study conducted in a cold seep, approximately 40 min of decompression from the 1,119 m depth resulted in 337 DEGs in the gill of the deep-sea mussel Gigantidas platifrons (formerly "Bathymodiolus" platifrons) (Chen et al., 2021). Therefore, we speculate that the large number of DEGs identified in this study might be induced by combined factors rather than a single factor during a sampling process of only ∼80 min.
Effect of the sampling stress caused differential gene expression on the deep-sea limpets
GO and KEGG enrichment analyses were performed on the identified DEGs to discover the biological activities and metabolic pathways influenced by the DEGs induced by sampling stress. GO enrichment analysis of the upregulated DEGs in the onboard fixed group identified 24 significantly enriched GO terms, including structural molecule activity (GO:0005198), transmembrane transporter activity (GO:0022857), ribosome (GO:0005840), translation (GO:0006412), cellular biosynthetic process (GO:0044249), and plasma membrane (GO:0005886) (Table 1). GO enrichment analysis of the downregulated DEGs in the onboard fixed group resulted in 27 GO terms, including catalytic activity (GO:0003824), hydrolase activity (GO:0016787), peptidase activity (GO:0008233), small molecule metabolic process (GO:0044281), lipid metabolic process (GO:0006629), and lysosome (GO:0005764) (Table 2). KEGG enrichment analysis of all the DEGs identified 14 significantly enriched pathways, which were involved in transport and catabolism (lysosome [ko04142]); glycan biosynthesis and metabolism (glycosaminoglycan degradation [ko00531], other glycan degradation [ko00511]); carbohydrate metabolism (pentose and glucuronate interconversions [ko00040], amino sugar and nucleotide sugar metabolism [ko00520]); amino acid metabolism (histidine metabolism [ko00340], glycine, serine, and threonine metabolism [ko00260]); digestive system (protein digestion and absorption [ko04974]); cell growth and death (ferroptosis [ko04216]); lipid metabolism (biosynthesis of unsaturated fatty acids [ko01040]); endocrine system (PPAR signaling pathway [ko03320], renin-angiotensin system [ko04614]); and metabolism of cofactors and vitamins (one carbon pool by folate [ko00670], retinol metabolism [ko00830]) (Figure 3). Many affected biological functions and metabolic pathways were identified, suggesting that the sampling stress during retrieval from the seafloor exerted a systemic influence on the vital activity of the sampled deep-sea limpets.
Table 1.
GO enrichment analysis of the upregulated transcripts in the onboard fixed group
| GO ID | GO description | Category | FDR |
|---|---|---|---|
| GO:0005198 | Structural molecule activity | Molecular Function | 7.86E-7 |
| GO:0003735 | Structural constituent of ribosome | Molecular Function | 7.53E-4 |
| GO:0022857 | Transmembrane transporter activity | Molecular Function | 7.69E-4 |
| GO:0005215 | Transporter activity | Molecular Function | 7.69E-4 |
| GO:0005840 | Ribosome | Cellular Component | 7.69E-4 |
| GO:0032501 | Multicellular organismal process | Biological Process | 4.96E-3 |
| GO:0006518 | Peptide metabolic process | Biological Process | 6.07E-3 |
| GO:0006412 | Translation | Biological Process | 6.07E-3 |
| GO:1901566 | Organonitrogen compound biosynthetic process | Biological Process | 6.07E-3 |
| GO:0034645 | Cellular macromolecule biosynthetic process | Biological Process | 6.07E-3 |
| GO:1901576 | Organic substance biosynthetic process | Biological Process | 6.07E-3 |
| GO:0044249 | Cellular biosynthetic process | Biological Process | 6.07E-3 |
| GO:0043043 | Peptide biosynthetic process | Biological Process | 6.07E-3 |
| GO:0043604 | Amide biosynthetic process | Biological Process | 6.07E-3 |
| GO:0043603 | Cellular amide metabolic process | Biological Process | 6.07E-3 |
| GO:0009059 | Macromolecule biosynthetic process | Biological Process | 6.07E-3 |
| GO:0044271 | Cellular nitrogen compound biosynthetic process | Biological Process | 6.07E-3 |
| GO:0043228 | Nonmembrane-bounded organelle | Cellular Component | 9.89E-3 |
| GO:0043232 | Intracellular nonmembrane-bounded organelle | Cellular Component | 9.89E-3 |
| GO:0005576 | Extracellular region | Cellular Component | 9.89E-3 |
| GO:0050877 | Nervous system process | Biological Process | 1.73E-2 |
| GO:0003008 | System process | Biological Process | 1.98E-2 |
| GO:0005886 | Plasma membrane | Cellular Component | 4.58E-2 |
| GO:0016020 | Membrane | Cellular Component | 4.58E-2 |
Table 2.
GO enrichment analysis of downregulated transcripts in the onboard fixed group
| GO ID | GO description | Category | FDR |
|---|---|---|---|
| GO:0003824 | Catalytic activity | Molecular Function | 1.47E-34 |
| GO:0016798 | Hydrolase activity, acting on glycosyl bonds | Molecular Function | 5.55E-24 |
| GO:0016491 | Oxidoreductase activity | Molecular Function | 4.48E-23 |
| GO:0016787 | Hydrolase activity | Molecular Function | 2.04E-21 |
| GO:0005975 | Carbohydrate metabolic process | Biological Process | 6.83E-15 |
| GO:0008233 | Peptidase activity | Molecular Function | 1.23E-12 |
| GO:0140096 | Catalytic activity, acting on a protein | Molecular Function | 1.23E-12 |
| GO:0044281 | Small molecule metabolic process | Biological Process | 1.13E-11 |
| GO:0006082 | Organic acid metabolic process | Biological Process | 5.24E-10 |
| GO:0006520 | Cellular amino acid metabolic process | Biological Process | 5.24E-10 |
| GO:0043436 | Oxoacid metabolic process | Biological Process | 5.24E-10 |
| GO:0005773 | Vacuole | Cellular Component | 5.24E-10 |
| GO:0019752 | Carboxylic acid metabolic process | Biological Process | 5.24E-10 |
| GO:0005576 | Extracellular region | Cellular Component | 1.60E-9 |
| GO:0022857 | Transmembrane transporter activity | Molecular Function | 9.62E-9 |
| GO:0005215 | Transporter activity | Molecular Function | 9.62E-9 |
| GO:0006629 | Lipid metabolic process | Biological Process | 2.85E-8 |
| GO:0005764 | Lysosome | Cellular Component | 1.45E-7 |
| GO:0000323 | Lytic vacuole | Cellular Component | 1.45E-7 |
| GO:0016810 | Hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds | Molecular Function | 4.89E-5 |
| GO:0009056 | Catabolic process | Biological Process | 5.04E-5 |
| GO:0006790 | Sulfur compound metabolic process | Biological Process | 1.24E-4 |
| GO:0005615 | Extracellular space | Cellular Component | 6.00E-4 |
| GO:0016829 | Lyase activity | Molecular Function | 6.23E-3 |
| GO:0071704 | Organic substance metabolic process | Biological Process | 1.26E-2 |
| GO:0044238 | Primary metabolic process | Biological Process | 1.26E-2 |
| GO:0043167 | Ion binding | Molecular Function | 1.26E-2 |
Figure 3.

A bubble diagram showing the enriched pathways obtained in the KEGG enrichment analysis of all the differentially expressed genes between the in situ and onboard fixed groups
Cell and tissue structure
GO enrichment analysis showed that cell structure maintenance, transmembrane transport, and biosynthetic process-related activities were upregulated (Table 1), whereas catabolism- and metabolic-process-related activities were downregulated potentially because of the sampling stress (Table 2). Collagen is the main structural protein in the extracellular matrix of diverse connective tissues, playing important roles in tissue morphogenesis and the maintenance of tissue structural integrity (Gelse et al., 2003). Previous studies uncovered that some collagens are positively selected in the deep-sea fish Aldrovandia affinis (Lan et al., 2018) and the deep-sea alvinocaridid shrimp Shinkaicaris leurokolos (Zhu et al., 2020), suggesting their potential roles in deep-sea adaptation. Herein, 86 collagen-encoding transcripts were identified, with 19 upregulated and 4 downregulated in the onboard fixed group (Table S2). The alteration of the expression levels of such a large number of collagen-encoding transcripts suggests that sampling stress, especially the pressure and temperature variations, will likely have an impact on the cell and tissue structure.
Lysosomal activity and fluid balance
KEGG enrichment analysis indicated that lysosome (ko04142) was the most affected pathway (Figure 3), implying that lysosomal activity was greatly altered in the cell of the limpets that suffered from sampling stress. Lysosome (ko04142) was also significantly enriched in the enrichment analysis of the downregulated DEGs (Figure S3A), suggesting that the lysosomal activity might be downregulated in onboard fixed limpets. In addition, the renin-angiotensin system (ko04614) pathway, which is mainly engaged in osmoregulation, was also significantly enriched (Figure 3), suggesting that the sampling stress might perturb the fluid balance of the deep-sea limpets (Salzet et al., 2001), which would be caused by decompression-associated osmotic pressure change or by dehydration when the remotely operated underwater vehicle (ROV) was retrieved on board (Nobata et al., 2013; Yancey et al., 2014).
Unsaturated fatty acid metabolism
High hydrostatic pressure and low temperature are the key limiting factors in the colonization of deep-sea organisms (Brown and Thatje, 2014). Increased hydrostatic pressure and decreased temperature can reduce the fluidity of biological membranes, which are mainly composed of lipid bilayers and various proteins, leading to their disfunction (Balny et al., 2002; Hazel, 1995; Kato et al., 2002; Marques et al., 2003). Many deep-sea organisms are known to rely on a large proportion of unsaturated fatty acids to cope with high hydrostatic pressure and low-temperature-caused rigidity of the membranes (Parzanini et al., 2018; Van Campenhout et al., 2016; Wang et al., 2019a). Deep-sea vent shrimps exposed to atmospheric pressure exhibit a lower level of unsaturated fatty acids than those kept under natural high pressure (Shillito et al., 2020). When the shallow-water amphipod Eogammarus possjeticus was exposed to high pressure, the expression of fatty acid desaturase and the elongation of the very long-chain fatty acids protein (ELOVL) involved in the production of unsaturated fatty acids increased (Chen et al., 2019b). As the biosynthesis of unsaturated fatty acids (ko01040) pathway regulating the production of unsaturated fatty acids was significantly enriched from all the DEGs (Figure 3), and the fatty acid metabolism (ko01212) pathway was significantly enriched from the downregulated DEGs as well (Figure S3A), the expression of fatty acid desaturase and fatty acid elongation protein was investigated. Four fatty acid desaturase-encoding transcripts and three fatty acid elongation protein-encoding transcripts were downregulated in the onboard fixed group (Figure 4A, Table S3), indicating that the biosynthesis of unsaturated fatty acids in the deep-sea limpets decreased during retrieval, which might have been caused by the decreased hydrostatic pressure along with the increased ambient temperature during the sampling process.
Figure 4.

Heatmaps showing the expression pattern of selected transripts
(A) Downregulated fatty acid desaturase- and fatty acid elongation protein-encoding transcripts. (B) Upregulated toxin and cysteine-rich venom protein-encoding transcripts.
Chemical defense reactions
Many marine animals, such as squids release secretions in defense against predator attacks (Wood et al., 2010), and sea anemones maintain their venom quality and quantity when suffering from environmental stressors (Hoepner et al., 2019). Among all the DEGs identified between the onboard and in situ fixed groups, 14 toxin-encoding transcripts and eight cysteine-rich venom protein-encoding transcripts were significantly upregulated during the retrieval process of deep-sea limpets from the seafloor (Figure 4B, Table S4). We speculated that sampling stress might also trigger their chemical defense reactions, resulting in the increased expression of toxin- and cysteine-rich venom protein-encoding transcripts. These reactions might be mediated by pathways involved in neural signal transduction, such as neuroactive ligand-receptor interaction (ko04080) and calcium signaling pathway (ko04020), which were significantly enriched from the upregulated DEGs (Figure S3B).
Insights into deep-sea adaptation through the in situ fixed limpet transcriptome
The highly expressed transcripts of a transcriptome are likely to play decisive roles. The top 10% most abundant transcripts (top 2,767 transcripts ranked by transcripts per million [TPM] values) of the in situ fixed B. lactea transcriptome were applied to the KEGG enrichment analysis to investigate the pathways involved in their environmental adaptation to the seep habitat. Results showed that 19 pathways were significantly enriched, several of which have been reported to play potential roles in deep-sea adaptation, such as lysosome (ko04142), metabolism of xenobiotics by cytochrome P450 (ko00980), glutathione metabolism (ko00480), and fatty acid elongation (ko00062) (Figure 5A).
Figure 5.

KEGG enrichment analysis of the top 10% most abundant transcripts in the in situ fixed limpet transcriptome
(A) Bubble diagram showing the enriched pathways of the top 10% most abundant transcripts.
(B) Heatmap showing the expression pattern of the top 10% highly expressed transcripts mapped to the lysosome (ko04142) pathway. Differentially expressed transcripts are in black and those not are in gray.
Lysosome (ko04142) is the most significantly enriched pathway in the KEGG enrichment analysis of the top 10% most abundant transcripts in the in situ fixed B. lactea transcriptome (Figure 5A). A total of 64 transcripts mapped to lysosome (ko04142), including lysosomal acid hydrolases, lysosomal membrane proteins, other lysosomal enzymes and activators, mannose-6-phosphate receptor, clathrins, adaptor protein complex 3 (AP-3), and V- ATPase, were highly expressed (Figure 5B and Table S5), suggesting high lysosomal activity in the limpet living in the natural cold seep environment. High lysosomal activity has been reported in the symbiotic organs of several cold-seep symbiotic species, including mussels (Yu et al., 2019; Zheng et al., 2017), clams (Ip et al., 2021; Lan et al., 2019), and tubeworms (Sun et al., 2021), and was deduced to digest the symbionts in the specialized bacteriocytes for nutrition and to control the symbiont populations as well. Nevertheless, the limpets of Bathyacmaea are considered to mainly graze the bacterial film on the substrates they attached to (i.e., mainly mussel shells) for nutrition (Chen et al., 2019a; Liu et al., 2020), implying that the high lysosomal activity detected in B. lactea might play different roles. However, 45 of the 63 highly expressed transcripts were significantly downregulated in the onboard fixed group (Figure 5B), indicating that the role of high lysosomal activity might be interrupted by sampling stress, which is consistent with the result that lysosome (ko04142) was the most significantly enriched pathway in the KEGG enrichment analysis of all the DEGs (Figure 2A) and downregulated DEGs (Figure S3A). Lysosomes are ubiquitous cellular organelles known as the waste disposal system involved in the degradation and recycling of the cellular waste derived from both extracellular and intracellular regions, and growing evidence shows that they are also involved in many other cellular processes, including secretion, metabolic signaling, plasma membrane repair, and response to environmental cues, playing vital roles in maintaining cellular and organismal homeostasis (Ballabio and Bonifacino, 2020; Settembre et al., 2013).
In cold seeps, stressful environmental conditions might increase the production of intracellular waste (e.g., damaged organelles, oxidized lipids, and misfolded proteins) and the encounter with the pathogens of local organisms. Therefore, the high lysosomal activity would be conducive to increasing the efficiency of cellular clearance and reusage of the breakdown products for imperative nutritional needs. We assume that the high lysosomal activity might be a normalcy in the cold-seep-adapted nonsymbiotic invertebrates, which is a universal strategy for maintaining cellular and organismal homeostasis under the harsh environmental conditions of seep areas. However, various stressors can influence the function of lysosomes. For example, chemical contaminant treatments significantly decrease the lysosomal stability in molluscan hepatopancreas (Shaw et al., 2019), and the acute thermal stress on the deep-sea sponge holobiont causes significant lysosomal destabilization (Strand et al., 2017). Deep-sea sampling stress may have perturbed the lysosomal stability of the deep-sea limpets and thus decreased the lysosomal activity.
In the Haima cold seep, B. lactea usually attaches to the shells of the bathymodioline mussel Gigantidas haimaensis harboring methane-oxidizing endosymbionts, which makes use of methane emitted from the seafloor as the energy resource (Xu et al., 2019). Under such a condition, these limpets are also exposed to methane along with other toxic substances from their ambient environment and thus need to evolve suitable strategies for detoxification. Considering that the metabolism of xenobiotics by cytochrome P450 (ko00980) and glutathione metabolism (ko00480) pathways were significantly enriched from the top 10% most abundant transcripts, we investigated the expression of the mixed-function oxygenase (MFO) system components cytochrome P450 (CYP), conjugating enzyme glutathione S-transferase (GST), antioxidant enzymes superoxide dismutase (SOD), and glutathione peroxidase (GPX) (Lee, 1981; Ramos and Garcia, 2007; Xiao et al., 2020). Seven transcripts of CYP, twelve transcripts of GST, three transcripts of SOD, and one transcript of GPX ranked in the top 10% (Figure 6 and Table S6), indicating high MFO system and antioxidant enzyme activities in the limpet B. lactea, which might be responsible for the xenobiotic detoxification.
Figure 6.

A histogram showing the expression levels of transcripts involved in xenobiotic detoxification of the in situ and onboard fixed groups
TPM: transcripts per million.
Conclusions
In this study, we successfully sampled the in situ fixed deep-sea limpet B. lactea and obtained its first in situ transcriptome. Comparative transcriptomic analysis of the in situ and onboard fixed samples revealed that the concomitant stress during conventional deep-sea sampling without in situ fixation affected their gene expression. Furthermore, sampling stress exerted systemic influences on the life of the sampled deep-sea limpets by perturbing the cell and tissue structure, lysosomal activity, fluid balance, and unsaturated fatty acid metabolism. These findings reveal that conventionally sampled samples from deep sea without RNA in situ fixation might lead to biased results in transcriptomic analyses and suggest that in situ fixed deep-sea samples are highly demanded for mRNA quantitative-analysis-based studies.
Limitations of the study
Due to the small size (0.5–0.8 cm) of B. lactea we collected, the whole body of each individual, rather than the dissected tissues, was used for transcriptome sequencing and downstream data analyses. This sampling strategy may bias the expression levels of some genes that actually exhibit opposite expression patterns in different tissues. Nevertheless, we consider such impacts to be limited and would not influence the main findings of this work.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ammonium sulfate | Xilong Scientific | Cat#12600901 |
| EDTA | Solarbio Life Sciences | Cat#E8040-100g |
| Sodium citrate | Xilong Scientific | Cat#10201601 |
| TRIzol | Thermo Fisher Scientific | Cat#15596018 |
| RNAlater | Thermo Fisher Scientific | Cat#AM7021 |
| Deposited data | ||
| Raw RNA-seq data of six limpet Bathyacmaea lactea | This study | NCBI under BioProject PRJNA765439 |
| Software and algorithms | ||
| Trimmomatic version 0.39 | Bolger et al. (2014) | N/A |
| Trinity version 2.8.5 | Grabherr et al. (2011) | N/A |
| Salmon version 1.2.1 | Patro et al. (2017) | N/A |
| TransDecoder version 5.5.0 | https://github.com/TransDecoder/TransDecoder/wiki | N/A |
| CD-HIT version 4.8.1 | Fu et al. (2012) | N/A |
| BUSCO version 3.0.2 | Waterhouse et al. (2018) | N/A |
| OmicsBox version 1.4.11 | Biobam | N/A |
| RNA-seq 2G | Zhang et al., 2017 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pei-Yuan Qian (boqianpy@ust.hk).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Experimental model and subject details
This study was based on wild deep-sea limpet B. lactea, and no experimental models were used.
Method details
Deep-sea sampling and fixation
Limpets of B. lactea were collected from a single colony in the Haima cold seep (∼1400 m depth) in South China Sea by the ROV Haima 2 onboard the R/V Haiyangdizhi6 during the HYDZ6-202005 cruise in August 2020. CTD (Sea-Bird, Bellevue, WA, USA) data showed that the seawater temperature increased from 3.0°C to 30.7°C, the dissolved oxygen increased from 1.8 mg/L to 5.2 mg/L, and the salinity changed from 34.6 to 33.4 at the sampling site (Figure S2). Two methods were used for sample fixation. 1) Onboard fixation: G. haimaensis mussels attached with limpets were wrapped up with a tuck net held by the ROV manipulator arm and placed into the sample basket of the ROV (Figure 1B). The tuck net was transferred into the laboratory on board after the ROV was retrieved, and the limpets were immediately fixed by RNAlater (Thermo Fisher Scientific, Waltham, MA, USA) and frozen with liquid nitrogen after 4°C overnight. The sampling process from the seafloor to the laboratory took approximately 80 min 2) In situ fixation: G. haimaensis mussels attached with limpet B. lactea were cracked slightly by the ROV manipulator arm before they were placed into the sampling chamber fully filled on board with ∼12 L in-house RNA stabilizing solution to preserve their RNA in situ (Figure 1C). The chamber was sealed by closing the lid and returned to the sample basket of the ROV. After the ROV was retrieved on board, the sampling chamber was transferred to the lab on board. The in situ fixed limpets were transferred to the RNAlater (Thermo Fisher Scientific, USA) immediately and frozen with liquid nitrogen after 4°C overnight. All the limpets were stored at −80°C until usage. The in-house RNA stabilizing solution was prepared as previously described (Mat et al., 2020) with 700 g of ammonium sulfate, 40 mL of 0.5 M EDTA, 25 mL of 1 M sodium citrate, and 935 mL of distilled water; the pH was adjusted to 5.2, and the solution was stored in a 4°C cold room in the ship before the ROV dive.
RNA extraction and sequencing
Four individuals of B. lactea with in situ fixation and three with onboard fixation were used in this study. The whole body tissue of each individual was used for RNA extraction using TRIzol Reagent (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol. The quality and quantity of the extracted RNA were measured by the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). The mRNA of each individual was enriched by Oligo-dT probes and used for cDNA synthesis and eukaryotic library construction. All cDNA libraries were sequenced on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) to yield paired-end reads with a length of 150 bp in Novogene (Beijing, China).
Transcriptome assembly and annotation
Adaptors and low-quality bases of the raw reads were trimmed by Trimmomatic version 0.39 (Bolger et al., 2014) with the following setting: “ILLUMINACLIP: TruSeq3-PE-2.fa:2:30:10:2:keepBothReads LEADING:3 TRAILING:3 MINLEN:36”. The obtained clean reads of all the individuals were used for the de novo assembly of the transcriptome by Trinity version 2.8.5 (Grabherr et al., 2011). Salmon version 1.2.1 (Patro et al., 2017) was used to quantify the expression of each assembled transcript. The transcripts with a transcript per million value below 0.1 were removed. TransDecoder version 5.5.0 (https://github.com/TransDecoder/TransDecoder/wiki) was used to predict the open reading frame of the transcripts. Potential isoforms of the protein sequences were removed using CD-HIT version 4.8.1 (Fu et al., 2012) with c set to 0.95. The completeness of the assembled transcriptome was assessed using BUSCO version 3.0.2 to search against the metazoa_odb10 database (Waterhouse et al., 2018). The predicted protein sequences were used for functional annotation by searching their predicted protein sequences against the NCBI Non-Redundant (NR) databases using BLASTp version 2.10.0+ with an E-value cut-off of 1 × 10−5, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database via KEGG Automatic Annotation Server (KAAS), and the Gene Ontology (GO) via OmicsBox version 1.4.11 (BioBam, Valencia, Spain).
Quantification and statistical analysis
Gene expression analysis
Differential expression analysis was performed between four in situ and three onboard fixed limpets using DESeq2 (Love et al., 2014) implemented in RNA-seq 2G (Zhang et al., 2017) on the basis of the mapped reads count. Genes with a significant false discovery rate (FDR) value less than 0.05 and a fold change larger than two were identified as DEGs. The script “abundance_estimates_to_matrix.pl” implemented in Trinity (Grabherr et al., 2011) was used to generate a matrix of TMM-normalized expression values. This matrix was used for PCA by Past version 4.03 (Hammer et al., 2001). The significance of the comparison in PCA was further examined using PERMANOVA on a Bray-Curtis dissimilarity matrix (implemented in Past version 4.03) calculated for TMM-normalized expression values.
Enrichment analyses
The GO terms of the DE-Gs were enriched using the Fisher Exact Test implemented in OmicsBox 1.4.11 (BioBam). The KEGG pathways were enriched with the cumulative hypergeometric distribution method implemented in OmicShare online tool version 6.3.0 (http://www.omicshare.com/tools).
Acknowledgments
The authors thank the crew of R/V Haiyangdizhi six and the operation team of ROV Haima two for their great support in sample collection during the HYDZ6-202005 cruise. This work was supported by grants from the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0409), the Major Project of Basic and Applied Basic Research of Guangdong Province (2019B030302004), the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC01), and a GRF grant from HKSAR government (16101219). Thanks to Miss Wai Chuen Wong for her help in preparing the graphical abstract.
Author contributions
P-YQ and JS conceived this project. GY and TX collected the samples. GY and YL performed the bioinformatics analyses and drafted the manuscript. TW helped to prepare the figures. All authors contributed to the manuscript writing and approved it for submission and publication.
Declaration of interests
The authors declare no competing interests.
Published: April 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104092.
Supplemental information
Data and code availability
-
•
All raw sequencing data and the transcriptome assembly of B. lactea were deposited to NCBI under BioProject PRJNA765439.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- Balny C., Masson P., Heremans K. High pressure effects on biological macromolecules: from structural changes to alteration of cellular processes. Biochim. Biophys. Acta. 2002;1595:3–10. doi: 10.1016/s0167-4838(01)00331-4. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A., Thatje S. Explaining bathymetric diversity patterns in marine benthic invertebrates and demersal fishes: physiological contributions to adaptation of life at depth. Biol. Rev. Camb Philos. Soc. 2014;89:406–426. doi: 10.1111/brv.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Watanabe H.K., Nagai Y., Toyofuku T., Xu T., Sun J., Qiu J.W., Sasaki T. Complex factors shape phenotypic variation in deep-sea limpets. Biol. Lett. 2019;15:20190504. doi: 10.1098/rsbl.2019.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Wang M.X., Li M.N., Lian C., Zhou L., Zhang X., Zhang H., Zhong Z.S., Wang H., Cao L., Li C.L. A glimpse of deep-sea adaptation in chemosynthetic holobionts: depressurization causes DNA fragmentation and cell death of methanotrophic endosymbionts rather than their deep-sea Bathymodiolinae host. Mol. Ecol. 2021;30:2298–2312. doi: 10.1111/mec.15904. [DOI] [PubMed] [Google Scholar]
- Chen J.W., Liang L.Y., Li Y.A., Zhang H.B. Molecular response to high hydrostatic pressure: time-series transcriptomic analysis of shallow-water sea cucumber apostichopus japonicus. Front Genet. 2020;11:355. doi: 10.3389/fgene.2020.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.W., Liu H.L., Cai S.Y., Zhang H.B. Comparative transcriptome analysis of eogammarus possjeticus at different hydrostatic pressure and temperature exposures. Sci. Rep. 2019;9:3456. doi: 10.1038/s41598-019-39716-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M.R., Consalvey M., Rowden A.A. John Wiley & Sons; 2016. Biological Sampling in the Deep Sea. [Google Scholar]
- Feng D., Qiu J.W., Hu Y., Peckmann J., Guan H.X., Tong H.P., Chen C., Chen J.X., Gong S.G., Li N., Chen D.F. Cold seep systems in the South China sea: an overview. J. Asian Earth Sci. 2018;168:3–16. doi: 10.1016/j.jseaes.2018.09.021. [DOI] [Google Scholar]
- Fu L.M., Niu B.F., Zhu Z.W., Wu S.T., Li W.Z. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z.M., Huang J.M., Cui G.J., Li W.L., Li J., Wei Z.F., Chen J., Xin Y.Z., Cai D.S., Zhang A.Q., Wang Y. In situ meta-omic insights into the community compositions and ecological roles of hadal microbes in the Mariana Trench. Environ. Microbiol. 2019;21:4092–4108. doi: 10.1111/1462-2920.14759. [DOI] [PubMed] [Google Scholar]
- Gelse K., Poschl E., Aigner T. Collagens--structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003;55:1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q.D., et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer Ø., Harper D., Ryan P. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001;4:9. [Google Scholar]
- Hazel J.R. Thermal adaptation in biological membranes: is homeoviscous adaptation the explanation? Annu. Rev. Physiol. 1995;57:19–42. doi: 10.1146/annurev.ph.57.030195.000315. [DOI] [PubMed] [Google Scholar]
- Hoepner C.M., Abbott C.A., da Silva K.B. The ecological importance of toxicity: sea anemones maintain toxic defence when bleached. Toxins. 2019;11:266. doi: 10.3390/toxins11050266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip J.C., Xu T., Sun J., Li R., Chen C., Lan Y., Han Z., Zhang H., Wei J., Wang H., et al. Host-endosymbiont genome integration in a deep-sea chemosymbiotic clam. Mol. Biol. Evol. 2021;38:502–518. doi: 10.1093/molbev/msaa241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Hayashi R., Tsuda T., Taniguchi K. High pressure-induced changes of biological membrane - study on the membrane-bound Na+/K+-ATPase as a model system. Eur. J. Biochem. 2002;269:110–118. doi: 10.1046/j.0014-2956.2002.02621.x. [DOI] [PubMed] [Google Scholar]
- Kiel S. A biogeographic network reveals evolutionary links between deep- sea hydrothermal vent and methane seep faunas. Proc. R. Soc. B Biol. Sci. 2016;283:20162337. doi: 10.1098/rspb.2016.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., Sun J., Xu T., Chen C., Tian R., Qiu J.W., Qian P.Y. De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genomics. 2018;19:394. doi: 10.1186/s12864-018-4720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., Sun J., Zhang W.P., Xu T., Zhang Y., Chen C., Feng D., Wang H.B., Tao J., Qiu J.W., Qian P.Y. Host-symbiont interactions in deep-sea chemosymbiotic vesicomyid clams: insights from transcriptome sequencing. Front Mar. Sci. 2019;6:680. doi: 10.3389/fmars.2019.00680. [DOI] [Google Scholar]
- Lee R.F. Mixed function oxygenases (MFO) in marine invertebrates. Mar. Biol. Lett. 1981;2:87–105. [Google Scholar]
- Levin L.A. Ecology of cold seep sediments: interactions of fauna with flow, chemistry and microbes. Oceanogr Mar. Biol. 2005;43:1–46. doi: 10.1201/9781420037449.ch1. [DOI] [Google Scholar]
- Liu R.Y., Wang K., Liu J., Xu W.J., Zhou Y., Zhu C.L., Wu B.S., Li Y.X., Wang W., He S.P., et al. De Novo genome assembly of limpet bathyacmaea lactea (gastropoda: pectinodontidae): the first reference genome of a deep-sea gastropod endemic to cold seeps. Genome Biol. Evol. 2020;12:905–910. doi: 10.1093/gbe/evaa100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques M.I., Borreguero J.M., Stanley H.E., Dokholyan N.V. Possible mechanism for cold denaturation of proteins at high pressure. Phys. Rev. Lett. 2003;91:138103. doi: 10.1103/PhysRevLett.91.138103. [DOI] [PubMed] [Google Scholar]
- Mat A.M., Sarrazin J., Markov G.V., Apremont V., Dubreuil C., Eche C., Fabioux C., Klopp C., Sarradin P.M., Tanguy A., et al. Biological rhythms in the deep-sea hydrothermal mussel Bathymodiolus azoricus. Nat. Commun. 2020;11:3454. doi: 10.1038/s41467-020-17284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullin E.R., Bergquist D.C., Fisher C.R. Metazoans in extreme environments: adaptations of hydrothermal vent and hydrocarbon seep fauna. Gravit. Space Biol. Bull. 2000;13:13–23. [PubMed] [Google Scholar]
- Motoki K., Watsuji T.O., Takaki Y., Takai K., Iwasaki W. Metatranscriptomics by in situ RNA stabilization directly and comprehensively revealed episymbiotic microbial communities of deep-sea squat lobsters. mSystems. 2020;5:e00551-20. doi: 10.1128/mSystems.00551-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobata S., Ando M., Takei Y. Hormonal control of drinking behavior in teleost fishes; insights from studies using eels. Gen. Comp. Endocr. 2013;192:214–221. doi: 10.1016/j.ygcen.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Parzanini C., Parrish C.C., Hamel J.F., Mercier A. Functional diversity and nutritional content in a deep-sea faunal assemblage through total lipid, lipid class, and fatty acid analyses. PLoS One. 2018;13:e0207395. doi: 10.1371/journal.pone.0207395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Llodra E., Brandt A., Danovaro R., De Mol B., Escobar E., German C.R., Levin L.A., Arbizu P.M., Menot L., Buhl-Mortensen P., et al. Deep, diverse and definitely different: unique attributes of the world's largest ecosystem. Biogeosciences. 2010;7:2851–2899. doi: 10.5194/bg-7-2851-2010. [DOI] [Google Scholar]
- Ramos R., Garcia E. Induction of mixed-function oxygenase system and antioxidant enzymes in the coral Montastraea faveolata on acute exposure to benzo(a)pyrene. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007;144:348–355. doi: 10.1016/j.cbpc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Rex M.A., Etter R.J. Harvard University Press; 2010. Deep-sea Biodiversity: Pattern and Scale. [Google Scholar]
- Salzet M., Deloffre L., Breton C., Vieau D., Schoofs L. The angiotensin system elements in invertebrates. Brain Res. Rev. 2001;36:35–45. doi: 10.1016/S0165-0173(01)00063-7. [DOI] [PubMed] [Google Scholar]
- Sanders J.G., Beinart R.A., Stewart F.J., Delong E.F., Girguis P.R. Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. Isme J. 2013;7:1556–1567. doi: 10.1038/ismej.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Bio. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J.P., Moore M.N., Readman J.W., Mou Z., Langston W.J., Lowe D.M., Frickers P.E., Al-Moosawi L., Pascoe C., Beesley A. Oxidative stress, lysosomal damage and dysfunctional autophagy in molluscan hepatopancreas (digestive gland) induced by chemical contaminants. Mar. Environ. Res. 2019;152:104825. doi: 10.1016/j.marenvres.2019.104825. [DOI] [PubMed] [Google Scholar]
- Shillito B., Desurmont C., Barthelemy D., Farabos D., Despres G., Ravaux J., Zbinden M., Lamaziere A. Lipidome variations of deep-sea vent shrimps according to acclimation pressure: a homeoviscous response? Deep-sea Res. Pt. 2020;161:103285. doi: 10.1016/j.dsr.2020.103285. [DOI] [Google Scholar]
- Strand R., Whalan S., Webster N.S., Kutti T., Fang J.K.H., Luter H.M., Bannister R.J. The response of a boreal deep-sea sponge holobiont to acute thermal stress. Sci. Rep-uk. 2017;7:1660. doi: 10.1038/s41598-017-01091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Zhang Y., Xu T., Zhang Y., Mu H.W., Zhang Y.J., Lan Y., Fields C.J., Hui J.H.L., Zhang W.P., et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017;1:0121. doi: 10.1038/s41559-017-0121. [DOI] [PubMed] [Google Scholar]
- Sun J., Chen C., Miyamoto N., Li R.S., Sigwart J.D., Xu T., Sun Y.A., Wong W.C., Ip J.C.H., Zhang W.P., et al. The Scaly-foot Snail genome and implications for the origins of biomineralised armour. Nat. Commun. 2020;11:1657. doi: 10.1038/s41467-020-15522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Sun J., Yang Y., Lan Y., Ip J.C., Wong W.C., Kwan Y.H., Zhang Y., Han Z., Qiu J.W., Qian P.Y. Genomic signatures supporting the symbiosis and formation of chitinous tube in the deep-sea tubeworm Paraescarpia echinospica. Mol. Biol. Evol. 2021;38:4116–4134. doi: 10.1093/molbev/msab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Feng D., Tao J., Qiu J.W. A new species of deep-sea mussel (Bivalvia: Mytilidae: Gigantidas) from the South China Sea: morphology, phylogenetic position, and gill-associated microbes. Deep-sea Res. Oceanogr Res. Pap. 2019;146:79–90. [Google Scholar]
- Van Campenhout J., Vanreusel A., Van Belleghem S., Derycke S. Transcription, signaling receptor activity, oxidative phosphorylation, and fatty acid metabolism mediate the presence of closely related species in distinct intertidal and cold-seep habitats. Genome Biol. Evol. 2016;8:51–69. doi: 10.1093/gbe/evv242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Shen Y.J., Yang Y.Z., Gan X.N., Liu G.C., Hu K., Li Y.X., Gao Z.M., Zhu L., Yan G.Y., et al. Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nat. Ecol. Evol. 2019;3:823–833. doi: 10.1038/s41559-019-0864-8. [DOI] [PubMed] [Google Scholar]
- Wang Y., Gao Z.M., Li J., He L.S., Cui G.J., Li W.L., Chen J., Xin Y.Z., Cai D.S., Zhang A.Q. Hadal water sampling by in situ microbial filtration and fixation (ISMIFF) apparatus. Deep-sea Res. Pt. 2019;144:132–137. doi: 10.1016/j.dsr.2019.01.009. [DOI] [Google Scholar]
- Waterhouse R.M., Seppey M., Simao F.A., Manni M., Ioannidis P., Klioutchnikov G., Kriventseva E.V., Zdobnov E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018;35:543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watsuji T.O., Yamamoto A., Takaki Y., Ueda K., Kawagucci S., Takai K. Diversity and methane oxidation of active epibiotic methanotrophs on live Shinkaia crosnieri. Isme J. 2014;8:1020–1031. doi: 10.1038/ismej.2013.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z.F., Li W.L., Li J., Chen J., Xin Y.Z., He L.S., Wang Y. Multiple in situ nucleic acid collections (MISNAC) from deep-sea waters. Front Mar. Sci. 2020;7:81. doi: 10.3389/fmars.2020.00081. [DOI] [Google Scholar]
- Weatherall P., Marks K.M., Jakobsson M., Schmitt T., Tani S., Arndt J.E., Rovere M., Chayes D., Ferrini V., Wigley R. A new digital bathymetric model of the world's oceans. Earth Space Sci. 2015;2:331–345. doi: 10.1002/2015ea000107. [DOI] [Google Scholar]
- Wood J.B., Maynard A.E., Lawlor A.G., Sawyer E.K., Simmons D.M., Pennoyer K.E., Derby C.D. Caribbean reef squid, Sepioteuthis sepioidea, use ink as a defense against predatory French grunts, Haemulon flavolineatum. J. Exp. Mar. Biol. Ecol. 2010;388:20–27. doi: 10.1016/j.jembe.2010.03.010. [DOI] [Google Scholar]
- Xiao Y., Xu T., Sun J., Wang Y., Wong W.C., Kwan Y.H., Chen C., Qian P.Y. Population genetic structure and gene expression plasticity of the deep-sea vent and seep squat lobster shinkaia crosnieri. Front Mar. Sci. 2020;7:587686. doi: 10.3389/fmars.2020.587686. [DOI] [Google Scholar]
- Yancey P.H., Gerringer M.E., Drazen J.C., Rowden A.A., Jamieson A. Marine fish may be biochemically constrained from inhabiting the deepest ocean depths. Proc. Natl. Acad. Sci. U S A. 2014;111:4461–4465. doi: 10.1073/pnas.1322003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Sun J., Sun Y.N., Kwan Y.H., Wong W.C., Zhang Y.J., Xu T., Feng D., Zhang Y., Qiu J.W., Qian P.Y. Genomic, transcriptomic, and proteomic insights into the symbiosis of deep-sea tubeworm holobionts. Isme J. 2020;14:135–150. doi: 10.1038/s41396-019-0520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.J., Wang M.X., Liu B.Z., Yue X., Li C.L. Gill symbionts of the cold-seep mussel Bathymodiolus platifrons: composition, environmental dependency and immune control. Fish Shellfish Immunol. 2019;86:246–252. doi: 10.1016/j.fsi.2018.11.041. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Zhang Y., Evans P., Chinwalla A., Taylor D. RNA-seq 2G: online analysis of differential gene expression with comprehesive options of statistical methods. bioRxiv. 2017:122747. doi: 10.1101/122747. [DOI] [Google Scholar]
- Zheng P., Wang M.X., Li C.L., Sun X.Q., Wang X.C., Sun Y., Sun S. Insights into deep-sea adaptations and host-symbiont interactions: a comparative transcriptome study on Bathymodiolus mussels and their coastal relatives. Mol. Ecol. 2017;26:5133–5148. doi: 10.1111/mec.14160. [DOI] [PubMed] [Google Scholar]
- Zhu F.C., Sun J., Yan G.Y., Huang J.M., Chen C., He L.S. Insights into the strategy of micro-environmental adaptation: transcriptomic analysis of two alvinocaridid shrimps at a hydrothermal vent. PLoS One. 2020;15:e0227587. doi: 10.1371/journal.pone.0227587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw sequencing data and the transcriptome assembly of B. lactea were deposited to NCBI under BioProject PRJNA765439.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.