

ABSTRACT
Breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death in women worldwide. Recent advances in the field of immuno-oncology demonstrate the beneficial immunostimulatory effects of the induction of immunogenic cell death (ICD). ICD increases tumor infiltration by T cells and is associated with improved prognosis in patients affected by triple negative breast cancer (TNBC) with residual disease. The aim of this study was to evaluate the antitumoral effect of PKHB1, a thrombospondin-1 peptide mimic, against breast cancer cells, and the immunogenicity of the cell death induced by PKHB1 in vitro, ex vivo, and in vivo. Our results showed that PKHB1 induces mitochondrial alterations, ROS production, intracellular Ca2+ accumulation, as well calcium-dependent cell death in breast cancer cells, including triple negative subtypes. PKHB1 has antitumor effect in vivo leading to a reduction of tumor volume and weight and promotes intratumoral CD8 + T cell infiltration. Furthermore, in vitro, PKHB1 induces calreticulin (CALR), HSP70, and HSP90 exposure and release of ATP and HMGB1. Additionally, the killed cells obtained after treatment with PKHB1 (PKHB1-KC) induced dendritic cell maturation, and T cell antitumor responses, ex vivo. Moreover, PKHB1-KC in vivo were able to induce an antitumor response against breast cancer cells in a prophylactic application, whereas in a therapeutic setting, PKHB1-KC induced tumor regression; both applications induced a long-term antitumor response. Altogether our data shows that PKHB1, a thrombospondin-1 peptide mimic, has in vivo antitumor effect and induce immune system activation through immunogenic cell death induction in breast cancer cells.
KEYWORDS: Breast cancer, immunogenic cell death, thrombospondin 1, PKHB1, tumor cell lysate, anticancer vaccine
Background
Breast cancer is the most frequent type of cancer among women; its innate and acquired treatment resistance to current therapies is the principal problem to treat it, causing the greatest number of cancer-related deaths.1 While systemic therapies have increased the survival rates of breast cancer patients, the dramatic variations in response rates of patients with distinct clinicopathologic parameters,2 as well as innate or acquired resistance to current therapies,3 make relevant the search for new effective treatments for the different molecular subtypes of breast cancer, in particular those associated with poor prognosis.
Recently, several clinical studies have demonstrated the beneficial immunostimulatory effects of inducing immunogenic cell death (ICD),4,5 recognized as a critical determinant for the efficiency of cancer therapies. Indeed, this peculiar type of cell death is capable of stimulating a long-term antitumor immune response against dead cancer cell antigens.6,7 Additionally, inducing ICD increases tumor tissue infiltration by T cells, which plays an essential role in mediating a positive response to chemotherapy and is associated with improving clinical outcomes in all subtypes of breast cancer.5,8
We recently designed PKHB1 through a structure–activity relationship studies around the C-terminal Binding Domain (CBD) of thrombospondin-1 (TSP-1). PKHB1 is a peptide mimic stable in the serum of mice and human and able to induce a cell death involving CD47 activation in different cancer cells, especially in hematological malignancies.9–12 The ability of PKHB1 to induce cell death was also observed in leukemic cells from patients with aggressive and chemo-resistant phenotypes, without affecting non-tumoral cells from humans or mice.9,11,13 Additionally, PKHB1 induces ICD in T cell acute lymphoblastic leukemia.11,14 If this peptide is currently under development in pre-clinical studies addressing CLL (χ-Pharma), little is known about the cell death capacity, mechanism, immunogenicity, and the antitumor effect of PKHB1 in solid cancers with different molecular characteristics and poor prognosis such as breast cancer (including the triple negative subtype).
Therefore, the aim of this study was to evaluate the antitumor potential of PKHB1 in breast cancer cells (in vitro and in vivo) including triple negative subtypes and to determine whether it induces antitumor immune system activation through ICD induction (ex vivo and in vivo).
Methods
Peptide synthesis
PKHB1 and 4NGG peptides were synthesized manually, using Fmoc-protected amino acids and standard solid phase peptide synthesis (SPPS) (supplemental material and methods), as described previously.6
Cell culture
MCF-7, MDA-MB-231, and 4T1 cell lines were obtained from the ATCC. MCF-7 and MDA-MB-231 cell lines were maintained in DMEM-F12 medium (GIBCO by Life Technologies, Grand Island, NY, USA), while 4T1 cell line was maintained in RPMI-1640 medium, both were supplemented with 10% of fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin-streptomycin (GIBCO by Life Technologies, Grand Island, NY, USA), and incubated at 37°C in a controlled humidified atmosphere with 5% CO2. Cell count was performed following the ATCC’s standard protocols.
Cell death induction and inhibition analysis
5 x 104 cells were plated in 24 wells dishes and left untreated or treated for 2 h with 100 μM, 200 μM, 300 μM, or 400 μM of PKHB1 (KRFYVVMWKK), or 300 μM of 4NGG (KRFYGGMWKK). Annexin-V-allophycocyanin (Ann-V-APC 0.1 μg/ml; BD Pharmingen, San Jose CA, USA), and propidium iodide (PI, 0.5 μg/ml Sigma-Aldrich) were used to assess phosphatidylserine exposure, cell death, and cell viability quantification, respectively, in a BD AccuryC6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) (total population 10,000 cells). Data was analyzed using FlowJo software (LLC, Ashland, OR, USA).
The calcium chelator BAPTA (5 mM), the pan-caspase inhibitor Z-VAD-FMK (Z-VAD, 50 μM), the antioxidant N-Acetyl Cysteine (NAC, 5 mM), the necroptotic inhibitor Necrostatin-1 (Nec-1, 50 μM), the phospholipase C (PLC) inhibitor U73122 (1.25 μM) and the ER receptor inhibitors dantrolene (50 μM) and 2-aminoethoxydiphenyl borate (2-APB, 40 μM) were incubated 30 minutes with the indicated agent, before treatment with PKHB1 (CC50), epirubicin (42.5 μM for MCF-7 and MDA-MB-231 and 5 μM for 4T1 for 24 h), or H2O2 (25 μM for all cell lines for 24 h) when indicated.
Intracellular Ca2+ levels assay
5 × 104 cells/well in 24 wells dishes (Life Science) were left untreated or pre-incubated with 2.5 mM BAPTA, and then treated for 2 h with PKHB1 (CC50) or left untreated in medium. Then, cells were detached, washed with RINGER buffer without Ca2+, and resuspended in 200 μL of the same RINGER buffer with 0.001 μg/mL of Fluo-4 AM (Life Technologies) and 0.001 μg/mL of Pluronic F-127 (Life Technologies), incubated 37°C for 30 min. Next, cells were washed with RINGER buffer w/o Ca2+ and assessed by BD Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) (total population 10,000 cells), and data was analyzed using FlowJo software (LLC, Ashland, OR, USA).
In vivo model
This study was approved by The Animal Research and Welfare Ethics Committee (CEIBA), of the College of Biological Sciences, number: CEIBA-2018-003. All experiments were conducted according to Mexican regulation NOM-062-ZOO-1999. Female BALB/c mice (6- to 8-week-old; 22 ± 2 g weight) were maintained in controlled environmental conditions (25°C and 12 h light/dark cycle) and supplied with rodent food (LabDiet, St. Louis, MO, USA) and water ad libitum, and they were monitored daily for health status. Mice were randomly assigned to different groups for all studies. All experiments were designed in accordance with the ARRIVE guidelines for animal care and protection (supplemental material).15
Tumor establishment
5 × 105 live 4T1 cells in 100 μL of PBS were injected subcutaneously in the left hind. Tumor volume and mice weight were measured three times per week using a caliper (Digimatic Caliper Mitutoyo Corporation, Japan) and a digital scale (American Weigh Scale-600-BLK, USA), respectively. Tumor volume was determined with the formula: tumor volume (mm3) = (Length × width2)/2. When tumor reached 70–120 mm3, 3 days after inoculation with tumor cells, mice were treated daily with 400 μg of PKHB1 in 200 μL of sterile water by intraperitoneal injection, control mice were treated with 200 μL of sterile water. Sixteen days after inoculation with tumor cells, mice were anesthetized with ketamine (i.p. 80 mg/kg body weight) and xylazine (i.p. 10 mg/kg body weight) and were euthanized by cervical dislocation. Tumors from Control or PKHB1-treated mice, were obtained and fixed in 3.7% neutral formalin, embedded in paraffin, sectioned (5 μm thickness) and stained with H&E (MERCK). Histopathological analyses were done by an external veterinarian pathologist (National professional certificate 2,593,012).
T cells evaluation
Sixteen days after tumor inoculation, mice treated (n = 6) or untreated (n = 6) were anesthetized and sacrificed as described above. Blood was obtained by cardiac puncture and isolation of the peripheral blood mononuclear cells (PBMCs) was performed by density gradient centrifugation using Ficoll-Hypaque-1119 (Sigma-Aldrich, St Louis, MO, USA). The spleen, lymph node, and tumor were harvested and filtered through a cell strainer (70 μM) with PBS (PBMCs were obtained from the spleen as described above), then 1 × 106 cells/mL were plated and the percent of CD3+, CD4+ and CD8 + T cells was observed by flow cytometry with the Mouse T lymphocyte subset antibody cocktail CD3 (clone 145–2C11), CD4 (clone RM4-5), and CD8 (clone 53–6.7) (from BD Bioscience) following the manufacturer’s instructions.
Myeloid-derived suppressor cells (MDSCs) and Tregs evaluation
For MDSCs assessment, PBMCs were obtained from the blood of mice as described above. Cells were labeled with a cocktail of CD11b‐PE (clone M1/70), Gr‐1‐APC (clone RB6/8C5), and Ly‐6 G‐FITC (clone 1A8) using the Mouse MDSC Flow kit (from Biolegend) following the manufacturer’s instructions.
For Tregs evaluation, PBMCs were obtained from the blood of mice as described above. Cells were labeled using a True Nuclear One Step Staining Mouse Treg Flow kit (FOXP3-AlexaFluor488, CD25-PE, CD4-PerCP; Biolegend) following the manufacturer’s instructions.
Cells were assessed in a BD Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and data was analyzed using FlowJo software (LLC, Ashland, OR, USA).
Calreticulin, HSP70, and HSP90 exposure
5 × 104 cells/well were plated in 24-well plates and treated with PKHB1 (CC50) for 2 h or epirubicin (42.5 μM for MCF-7 and MDA-MB-231 and 5 μM for 4T1 for 24 h). Then, cells were detached, washed, and incubated for 1 h at room temperature (RT) with 2 μg/mL of anti-Calreticulin (FMC-75, Enzo Life Science), 0.8 μg/mL anti-HSP70 (F-3, Santa Cruz Biotechnology), and 0.8 μg/mL anti-HSP90 (F-8, Santa Cruz Biotechnology) in FACS buffer; cells were washed and incubated for 30 min in darkness at RT with goat anti-mouse IgG (Alexa Fluor 488) (H + L, Life Technologies) (1:1500) in FACS buffer; cells were then washed and incubated in the dark for 10 min at RT with 7-AAD (Life Technologies) (1:1000) in FACS buffer. The surface exposure of CALR, HSP70 and HSP90 was determined by flow cytometry in non-permeabilized (7-AAD-negative) cells.
Immunofluorescence microscopy
2.5 × 105 cells/well in 6-well dishes were left untreated (Control) or treated with PKHB1 (CC50) and incubated for 2 h. Then, cells were washed with PBS and stained with Calreticulin-PE antibody (FMC-75, 2 μg/ml) and Hoechst 33,342 (0.5 μg/ml) (Thermo Scientific Pierce, Rockford, IL, USA), incubated for 1 h in FACS buffer at RT, washed twice, maintained in PBS, and assessed by confocal microscopy (Olympus X70; Olympus, Tokyo, Japan).
ATP and High-mobility group box 1 release assay
2.5 × 105 cells/well in 6-well dishes were left untreated (Control) or treated with PKHB1 (CC50) for 2 h. Supernatants were recovered, centrifuged at 1600 rpm/10 minutes and used to assess extracellular ATP by a luciferase assay (ENLITEN kit, Promega, Madison, WI, USA), or HMGB1 using the HMGB1 ELISA kit for MDA-MB-231, MCF-7 and 4T1 cells (BioAssay ELISA kit human or mouse, respectively; US Biological Life Science Salem, MA, USA) following the manufacturer’s instructions. Bioluminescence was assessed in a microplate reader (Synergy HT, Software Gen5; BioTek, Winooski, VT, USA) at 560 nm, and absorbance was assessed at 450 nm.
T cell isolation
Mice were anesthetized and sacrificed as described above, and blood was obtained by cardiac puncture. PBMCs isolation was performed as described above. Murine CD3+ cells were isolated from total PBMCs by positive selection using magnetic-activated cell sorting (MACS) microbead technology with anti-CD3ε-biotin and anti-biotin microbeads (Miltenyi Biotech; >98% purity and >98% viability), as stated by manufacturer’s instructions.
Differentiation of bone marrow-derived dendritic cells (BMDCs)
After sacrifice of anesthetized mice (n = 6), bone marrow was removed from the femur and tibia by flushing into RPMI-1640. Eluted cells were cultured for 5 days with 20 ng/mL of IL-4 and GM-CSF (R&D Systems, Minneapolis, MN, USA) until approximately 70% of the cells were CD11c+.
Evaluation of DCs maturation
CD11c, MHC-II, CD80, and CD86 were evaluated by flow cytometry with the fluorescent label-conjugated antibodies, antiCD11c-Alexa-fluor 488 (N418, R&D Systems), anti-MHC Class II-PE (REA813, Miltenyi Biotec), anti-CD80-FITC (16–10A1, R&D Systems), and antiCD86-APC (GL1) from BD Biosciences (San Jose, CA, USA). In brief, 1 × 106 DCs /mL were stained in 100 μL of FACS buffer with the indicated antibodies at RT for 30 minutes and then were washed twice with PBS, centrifugated at 1600 rpm/10 min, resuspended in 100 μl of FACS buffer and assessed by Flow Cytometer as described previously. For MHC-II and CD80 evaluation by flow cytometry, CD11c was added with MHC-II or CD86, we then gated CD11c+ cells and we next assessed MHC-II or CD86 MFI.
PKHB1-KC and EPI-KC preparation
4T1 cells (1.5 × 106 cells/mL per mice) were plated and, after adherence, cells were then treated with 400 μM of PKHB1 for 2 h or 10 μM of EPI for 24 h, to obtain 80–90% of killed cells (KC). After treatments, cells were obtained, centrifuged at 1600 rpm/10 min and resuspended in 100 μL of serum-free medium/mice. Cell death was confirmed using Trypan blue staining and flow cytometry. Finally, the PKHB1-KC or EPI-KC were inoculated by subcutaneous injection in the right flank.
Freeze and thaw-killed cells preparation
4T1 cells (3 × 106 cells/mL per mice) were first frozen at −80°C for 15 min, then thawed 10 min at 37°C in a water bath. The freeze–thaw (F-T) cycles were repeated three times in rapid succession. After the final thaw, killed cells were resuspended in PBS.
DCs’ co-culture with PKHB1-KC, EPI-KC, or FT-KC
DCs were resuspended in fresh medium (1 × 106 cells/mL), left untreated (control) or incubated with 3 × 106 4T1 killed cells/mL obtained after treatment with PKHB1, EPI, or FT, to give a range of 1:3 (DCs to killed cells); co-culture was left for 24 h. Then the supernatant was obtained, and the well was washed twice with PBS before the next co-culture.
DCs + T lymphocytes co-culture
Control DCs or DCs previously co-cultured with PKHB1-KC, EPI-KC, or FT-KC were maintained in fresh medium at 1 × 106 cells/mL. Then, allogeneic BALB/c mCD3+ cells were added at 3 × 106 cells/mL to give a range of 1:3 (DCs to CD3+ cells), co-culture was left for 96 h. Then, lymphocytes were collected (in the supernatant), washed with PBS, and resuspended in fresh medium at 5 × 106 cells/mL for their use in the next co-culture.
T-Lymphocytes + 4T1 cells co-culture
1 × 105 cells/mL viable 4T1 cells were plated. Then, allogeneic BALB/c mCD3+ cells were added to each well at 5 × 105 cells/mL, unprimed (previously co-cultured with control DCs) or primed (previously co-cultured with DCs-PKHB1-KC, EPI-KC, or FT-KC) to give a range of 1:5 (tumor to effector). Co-culture was left for 24 h.
Cytokine release assay
Supernatants from the indicated co-cultures were obtained for the assessment of IL-2, IL-4, IL5, and TNFα (BD CBA Mouse Th1/Th2 Cytokine Kit, San Jose, CA, USA) by flow cytometry following manufacturer’s instructions. IFNγ was assessed using an ELISA kit (Sigma-Aldrich) and the Synergy HTTM (BioTek Instruments, Inc., Winooski, VT, USA) plate reader at 570 nm wavelength, following manufacturer’s instructions.
Calcein assay
4T1 cells (1 × 106 cells/mL) were stained with (0.1 mL/mL) Calcein-AM from BD Biosciences (San Jose, CA) in FACS buffer at 37°C and 5% CO2 for 30 min, washed twice with PBS. Thus, primed or unprimed T cells were added in a 1:5 (tumor to effector) ratio. Co-culture was incubated at 37°C and 5% CO2 for 24 h. Finally, calcein positive or negative 4T1 cells were assessed in a BD AccuryC6 flow cytometer (BD Biosciences) (total population 10,000 cells). Data was then analyzed using FlowJo software.
Prophylactic vaccination
Vaccination was carried out as follows: PKHB1-KC (n = 10) or EPI-KC (n = 10) were obtained as previously described, and then inoculated s.c. in 100 μL of serum-free medium into the right hind leg (day −7), 7 days later, viable (5 × 105) 4T1 cells were inoculated into the left hind leg (day 0). Tumor volume and weight were measured as described above.
PKHB1-KC and EPI-KC treatment
Tumor was established by subcutaneous injection of 5 × 105 4T1 cells in 100 μL of PBS, in the left hind. Tumor volume and mice weight were measured as described above. When tumor reached 70–120 mm3 the first treatment of PKHB1-KC (n = 10) or EPI-KC (n = 10) was applied. Killed cells were inoculated subcutaneously in 100 µl of serum-free medium, in the right hind, twice a week for a total of four applications in a 2-week period. Control mice were treated with 100 µl of serum-free medium.
Long-term antitumor effect evaluation
Mice in complete remission after prophylactic (n = 9) or therapeutic (n = 9) 4T1-PKHB1-KC application were re-challenged with 5 × 105 4T1 viable cells in 100 μL of PBS in the left hind and tumor volume was measured as described above.
Long-term splenocytes-cytotoxicity
Mice in complete remission after prophylactic (n = 4) or therapeutic (n = 4) PKHB1-KC application were re-challenged with 5 × 105 4T1 viable cells in 100 μL of PBS in the left hind. Three days after tumor inoculation mice were sacrificed, spleens were harvested, filtered through a cell strainer (70 μM) with PBS, and PBMCs were obtained as described above. Splenocytes were recovered and co-cultured with 4T1 cells (previously stained with calcein-AM) at 44:1 ratio (respectively). Finally, calcein positive or negative cells were assessed as described above.
Statistical Analysis
Mice were randomly assigned to different groups for all in vivo studies. At least three independent experiments were repeated three independent times. Mann-Whitney tests and two-tailed unpaired Student’s t-tests were performed using GraphPad Prism Software (San Diego CA, USA) and presented as mean values ± SD. The p values were considered significant as follows: p< 0.05.
Results
PKHB1 induces breast cancer cell death
Although the potency of PKHB1was previously demonstrated in hematopoietic malignancies,9,11 its effectiveness was not yet evaluated for solid tumors in vivo. Thus, after peptide synthesis and characterization (Supplemental material and methods, table sup.1 and figure sup.1), we evaluated here its effect in two types of human breast cancer cell lines, I) MCF-7 (luminal subtype) and II) MDA-MB-231 (triple negative subtype), as well as on the murine 4T1 cell line (mimics triple negative subtype).(16) We observed that PKHB1 induces cell death in a concentration‐dependent way in MCF-7 (Figure 1(a)), MDA-MB-231 (Figure 1(b)), and 4T1 (Figure 1(c)) cells, as they showed an increase in the percentage of double-positive Ann‐V‐APC/PI staining (figure sup. 2). We determined that the cytotoxic concentration that induces approximately 50% of cell death (CC50) in MDA-MB-231 and MCF-7 is 200 μM whereas in 4T1 is 300 μM.
Figure 1.
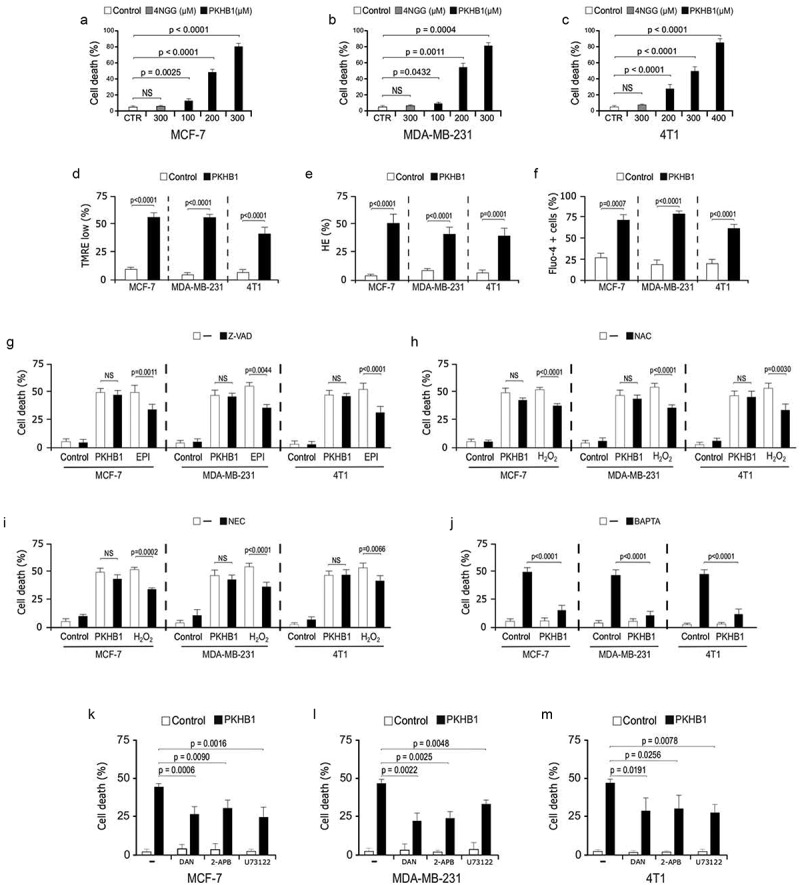
PKHB1 induces cell death in breast cancer cell lines. Cell death analysis in (A) MCF-7, (B) MDA-MB-231 and (C) 4T1 cells, without treatment (Control), treated with the control peptide 4NGG (300 μM) or PKHB1 (100, 200, 300, or 400 μM) for 2 h. (D) Representative graphs and quantification of the loss of ΔΨm measured through TMRE, (E) ROS levels measured through Hydroethidine staining and (F) intracellular Ca2+ by Fluo-4 staining, by flow cytometry in cells left alone or treated with the CC50 of PKHB1 (200 μM for MCF-7 and MDA-MB-231 and 300 μM for 4T1) for 2 h. (G-I) Cell death induced by PKHB1 (CC50), Epirubicin (42.5 μM for MCF-7 and MDA-MB-231 and 5 μM for 4T1) and H2O2 (25 μM for all cell lines) was assessed in cells left without pre-treatment (-) or pre-treated (30 minutes) with Z-VAD-FMK (Z-VAD), N-Acetyl Cysteine (NAC) and Necrostatin-1 (NEC-1). (J) Cell death induced by PKHB1 (CC50) was assessed in cells left without pre-treatment (-) or pre-treated (30 minutes) with BAPTA. Cell death induced by PKHB1 (CC50) was assessed as in J in cells left without pre-treatment (-) or pre-treated (30 minutes) with dantrolene, 2-APB or U73122. Graph represents the means (±SD) of triplicates of three independent experiments. NS = Not significant.
Next, we evaluated mitochondrial damage and cytosolic Ca2+ augmentation, and observed that PKHB1 (CC50) induced loss of mitochondrial membrane potential (Figure 1(d)), ROS production (Figure 1(e)) and increase of intracellular Ca2+ (Figure 1(f)) in all cell lines. Afterwards, we searched to determine the cell death effectors and evaluated ROS-dependence, also, we used inhibitors of caspases (Z-VAD), necroptosis (Nec-1), and the Ca2+-chelator (BAPTA) and assessed cell death. We used epirubicin (EPI) and H2O2 as controls for inhibition. We found that Z-VAD inhibited the cell death induced by EPI but not PKHB1-induced cell death (Figure 1(g)), while NAC and NEC-1 inhibited the cell dead induced by H2O2 but not PKHB1-mediated cell death (Figure 1(h,i)). Finally, we observed that the Ca2+ chelator BAPTA inhibited the Ca2+ augmentation (figure sup. 3) and cell death (Figure 1(j)), induced by PKHB1. This Ca2+ dependence was previously observed for leukemic cells,9,11 suggesting a similar cell death mechanism and common signaling pathway among solid and liquid cancers.
To assess this hypothesis, we used a phospholipase C (PLC) inhibitor (U73122) and ER receptor inhibitors (dantrolene, for ryanodine receptors, and 2-APB, for IP3 receptors). We determined that PKHB1-cell death is significatively inhibited when blocking the ER-Ca2+-channels with U73122, dantrolene and 2-APB (Figure 1(j,k) and Figure 1(l)) in breast cancer cells, confirming a similar cell death pathway induced by PKHB1 in breast cancer cells, as previously found in leukemic cells.
PKHB1 has antitumor effects in breast cancer and promotes intratumorally CD8 + T cell infiltration
To evaluate in vivo the potential antitumor effect of PKHB1, 4T1 breast cancer cells were grafted into BALB/c mice. Daily treatments were initiated when tumor volume reached approximately 100 mm3, and 16 days after the cell transplant, tumor volume of the control mice had reached 1500 mm3, requiring the sacrifice of the animals, while the tumor volume of the PKHB1-treated mice reached a maximum volume of 890 mm3 (at day 8) which started to decrease, reaching a volume of 570 mm3 at day 16 (Figure 2(a)) (individual growth curves in figure sup. 4). Also, daily treatment with PKHB1 did not affect mice weight (figure sup. 5). The decrease in tumor volume was correlated with the decrease in tumor weight, going from 1.5 grams in the controls to 0.40 grams in PKHB1-treated mice (Figure 2(b)). The decrease of tumor volume in mice treated with PKHB1, led us to evaluate the involvement of T cells in the observed effect. First, we analyzed histological sections of tumors from control (Figure 2(c)) or PKHB1-treated mice (Figure 2(d)); results revealed that tumors from control mice showed tumor cells (black arrows) with moderate mitotic activity (blue arrows), whereas the PKHB1-treated mice showed sporadic mitotic activity, extensive necrosis with abundant accumulation of cellular debris (green arrows), and abundant inflammatory exudate, composed of polymorphonuclear elements, eosinophils, and lymphoplasmacytic cells (red arrows).
Figure 2.
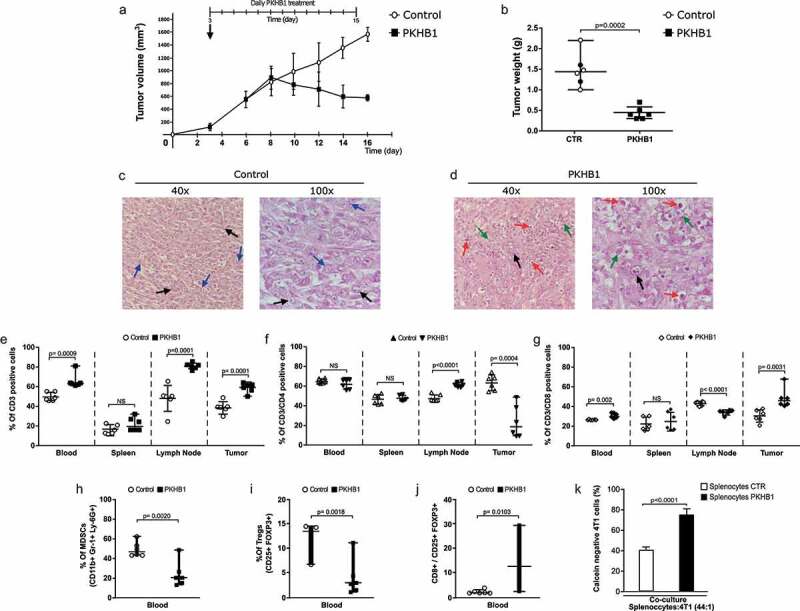
PKHB1 treatment induces tumor reduction and T cells distribution. Mice were inoculated s.c. with 5 × 105 4T1 viable cells and when tumor reached 70–120mm3 were treated with sterile water (Control, n = 6) or PKHB1 (400 μg of PKHB1 daily, n = 6), tumor volume was measured three times per week. (A) Graphs indicate the mean of the tumor volume in PBS-treated group (Control, n = 6) or PKHB1-treated group (PKHB1, n = 6). (B) Graphs indicate the tumor weight of control and treated mice at day 16. (C,D) Histology from tumors of control or PKHB1-treated mice stained with H&E. Tumor cells (black arrows), mitotic cells (blue arrows), cellular debris (green arrows) and immune system cells infiltration (red arrows). (E, F, and G) Graphs show the percent of CD3+, CD4+, and CD8+ cells in blood, spleen, lymph node, and tumor of control or PKHB1-treated mice at day 16. (H-I) Graphs show the percent of MDSCs and Tregs in blood of control or PKHB1-treated mice at day 16. (J) Graph show the ratio of CD8/Tregs cells of control or PKHB1-treated mice at day 16. (K) Graphs show the percent of calcein negative 4T1 cells after the co-culture with splenocytes of control (n = 4), or PKHB1-treated mice (n = 4). NS = Not significant.
Additionally, we evaluated if the cell number and distribution of T lymphocytes in peripheral blood, spleen, lymph nodes, and tumor site, changed after PKHB1 treatment. We observed (Figure 2(e)) that the percentage of CD3+ cells increased in blood, lymph nodes, and tumors of PKHB1-treated mice, while it was maintained in spleen. When we assessed CD4+ cells, the percentage of cells significantly augmented in lymph nodes, whereas it was significantly diminished in the tumor site (Figure 2(f)). Furthermore, CD8 + T cells significantly increased in peripheral blood and specially in tumor site (gating strategy in figure sup. 6), while they significantly diminished in lymph nodes (Figure 2(g)). To extend this analysis, we evaluated the proportion of myeloid-derived suppressor cells (MDSCs) and Tregs in peripheral blood of control and PKHB1-treated mice. We found a significative decrease in the percent of MDSCs (mean from 48% to 23%) and Tregs (mean from 12% to 3%) in mice treated with PKHB1, when compared with control mice (Figure 2(h,i)). Additionally, we determine a significative increase in the ratio of CD8/Tregs (mean 2% to 14%) in mice treated with PKHB1, when compared with control mice (Figure 2(j)). Finally, we determined whether splenocytes from PKHB1-treated mice could induce an antitumor cell cytotoxicity. For this purpose, we evaluated the calcein negative 4T1 cells after co-culture with splenocytes obtained from control or PKHB1-treated mice. In Figure 2(k), results show that splenocytes from PKHB1-treated mice induced a significant increase in calcein negative 4T1 cells (75%) in comparison with control mice (40%). These results improve the knowledge of the immune system-involvement in the antitumor effect mediated by PKHB1 treatment.
PKHB1 induces DAMPs exposure and release in breast cancer cell lines
As we observed that PKHB1 induced cell death in breast cancer cell lines and CD8 + T lymphocyte-recruitment in tumor site and the decrease of immunosuppressive cells, we wondered if cell death induced by PKHB1 was able to induce DAMPs’ exposure/release in breast cancer cells. The first step was to evaluate the exposure of CALR (one of the principal DAMPs related with ICD).17 Our results show that PKHB1-treatment and EPI-treatment were able to induce a significative increase of CALR positive cells in MCF-7 (Figure 3(a)), MDA-MB-231 (Figure 3(b)), and 4T1 (Figure 3(c)) cells. The CALR exposure induced by PKHB1 was confirmed by immunofluorescence microscopy, where we observed that PKHB1 induced CALR exposure in all the cases Figure 3(d,e) and Figure 3(f). Additionally, PKHB1 treatment induced 24 ± 3, 3.2 ± 1.3 and 4.23 ± 2-fold of HSP70 exposure (Figure 3(g)), 2.7 ± 0.6, 2 ± 0.6, and 7 ± 1.85-fold of HSP90 exposure (Figure 3(h)) in MCF-7, MDA-MB-231 and 4T1 cells, respectively, when compared with untreated cells.
Figure 3.
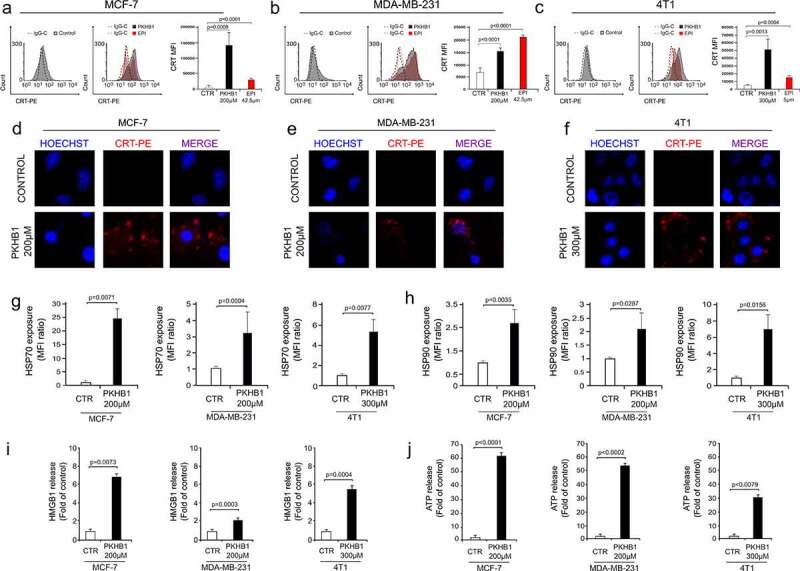
PKHB1 induces exposure and release of DAMPs in breast cancer cells. Representative FACS histograms of CALR exposure (filled histograms) and IgG isotype antibodies (open histograms) in non-permeabilized (7-AAD negative cells) (A) MCF-7, (B) MDA-MB-231, and (C) 4T1 cells, untreated (Control) or treated with the CC50 of PKHB1 (in gray) (200 μM for MCF-7 and MDA-MB-231 and 300 μM for 4T1) for 2 h or EPI (in red) (42.5 μM for MCF-7 and MDA-MB-231 and 5 μM for 4T1) for 24 h. Calreticulin exposure observed by confocal microscopy in (D) MCF-7, (E) MDA-MB-231, and (F) 4T1 cells untreated (Control) or treated with PKHB1 (CC50) using CALR-PE staining and Hoechst 33,342. Representative graphs of the ratio of HSP70 (G) or HSP90 (H) exposure in non-permeabilized cells (7-AAD negative cells), untreated (Control) or treated with PKHB1 (CC50) for 2 h. Representative graphs of the (I) HMGB1 or (J) ATP release in the supernatants of control or PKHB1 (CC50) treated cells. Graphs shown are means (± SD) of triplicates of three independent experiments. CALR-PE = Calreticulin-PhycoErythrin.
Finally, we wondered if PKHB1 induced the release of HMGB1 and ATP, two important DAMPs related with ICD.7 Therefore, the presence of HMGB1 and ATP was assessed in the supernatants of treated and untreated breast cancer cells. In Figure 3, results showed a significant release of HMGB1 (Figure 3(i)) and ATP (Figure 3(j)) in the supernatants of PKHB1-treated cells, when compared with untreated cells.
PKHB1-KC induce maturation of bone marrow-derived DCs and antitumor T cell responses
To assess the immunogenicity of the dead cells obtained upon treatment with PKHB1, 4T1 cells were treated with 400 μM of PKHB1, epirubicin (EPI) or freeze and thaw cycles (FT) as positive or negative controls (respectively) of ICD. The PKHB1-killed cells (PKHB1-KC), EPI-KC, or FT-KC were then prepared as described in the methods section, and its ability to induce DCs maturation was evaluated as we show in the schema of Figure 4(a). Thus, bone marrow-derived murine DCs were left untreated (Control) or pulsed for 24 h with the PKHB1-KC, EPI-KC, FT-KC, or LPS (1 μg/mL). After co-culture, DCs pulsed with the different stimulus (killed cells or LPS) maintained the expression of the DCs marker CD11c (Figure 4(b)), while only LPS induced a significative increase of the MHC-II in cell surface (Figure 4(c)). However, the PKHB1-KC, EPI-KC, and LPS induced a significant increase of the co-stimulatory molecule CD86 while no difference was observed in the DCs stimulated with the FT-KC (Figure 4(d)). Additionally, DCs pulsed with PKHB1-KC show a significant increase in CD80 cell surface expression and TNFα release in comparison with unstimulated DCs (figure sup. 7A-C).
Figure 4.
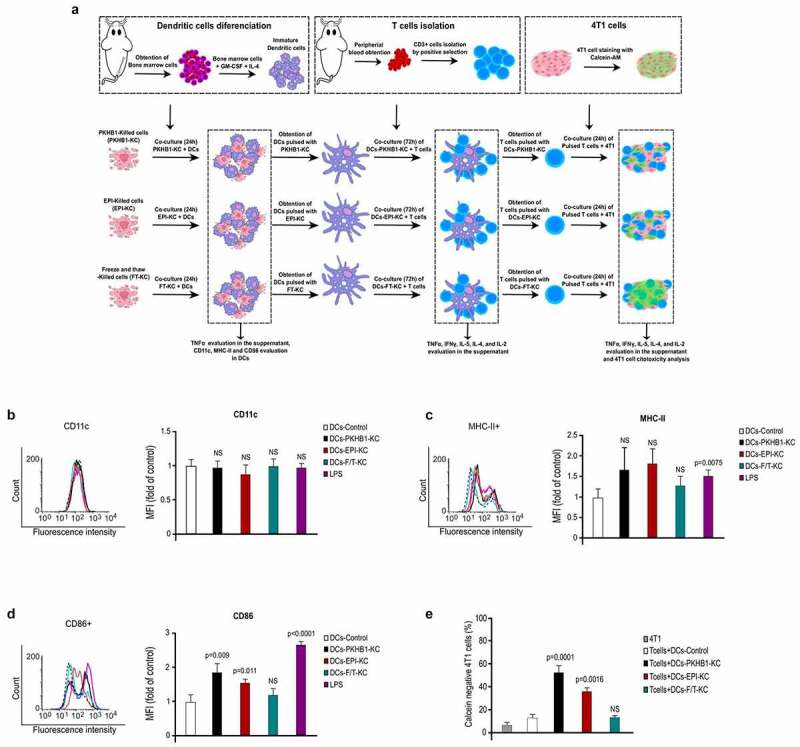
PKHB1-Killed cells induce antitumor immune responses ex vivo. (A) Schema of the ex vivo experiments. (B) Representative histograms from flow cytometry analyses of CD11c expression on DCs left with medium (CTR) or pulsed 24 h with a PKHB1-KC, EPI-KC, FT-KC, or LPS, graphs of the means obtained by FACS (right side). (C) Representative histograms from flow cytometry analyses of MHC-II expression on DCs treated as in A, graphs of the means obtained by FACS (right side). (D) Representative histograms from flow cytometry analyses of CD86 expression on DCs treated as in A, graphs of the means obtained by FACS (right side). (E) Graphs shown are means (± SD) of triplicates of three independent experiments from flow cytometry analyses of 4T1 cells stained with calcein-AM and co-cultured with T lymphocytes (unprimed or primed). NS = Not significant.
Once we determined DCs markers after co-culture with killed cells, we assessed if the DCs pulsed with the different killed cells (PKHB1-KC, EPI-KC or FT-KC) were able to prime T cells. First, primary T lymphocytes (CD3+ cells) were co-cultured for 96 h with pulsed or unpulsed DCs, and we observed the release of TNFα, IFNγ, and IL-2 in the co-culture of CD3+ and DCs-PKHB1-TCL (table sup. 2). Next, primed (co-cultured with pulsed DCs-PKHB1-KC, EPI-KC, or FT-KC) or unprimed (co-cultured with unstimulated DCs) T lymphocytes were collected and co-cultured during 24 h with viable 4T1 cells (previously stained with calcein-AM). To assess antitumor cell cytotoxicity, we evaluated the increase in calcein negative 4T1 cells after co-culture with primed or unprimed T lymphocytes. Results showed that only T lymphocytes co-cultured with pulsed DCs-PKHB1-KC and DCs-EPI-KC induced a significant increase in calcein negative 4T1 cells (Figure 4(e)), in comparison with the lymphocytes co-cultured with DCs-FT-KC or unprimed T lymphocytes. Additionally, lymphocytes stimulated with DCs-PKHB1-KC show a significant increase in IFNγ and IL-2 release in the co-culture with 4T1 cells in comparison with lymphocytes stimulated with control DCs (figure sup. 7D and E).
Prophylactic vaccination with PKHB1‐KC prevented tumor establishment of 4T1 cells
Considering that PKHB1 treatment induces tumor decline, infiltration of CD8+ cells into the tumor, DAMPs’ exposure and release, and the antitumor immune response ex vivo, the next step was to carry out the Gold Standard of ICD (prophylactic vaccination)18,19 to confirm whether PKHB1 induced ICD. The vaccine was based in the subcutaneous inoculation of the 4T1-PKHB1-KC, 7 days before the transplantation of viable 4T1 cells, while mice were inoculated with 4T1-EPI-KC used as a positive control, and controls without KC were injected with serum-free medium (Figure 5(a)). Results showed that vaccination with PKHB1‐KC prevented tumor establishment in 80% (8/10) of mice compared to 70% (7/10) of mice treated with EPI-KC. No survival (0%) was observed in the Control group inoculated with serum-free medium (Figure 5(b)) (individual growth curves in figure sup. 8 A-C). Additionally, survival rates of mice in each group were consistent with tumor growth, observing, respectively, 80% and 70% of survival in mice vaccinated with PKHB1-KC and EPI-KC by day 60, while control mice perished by day 21 (Figure 5(c)).
Figure 5.
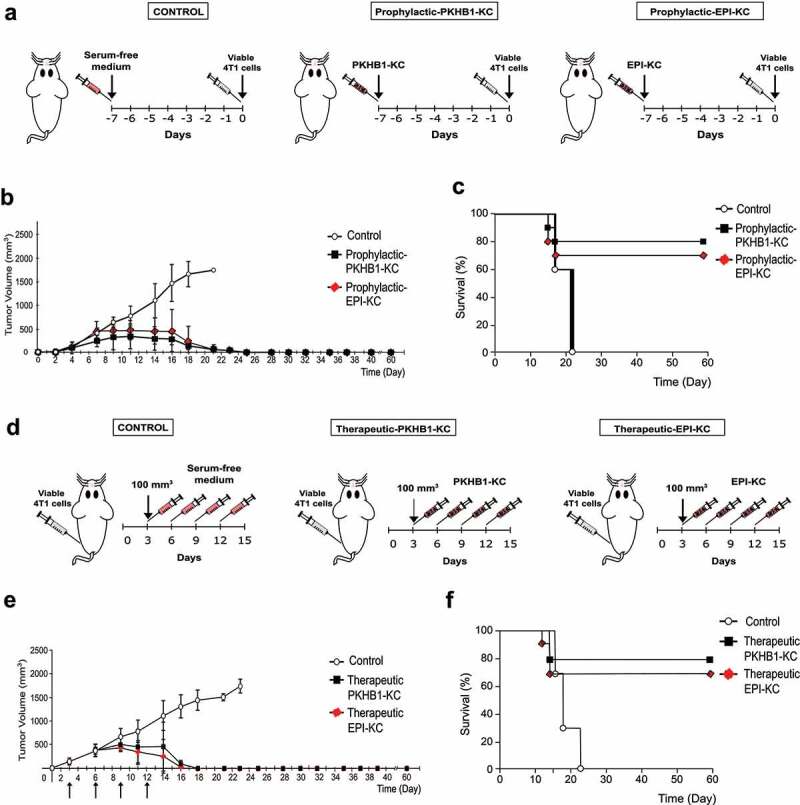
PKHB1-KC induce tumor elimination in a prophylactic and therapeutic application. (A) Schema of PKHB1-KC or EPI-KC prophylactic application. (B) Graph indicates the mean of the tumor growth in mice treated with serum-free medium (Control; n = 10) or mice receiving a prophylactic vaccination with PKHB1-KC (Prophylactic-PKHB1-KC, n = 10) or EPI-KC (Prophylactic-EPI-KC, n = 10). (C) Kaplan–Meier survival graph of mice treated as in B over time. (D) Schema of PKHB1-KC or EPI-KC therapeutic application. (E) Graph indicates the mean of the tumor growth in mice treated with serum-free medium (Control; n = 9), PKHB1-KC (Therapeutic-PKHB1-KC, n = 9) or EPI-KC (Therapeutic-EPI-KC, n = 9), arrows indicate days of KC or serum-free medium inoculation. (F) Kaplan–Meier survival graph of mice treated as in E over time.
Treatment with PKHB1-KC induces tumor regression
After ex vivo and in vivo results, we evaluated if the immunogenicity of PKHB1-KC was able to diminish tumor growth and improve overall survival in syngeneic mice bearing 4T1 tumors. First, 4T1 viable cells were inoculated in BALB/c mice. When tumor reached 70–120 mm3, a control group was treated with serum-free medium, a second group was treated with PKHB1-KC and the third group was treated with EPI-KC. All mice were treated two times per week for a total of four treatments (Figure 5(d)). Tumor growth measurements show that PKHB1-KC-treated mice had diminished tumor growth after day 10 (7 days after the first treatment), which continued to decrease until no tumor was detected by day 18, in the group of EPI-KC the tumor diminished after day 10 (7 days after the first treatment) which continued to decrease until no tumor was detected by day 16 (Figure 5(e)). Tumor growth diminution was reflected in overall mice survival, as PKHB1-KC-treated mice presented a 78% (7/9) of survival, while the EPI-KC-treated mice presented 67% (6/9) of survival, and all control mice perished by day 23 (Figure 5(f)) (individual growth curves in figure sup. 8D-F).
PKHB1-KC prophylactic and therapeutic vaccinations induce long-term antitumor effect
To assess the long-term antitumor response against 4T1 breast cancer cells induced by PKHB1-KC in a prophylactic or therapeutic application, mice in complete remission (tumor free >60 days) were re-challenged with living 4T1 cells. Tumor volume analysis showed that, contrary to naïve mice (Control), which showed a correct 4T1 tumor establishment, mice in remission after PKHB1-KC prophylactic or therapeutic application showed a slight increase in tumor volume at day 3, which immediately disappears by day 6 (Figure 6(a)). These results correlate with mice survival, where we observed that compared to naïve mice, in which a primary 4T1 cells challenge resulted in a 0% (0/9) of survival by day 23, those that were in remission after prophylactic or therapeutic application of PKHB1-KC were completely resistant to a re-challenge with 4T1 cells, resulting in a 100% (9/9) of survival (Figure 6(b)). Furthermore, we determined if splenocytes from re-challenged mice can induce an antitumor cell cytotoxicity. For this purpose, we evaluated the increase in calcein negative 4T1 cells after co-culture with splenocytes obtained from naïve or re-challenged mice. In Figure 6 (d), results showed that splenocytes from re-challenged mice induced a significant increase in calcein negative 4T1 cells (60%) in comparison with naïve mice (30%).
Figure 6.
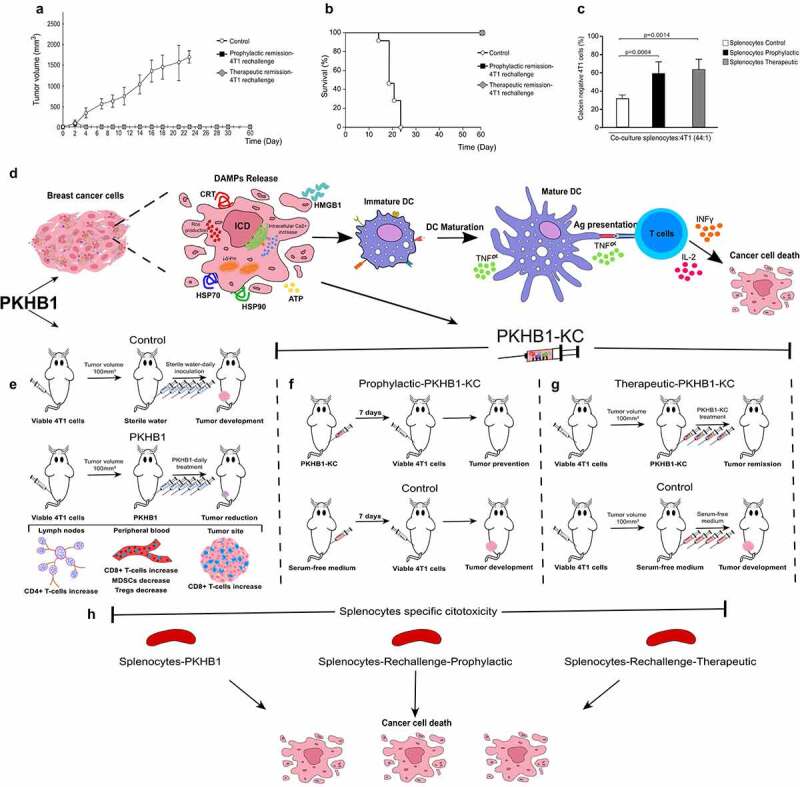
PKHB1-KC prophylactic and therapeutic application induces long-term antitumor effect, schematic representation of the PKHB1-effect. Mice in remission (30 days) after PKHB1 prophylactic or therapeutic PKHB1-KC application were re-challenged with 5 × 105 4T1 viable cells. (A) Graph indicates the mean of the tumor growth in PBS-treated group (Control; n = 9), prophylactic re-challenged group (Prophylactic remission-4T1 re-challenge, n = 9) or therapeutic re-challenged group (Therapeutic remission-4T1 re-challenge, n = 9). (B) Kaplan–Meier survival graph of mice treated as in A over time. (C) Graphs show the percent of calcein negative 4T1 cells after co-culture with splenocytes control (n = 4), prophylactic (n = 4) or therapeutic (n = 4) re-challenged mice. (D) PKHB1 induces ROS production, intracellular Ca2+ accumulation, loss of mitochondrial membrane potential (ΔΨm), leading to DAMPs exposure and release in breast cancer cells. Neoantigens and DAMPs exposure/release induced by PKHB1 promotes DCs maturation, which triggers T cell activation to induce cancer cytotoxicity. (E) However, PKHB1-treatment in vivo induces T cell redistribution in lymph nodes, peripheral blood and intratumorally, leading to tumor reduction. (F) PKHB1-KC prophylactic vaccination prevented tumor establishment. (G) PKHB1-KC therapeutic application induced tumor remission. (H) PKHB1-treatment, PKHB1-KC prophylactic or therapeutic application induced tumor-specific splenocytes’ cytotoxicity.
Discussion
Here, we assessed for the first time the characteristics of the cell death induced by PKHB1 in breast cancer cells, including the triple negative phenotype, which conserves the principal molecular characteristics of cell death (caspase-independent, calcium-dependent, PLC-dependent, and IP3R and RYR receptor-dependent cell death with the presence of ROS, loss of mitochondrial membrane potential and the intracellular accumulation of Ca2+) reported mainly in leukemic cells.9,11,12,14 We recently reported the overexpression of PLCγ1 and its importance in the cell death induced by PKHB1 in CLL cells,9 but although here we did not assess specially this isoform, it has been demonstrated the the overexpression of PLCγ1 in breast cancer patients is correlated with poor clinical outcome,20 and the overexpression of PLC-β,¹¯²PLC-ε, and PLC-δ has negative outcomes.21 Thus, the involvement of PLC in the mechanism of PKHB1-cell death, might have an advantage in the cancer cells that overexpress these proteins.
Our results also revealed the antitumor effect of PKHB1 against 4T1-breast cancer cells in vivo, as PKHB1-treatment diminished tumor volume and weight. Furthermore, we observed the distribution of T cells in PKHB1-treated mice that involves the increase of T cells (in blood, lymph node, and tumor), trafficking of CD4+ cells to lymph nodes, and tumor CD8+ cells infiltration that its associated to an antitumor response.22 Additionally, PKHB1 induced the decrease of immunosuppressive cells such as MDSCs and Tregs in blood, also, we observed an increase in the ratio of CD8+/Tregs, which has been related with an enhanced antitumor immune activity.23,24 Overall, our results are promising, and indicate that PKHB1 promote a robust antitumor immune response as it has been reported that extensive tumor infiltration by cytotoxic CD8 + T cells, the decrease of MDSCs and Tregs, and the increase in the ratio CD8+/Tregs are strongly associated with patient’s survival and response to therapy, even in different phenotypes of breast cancer.23–27 Finally, splenocytes from PKHB1-treated mice were more cytotoxic against breast cancer cells than splenocytes from control mice, probably due to the immunogenicity of the cell death triggered by PKHB1.22,28
The low immunogenicity of tumor cells is a main obstacle of antitumor therapies; therefore, a way to reactivate potent antitumor immune responses is through the emission of DAMPs 29 and dead cells-derived antigens, which can be achieved in the ICD.5,30 Here, we demonstrated that PKHB1 is capable of inducing CALR, HSP70, and HSP90 exposure, HMGB1 and ATP release, which can promote the uptake of dying cells, and the recruitment, maturation and cross-presentation activity of antigen-presenting cells (APCs).28,31 In this sense, we demonstrated that the cell death induced by PKHB1 and epirubicin (our positive control of ICD)32–34 are able to promote a mature phenotype of DCs,35 both induced a significant increase in the co-stimulatory molecule CD86, such as different ICD inductors.36 Additionally, we observed that DCs pulsed with the PKHB1-KC and EPI-KC promote the antitumor specific cytotoxicity of T cells, which confirm the phenotypic and functional maturation of DCs. Additionally, we observed that the freeze and thaw-killed cells do not stimulate the DCs, as other studies have demonstrated that freeze and thaw-killed cells suppress DCs maturation and function.37
However, vaccination assays involving syngeneic models are the gold standard to formally identify ICD inducers, since this demonstrates the tumor rejection capacity of the immunized host.29,38 Our results show that the prophylactic application of EPI-KC and PKHB1-KC prevented tumor establishment and increased survival in 70 and 80% (respectively) of the mice, without using adjuvants and with only one vaccination, in comparison with other strategies of prophylactic KC-vaccines.39–41 Also, we used Epirubicin, a well-known ICD inductor with major side-effects in human32,42 as a positive control, highlighting its ability to prevent the establishment of breast cancer in 70% of the mice. Our results are in line with the protective potential of established ICD inducers including oxaliplatin, doxorubicin, idarubicin, mitoxantrone, and specific forms of radiotherapy (in colon cancer) which presented between 80% and 90% of tumor-free mice17,43,44 while the antibody 7A7 (anti-EGFR) induced 50% of survival (lung cancer),45 and especially with the fact that an ICDinductor should display elevated tumor-free survival (>50%).19
The therapeutic application of the dead cells killed by a potential ICD inducer can be used as a confirmatory trial for ICD inductors, to evaluate their ability to mediate therapeutic effects depending on the immune system against established neoplasms.38 In this sense, our results show that when we treated tumor bearing mice with only four applications of PKHB1-KC, tumor volume decreased 7 days after the first administration, reaching tumor regression on day 18 in approximately 80% of mice, while in the group of EPI-KC the tumor diminished 7 days after the first treatment which continued to decrease until tumor regression on day 16. Our results highlight the immunogenicity of the PKHB1-induced cell death, because the therapeutic application of the PKHB1-KC induced tumor remission even in the absence of adjuvants. Additionally, we determined the therapeutic potential of the EPI-KC for the first time, as a novel strategy for the application of chemotherapy-ICD inductors. These results differentiate the PKHB1-KC from other therapeutic strategies with tumor lysates against melanoma, prostate and ovarian cancer, which have been poorly evaluated and were mainly used in combination with adjuvants.46,47 The success of the therapeutic application of PKHB1-KC in breast cancer was similar to the T-ALL model,14 despite their intrinsic molecular differences.16,48–50
The perspectives for cell therapy against cancer are based on the development of T cell responses, resulting in effective rejection of tumors and long-term protection.51,52 From this fact, the induction of ICD eventually results in long-lasting protective antitumor immunity.53 Our results demonstrate that PKHB1-KC induces long-term antitumor effect since 100% of mice in remission after PKHB1-KC prophylactic or therapeutic application survived at the re-challenge with 4T1 cells. Although immunotherapy with pulsed DCs, primed T lymphocytes or CAR-T cells is the main approach used to stimulate antitumor immune responses, they represent greater technical complexity, higher cost, among other disadvantages regarding the use of crude PKHB1-KC.54–56
Overall, our results demonstrate that PKHB1 is an ICD inductor in breast cancer cells and highlight a new approach for TSP-1 peptides mimic, which could induce ICD as a conserved mechanism of cell death in different types of tumor cells, including solid cancers, additionally, our results provide evidence for a novel strategy in the obtention and application of killed cancer cells against breast cancer.
Supplementary Material
Acknowledgments
We thank the SEP-CONACYT-ECOS-ANUIES grant 291297, PAICYT, the Laboratory of Immunology and Virology of the Faculty of Biological sciences, UANL, Kaybiotix and the DRUG Lab from Sorbonne University for the financial support and the facilities provided to achieve this work. KMCR, LGM, and ACUP thank CONACYT for their scholarship. LGM thanks SU/LBM/DRUG Lab/Kaybiotix for scholarship. PK is grateful to SATT-Lutech and DRI from SU, Kayvisa/Kaybiotix/χ-Pharma for financial support and Oncodesign for hosting the LBM DRUG Lab. We thank Alejandra Reyes-Ruiz and Martin Herrick Ramón Kane for technical help, Moises Armides Franco-Molina for the facilities and for his help, and Alejandra Arreola-Triana for editing and proofreading.
Funding Statement
This work was supported by SEP-CONACYT-ECOS-ANUIES, Grant/Award Number: 291297; the Laboratory of Immunology and Virology of the College of Biological Sciences; UANL; Sorbonne-Université, Laboratoire des Biomolécules, DRUGLAB, Kaybiotix. KMCR, LGM, and RMR hold a Consejo Nacional de Ciencia y Tecnología (CONACYT) scholarship. LGM hold a Kaybiotix grant; SATTLutech; Kayvisa/Kaybiotix/-Pharma.
Authors’ contributions
KMCR, ACUP and RMR carried out cell death, TMRE, and ROS assessment. KMCR and RMR carried out Ca2+ assessment. LGM carried out peptide synthesis. KMCR performed ex vivo and in vivo experiments. ACMT and PK conceived and supervised the work. KMCR and ACMT prepared the figures and wrote the manuscript. KMCR, LGM, RMR, ACUP, PK, ACMT, and CRP designed experiments, analyzed and interpreted data, and read and approved the final manuscript.
Availability of data and material
The data used to support the findings of this study are available from the corresponding authors upon request.
Ethics approval
The Animal Research and Welfare Ethics Committee (CEIBA), of the School of Biological Sciences approved this study: CEIBA-2018-003. All experiments were conducted according to Mexican regulation NOM-062-ZOO-1999.
Disclosure statement
The authors declare the following competing financial interest(s): a patent including results from this paper has been filed. The authors declare that no other competing interests exist.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021 May 1 Internet]. [cited 2022 Feb 7];71(3):209–15. Available from: https://onlinelibrary.wiley.com/doi/full/ 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2.Fortis SP, Sofopoulos M, Sotiriadou NN, Haritos C, Vaxevanis CK, Anastasopoulou EA, Janssen N, Arnogiannaki N, Ardavanis A, Pawelec G, et al. Differential intratumoral distributions of CD8 and CD163 immune cells as prognostic biomarkers in breast cancer. J Immunother Cancer. 2017 Apr 18 Internet]. [cited 2020 Jul 15];5(1):39. Available from: http://jitc.bmj.com/lookup/doi/ 10.1186/s40425-017-0240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groenendijk FH, Bernards R. Drug resistance to targeted therapies: déjà vu all over again [Internet]. Vol. 8:Molecular Oncology. Elsevier. 2014. cited 2020 Jul 15 1067–1083. Available from: https://febs.onlinelibrary.wiley.com/doi/full/ 10.1016/j.molonc.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020. Internet]. 2020 Aug 5 [cited 2022 Feb 7];17(12):725–741. Available from: https://www.nature.com/articles/s41571-020-0413-z [DOI] [PubMed] [Google Scholar]
- 5.Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. OncoImmunology. 2017. Oct 4;6(12):e1386829. doi: 10.1080/2162402X.2017.1386829. PMID: 29209573 Available from: https://www.tandfonline.com/doi/full/ 10.1080/2162402X.2017.1386829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017. Feb;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 7.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 8.Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer [Internet]. Journal for ImmunoTherapy of Cancer BioMed Central Ltd. 2016. [cited 2020 Jul 15];4(1):59. Available from http://jitc.bmj.com/lookup/doi/ 10.1186/s40425-016-0165-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Torres AC, Quiney C, Attout T, Boullet H, Herbi L, Vela L, Barbier S, Chateau D, Chapiro E, Nguyen-Khac F, et al. CD47 agonist peptides induce programmed cell death in refractory chronic lymphocytic leukemia B cells via PLCγ1 activation: evidence from mice and humans. PLoS Med. 2015;12(3). doi: 10.1371/journal.pmed.1001796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denèfle T, Boullet H, Herbi L, Newton C, Martinez-Torres A-C, Guez A, Pramil E, Quiney C, Pourcelot M, Levasseur MD, et al. Thrombospondin-1 mimetic agonist peptides induce selective death in tumor cells: design, Synthesis, and structure–activity relationship studies. J Med Chem. 2016 Sep 22 Internet]. [cited 2018 Dec 21];59(18):8412–8421. Available from http://pubs.acs.org/doi/ 10.1021/acs.jmedchem.6b00781 [DOI] [PubMed] [Google Scholar]
- 11.Uscanga‐Palomeque AC, Calvillo‐Rodríguez KM, Gómez‐Morales L, Lardé E, Denèfle T, Caballero‐Hernández D, Merle-Béral H, Susin SA, Karoyan P, Martínez-Torres AC, et al. CD 47 agonist peptide PKHB 1 induces immunogenic cell death in T‐cell acute lymphoblastic leukemia cells. Cancer Sci. 110:1. 256–268. Internet]. 2018 Dec 14 [cited 2018 Dec 21];cas.13885. Available from https://onlinelibrary.wiley.com/doi/abs/ 10.1111/cas.13885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denèfle T, Pramil E, Gómez-Morales L, Levasseur MD, Lardé E, Newton C, Herry K, Herbi L, Lamotte Y, Odile E, et al. Homotrimerization approach in the design of thrombospondin-1 mimetic peptides with improved potency in triggering regulated cell death of cancer cells. J Med Chem. 2019 Sep 12 Internet]. [cited 2019 Sep 26];62(17):7656–7668. Available from: https://pubs.acs.org/doi/ 10.1021/acs.jmedchem.9b00024 [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Torres A-C, Quiney C, Attout T, Boullet H, Herbi L, Vela L, Barbier S, Chateau D, Chapiro E, Nguyen-Khac F, et al. CD47 agonist peptides induce programmed cell death in refractory chronic lymphocytic leukemia B cells via PLCγ1 activation: evidence from mice and humans. PLOS Med. 12:3. e1001796. Internet]. 2015 Mar 1 [cited 2021 Aug 26];():. Available from https://journals.plos.org/plosmedicine/article?id= 10.1371/journal.pmed.1001796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez-Torres AC, Calvillo-Rodríguez KM, Uscanga-Palomeque AC, Gómez-Morales L, Mendoza-Reveles R, Caballero-Hernández D, Karoyan P, Rodríguez-Padilla C. PKHB1 tumor cell lysate induces antitumor immune system stimulation and tumor regression in syngeneic mice with tumoral T lymphoblasts. J Oncol. Internet]. 2019 Jun 4 [cited 2019 Sep 23];2019:1–11. Available from:. https://www.hindawi.com/journals/jo/2019/9852361/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kilkenny C, Brown WJ, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010. Aug;160(7):1577–9. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Current Protocols in Immunology. 2001;Chapter 20. doi: 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- 17.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 18.Garg AD, Krysko D V, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJM, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31(5):1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humeau J, Lévesque S, Kroemer G, Pol JG. Gold Standard Assessment of Immunogenic Cell Death in Oncological Mouse Models. In: López-Soto A, Folgueras A. (eds) Cancer Immunosurveillance. Methods in Molecular Biology, vol 1884. Humana Press, New York, NY; 2019. 10.1007/978-1-4939-8885-3_21 [DOI] [PubMed] [Google Scholar]
- 20.Lattanzio R, Iezzi M, Sala G, Tinari N, Falasca M, Alberti S, Buglioni S, Mottolese M, Perracchio L, Natali PG, et al. PLC-gamma-1 phosphorylation status is prognostic of metastatic risk in patients with early-stage luminal-A and -B breast cancer subtypes. BMC Cancer. 2019 Dec 30 Internet]. [cited 2021 Mar 4];19(1):747. Available from: https://bmccancer.biomedcentral.com/articles/ 10.1186/s12885-019-5949-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai S, Sun P-H, Resaul J, Shi L, Jiang A, Satherley LK, Davies EL, Ruge F, Douglas-Jones A, Jiang WG, et al. Expression of phospholipase C isozymes in human breast cancer and their clinical significance. Oncol Rep. 2017 Mar 1 Internet]. [cited 2021 Aug 10];37(3):1707–1715. Available from: http://www.spandidos-publications.com/ 10.3892/or.2017.5394/abstract [DOI] [PubMed] [Google Scholar]
- 22.Krekorian M, Fruhwirth GO, Srinivas M, Figdor CG, Heskamp S, Witney TH, Aarntzen EHJG. Imaging of T-cells and their responses during anti-cancer immunotherapy. Theranostics 2019; 9(25):7924–7947. doi: 10.7150/thno.37924. Available from https://www.thno.org/v09p7924.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020 Feb 27 Internet]. [cited 2022 Jan 27];9(3). Available from:561. /pmc/articles/PMC7140518/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markowitz J, Wesolowski R, Papenfuss T, Brooks TR, Carson WE. Myeloid derived suppressor cells in breast cancer. Breast Cancer Res Treat. 2013; Jul Internet]. [cited 2022 Jan 27];140(1):13. Available from: /pmc/articles/PMC3773691/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng GL, Li L, Guo YW, Yu P, Yin XJ, Wang S, Liu CP . CD8+ cytotoxic and FoxP3+ regulatory T lymphocytes serve as prognostic factors in breast cancer. Am J Transl Res. 2019. Aug 15;11(8):5039–5053. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6731430/ [PMC free article] [PubMed]
- 26.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Annal Oncol. 2015;26(2):259–271. 10.1093/annonc/mdu450. Available from: https://www.sciencedirect.com/science/article/pii/S0923753419313699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catacchio I, Silvestris N, Scarpi E, Schirosi L, Scattone A, Mangia A. Intratumoral, rather than stromal, CD8+ T cells could be a potential negative prognostic marker in invasive breast cancer patients. Transl Oncol. Internet]. 2019 Mar 1 [cited 2020 Jul 15];12(3):585–595. Available from: /pmc/articles/PMC6350084/?report=abstract: 10.1016/j.tranon.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 29.Garg AD, Dudek-Peric AM, Romano E, Agostinis P. Immunogenic cell death. Int J Dev Biol. Internet]. 2015. Sep 2 [cited 2018 Dec 21];59(1–3):131–140. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26374534 [DOI] [PubMed] [Google Scholar]
- 30.Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6. doi: 10.3389/fimmu.2015.00588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020. Mar;8(1):e000337. doi: 10.1136/jitc-2019-000337. Available from: https://jitc.bmj.com/content/8/1/e000337 [DOI] [PMC free article] [PubMed]
- 32.Li L, Li Y, Yang C, Radford DC, Wang J, Janát‐Amsbury M, Kopeček J, Yang J. Inhibition of immunosuppressive tumors by polymer‐assisted inductions of immunogenic cell death and multivalent PD‐L1 crosslinking. Adv Funct Mater. 2020 Mar 3 Internet]. [cited 2021 Mar 4];30(12):1908961. Available from: https://onlinelibrary.wiley.com/doi/abs/ 10.1002/adfm.201908961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L, et al. Trial watch: chemotherapy-induced immunogenic cell death in immuno-oncology. OncoImmunology. 9:1. 10.1080/2162402X20191703449 Internet]. 2020 Jan 1 [cited 2022 Jan 27];9(1):1703449. Available from. https://www.tandfonline.com/abs/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reyes-Ruiz A, Calvillo-Rodriguez KM, Martínez-Torres AC, Rodríguez-Padilla C. The bovine dialysable leukocyte extract IMMUNEPOTENT CRP induces immunogenic cell death in breast cancer cells leading to long-term antitumour memory. Br J Cancer. 2021. Internet]. 2021 Feb 3 [cited 2021 Aug 26];124(8):1398–1410. Available from: https://www.nature.com/articles/s41416-020-01256-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudek AM, Martin S, Garg AD, Agostinis PI. Semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol. Internet]. 2013 Dec 11 [cited 2018 Dec 21];4:438. Available from:. http://www.ncbi.nlm.nih.gov/pubmed/24376443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montico B, Nigro A, Casolaro V, Dal Col J. Immunogenic apoptosis as a novel tool for anticancer vaccine development [Internet]. International Journal of Molecular Sciences MDPI AG. 2018. [cited 2021 Jun 22]. Available from; 19. /pmc/articles/PMC5855816/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdellateif MS, Shaarawy SM, Kandeel EZ, El-Habashy AH, Salem ML, El-Houseini ME. A novel potential effective strategy for enhancing the antitumor immune response in breast cancer patients using a viable cancer cell-dendritic cell-based vaccine. Oncol Lett. 2018 Jul 1 Internet]. [cited 2022 Jan 27];16(1):529–535. Available from: http://www.spandidos-publications.com/ 10.3892/ol.2018.8631/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kepp O, Tartour E, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3(9):e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ, Moon JJ. Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett. 2017 Dec 13 Internet]. [cited 2021 Aug 10];17(12):7387–7393. Available from: https://pubs.acs.org/doi/abs/ 10.1021/acs.nanolett.7b03218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin B, Zhao H, Fan J, Xie F, Wang W, Ding X. B16 cell lysates plus polyinosinic-cytidylic acid effectively eradicate melanoma in a mouse model by acting as a prophylactic vaccine. Mol Med Rep. 2014 Aug 1 Internet]. [cited 2021 Aug 10];10(2):911–916. Available from: http://www.spandidos-publications.com/ 10.3892/mmr.2014.2241/abstract [DOI] [PubMed] [Google Scholar]
- 41.Salewski I, Gladbach YS, Kuntoff S, Irmscher N, Hahn O, Junghanss C, Maletzki C. In vivo vaccination with cell line-derived whole tumor lysates: neoantigen quality, not quantity matters. J Transl Med. 2020. Oct 21;18(1):402. doi: 10.1186/s12967-020-02570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Azambuja E, Ameye L, Diaz M, Vandenbossche S, Aftimos P, Hernández SB, Shih-Li C, Delhaye F, Focan C, Cornez N, et al. Cardiac assessment of early breast cancer patients 18 years after treatment with cyclophosphamide-, methotrexate-, fluorouracil- or epirubicin-based chemotherapy. Eur J Cancer. 2015 Nov 1, 51(17):2517–2524. doi: 10.1016/j.ejca.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 44.Gorin JB, Ménager J, Gouard S, Maurel C, Guilloux Y, Faivre-Chauvet A, Morgenstern A, Bruchertseifer F, Chérel M, Davodeau F, Gaschet J. Antitumor immunity induced after α irradiation. Neoplasia. 2014. Apr;16(4):319–328. doi: 10.1016/j.neo.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garrido G, Rabasa A, Sánchez B, López MV, Blanco R, López A, Hernández DR, Pérez R, Fernández LE. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J Immunol. 2011. Nov 15;187(10):4954–4966. doi: 10.4049/jimmunol.1003477. [DOI] [PubMed] [Google Scholar]
- 46.Chiang CL, Coukos G, Kandalaft LE . Whole Tumor Antigen Vaccines: Where Are We? Vaccines (Basel). 2015 Apr 23;3(2):344–72. doi: 10.3390/vaccines3020344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu A, Oh S, Gharagozlou S, Vedi RN, Ericson K, Low WC, Chen W, Ohlfest JR. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J Immunother. 2007. Nov-Dec;30(8):789–797. doi: 10.1097/CJI.0b013e318155a0f6. [DOI] [PubMed] [Google Scholar]
- 48.Irvin BJ, Williams BL, Nilson AE, Maynor HO, Abraham RT. Pleiotropic contributions of phospholipase C-gamma1 (PLC-gamma1) to T-cell antigen receptor-mediated signaling: reconstitution studies of a PLC-gamma1-deficient Jurkat T-cell line. Mol Cell Biol. 2002. Dec;20(24):9149–9161. doi: 10.1128/MCB.20.24.9149-9161.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10(3):448–460. doi: 10.1158/1535-7163.MCT-10-0923. [DOI] [PubMed] [Google Scholar]
- 50.Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, Iorio E, Molinari A, Ramoni C, Podo F, et al. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res. 2012;14(2). doi: 10.1186/bcr3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henry F, Bretaudeau L, Hequet A, Barbieux I, Lieubeau B, Meflah K, Grégoire M. Role of antigen-presenting cells in long-term antitumor response based on tumor-derived apoptotic body vaccination. Pathobiology. 1999;67(5–6):306–310. doi: 10.1159/000028086. [DOI] [PubMed] [Google Scholar]
- 52.Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14(1). doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: present and emerging inducers. [Internet]. Vol. 23, Journal of Cellular and Molecular Medicine Blackwell Publishing Inc; 2019. [cited 2020 Jul 21]. p. 4854–4865. Available from: /pmc/articles/PMC6653385/?report=abstract. ;23:8. 10.1111/jcmm.14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calmeiro J, Carrascal MA, Tavares AR, Ferreira DA, Gomes C, Falcão A, et al. Dendritic cell vaccines for cancer immunotherapy: the role of human conventional type 1 dendritic cells [Internet]. Vol. 12:Pharmaceutics. MDPI AG. 2020. [cited 2020 Jul 22]. Available from: /pmc/articles/PMC7076373/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. 2016. Jul;4(14):261. doi: 10.21037/atm.2016.04.01 Available from: https://atm.amegroups.com/article/view/10188/11634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Ventola C. Cancer immunotherapy, part 1: current strategies and agents. P T. Internet]. 2017 Jun 1 [cited 2020 Jul 22];42(6):375–383. Available from: /pmc/articles/PMC5440098/?report=abstract [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to support the findings of this study are available from the corresponding authors upon request.