
Figure 1.
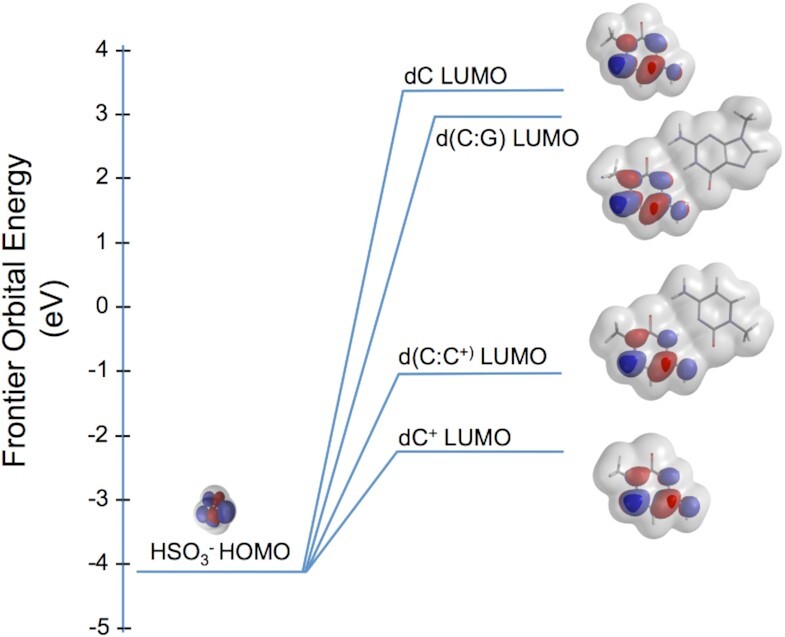
Frontier orbitals in the reaction between the bisulfite anion and cytosine in nucleic acids. The equilibrium geometry highest occupied molecular orbital (HOMO) for the bisulfite anion (lower left) is shown at its energy in eV under the transparent electron density that envelops of the molecule. Similar equilibrium geometry calculations, all at the Hartree-Fock 6–31G* level of theory, are depicted for the lowest unoccupied molecular orbital (LUMO) of models of cytosine in its several possible configurations in DNA. In each model the deoxyribose moiety at N1 of cytosine or guanine is modeled by a simple methyl group. Hence, the bases used in the electronic structure calculations are 1-methyl-cytosine, 1-methyl_cytoisne+ or 1-methyl-guanine. The geometry of the LUMO’s identify C6 of cytosine as the point of nucleophilic attack by the anion (blue center above the transparent electron density) in each model, and order the reactivity of each based on the LUMO energies in eV: dC+>d(C:C+)>>d(C:G)>dC.