
Fig. 1.
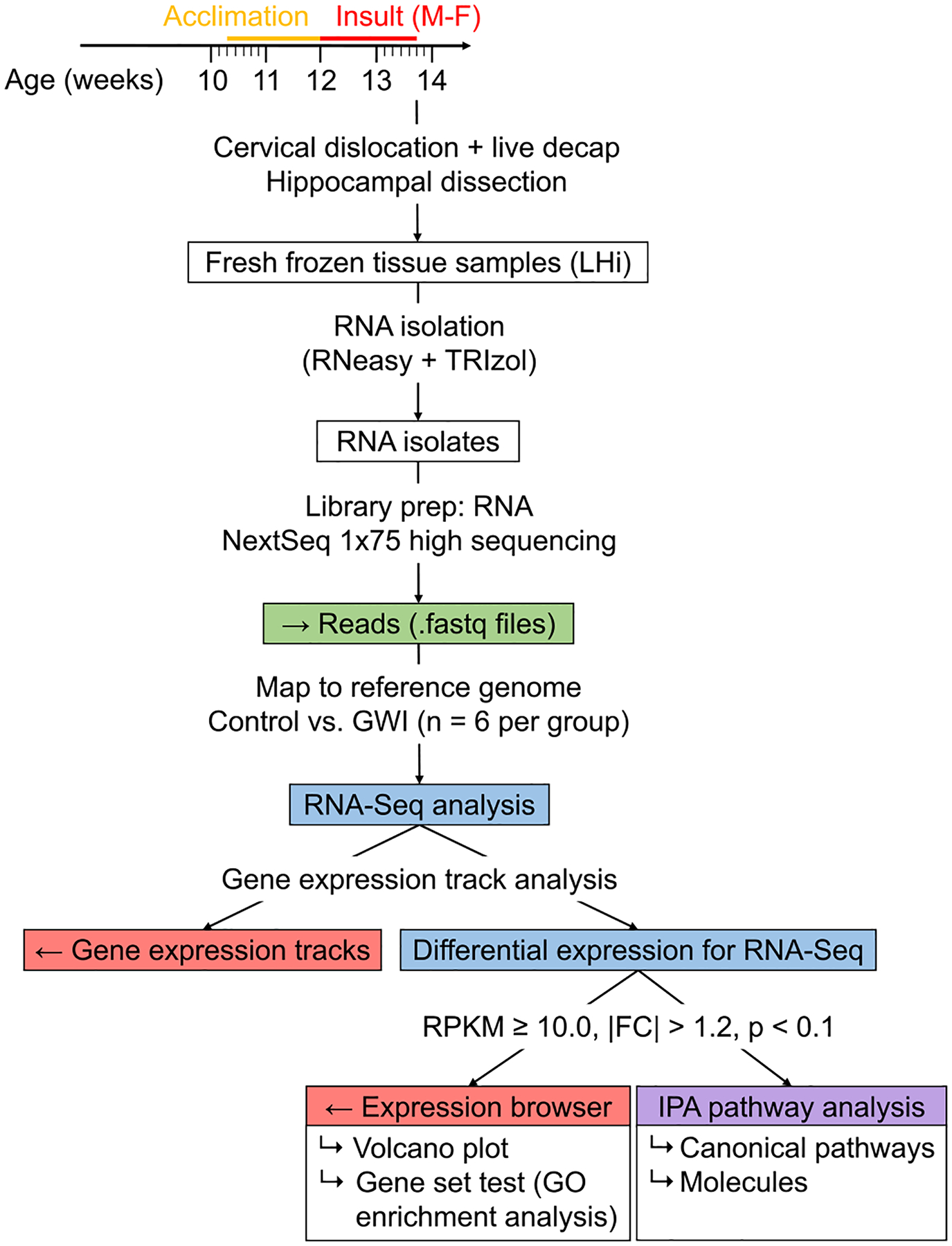
RNA-Seq analysis workflow with CLC Genomics Workbench and Ingenuity Pathway Analysis. Whole transcriptome sequencing was performed using mouse hippocampal RNA isolates collected 2–4 h after final exposure. Gene expression tracks were analyzed using the Differential Expression for RNA-Seq tool with RPKM >10.0, |FC| ≥ 1.2, and p < 0.1 as criteria for significance. GO enrichment analysis was performed on subset of genes that were significantly dysregulated. Data for significant genes was exported to Ingenuity Pathway Analysis to assess canonical pathways, molecules, diseases and functions, and other relevant information.