
FIGURE 3.
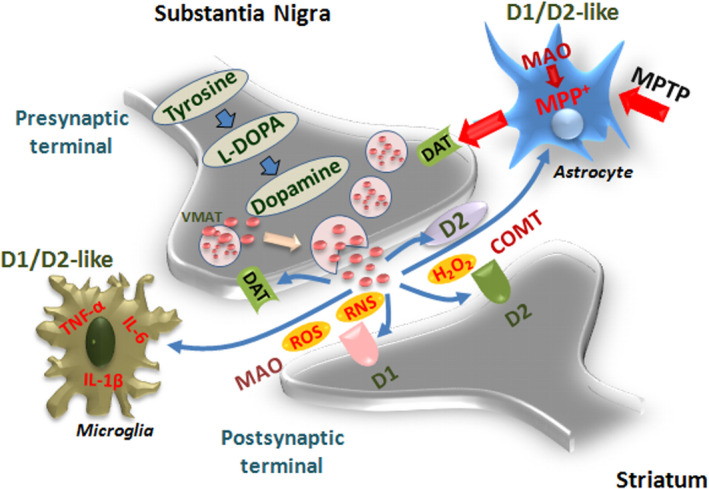
Dopamine metabolic pathways and astrocyte–microglial oxidative/inflammatory network. A schematic view of DA pre/postsynaptic regulatory functions. DA biosynthetic steps start with the action of the enzyme tyrosine hydroxylase (TH), the rate‐limiting step in the biosynthesis of DA in the presynaptic terminals to form the DA precursor, L‐DOPA, the principal drug in the therapeutic management of PD. Next, L‐DOPA is decarboxylated to form DA. DA is next incorporated into synaptic vesicles, via the vesicular monoamine transporter 2 (VMAT2), permitting its protection from metabolic inactivation, and its storage until stimulation, when DA released by exocytosis then reaches postsynaptic neurons and binds to cognate D1‐ and D2‐like receptors. D2 presynaptic (inhibitory) receptor can stop the further production and release of DA. The reuptake of DA by presynaptic terminals through the actions of the high‐affinity DA transporter (DT) represents another key step whereby DA is recycled back into the storage vesicles, responsible for modulating the concentration of extraneuronal DA in the brain. Two enzymes are responsible for DA inactivation, monoamine oxidases (MAOs) and catechol‐O‐methyl transferase (COMT), predominantly expressed by astrocytes. During DA metabolic steps, reactive oxygen (ROS) and nitrogen (RNS) species can be produced, which may further engender a neurotoxic cycle capable of causing cell death (for details, see the text). Astrocyte–neuron dialogue may be harmful upon exposure to 1‐methyl‐4‐phenyl‐1,2,3,6‐ tetrahydropyridine (MPTP), as the neurotoxin is converted to its active metabolite in astrocytes, MPP+, then specifically transported by DAT and concentrated within the nigral DA neurons where it inhibits complex I of the mitochondrial electron transport chain, resulting in ATP depletion and subsequent neuronal cell death. This process associated with a robust microgliosis and proinflammatory cytokines, tumor necrosis factor α (TNF‐α), and interleukin‐1β (IL‐1 β) production can be counter‐modulated by DA anti‐inflammatory effects via D1/D2‐like receptors in glial cells, as discussed in Sections 1.3–1.5