


Abstract
The DEAD-box protein Dbp5 is essential for RNA export, which involves regulation by the nucleoporins Gle1 and Nup159 at the cytoplasmic face of the nuclear pore complex (NPC). Mechanistic understanding of how these nucleoporins regulate RNA export requires analyses of the intrinsic and activated Dbp5 ATPase cycle. Here, kinetic and equilibrium analyses of the Saccharomyces cerevisiae Gle1-activated Dbp5 ATPase cycle are presented, indicating that Gle1 and ATP, but not ADP-Pi or ADP, binding to Dbp5 are thermodynamically coupled. As a result, Gle1 binds Dbp5-ATP > 100-fold more tightly than Dbp5 in other nucleotide states and Gle1 equilibrium binding of ATP to Dbp5 increases >150-fold via slowed ATP dissociation. Second, Gle1 accelerated Dbp5 ATPase activity by increasing the rate-limiting Pi release rate constant ∼20-fold, which remains rate limiting. These data show that Gle1 activates Dbp5 by modulating ATP binding and Pi release. These Gle1 activities are expected to facilitate ATPase cycling, ensuring a pool of ATP bound Dbp5 at NPCs to engage RNA during export. This work provides a mechanism of Gle1-activation of Dbp5 and a framework to understand the joint roles of Gle1, Nup159, and other nucleoporins in regulating Dbp5 to mediate RNA export and other Dbp5 functions in gene expression.
INTRODUCTION
DEAD-box proteins (DBPs) are ATPases that typically unwind and structurally reorganize RNA and ribonucleoprotein (RNP) complexes. As a large protein family in eukaryotes, bacteria, and archaea, DBPs support nearly all aspects of RNA metabolism (1), including ribosome biogenesis, mRNA splicing (2), RNA transport, translation (1) and RNA decay. DBPs are members of the SF2 family of helicases and defined by an enzymatic core formed by two RecA-like domains and a conserved Asp-Glu-Ala-Asp (D-E-A-D) amino acid sequence motif. The two RecA-like domains form a unit capable of binding ATP, RNA, and ATP hydrolysis, which is supported by the DEAD motif and 10 other characteristic sequence motifs within the helicase core (1).
Many DBPs are activated by RNA binding with an overall RNA-activated ATPase reaction cycle mechanism that appears to be conserved among DBP family members (1). During an ATPase cycle of ATP binding, hydrolysis, and product release there are a series of conformational states with distinct RNA binding properties (e.g. affinity, specificity). The functional diversity of DBPs is achieved through enzymatic adaptation of ATPase cycle kinetics and through interactions with specific regulatory proteins (2), which is coupled to specific biochemical activities (e.g. RNA duplex unwinding, protein displacement) and the individual physiological functions of the DBP.
Dbp5 (DDX19 in humans) is an essential Saccharomyces cerevisiae DBP required for mRNA export (3–6), with less defined roles in both ncRNA export and translation (7–9). Dbp5 is dynamically bound to the cytoplasmic face of a nuclear pore complex (NPC) via interaction with the nucleoporins Nup159 (Nup214 in humans) and Gle1, with a significant fraction of Dbp5 present in the cytoplasm and nucleoplasm (3,4,6,10). The interaction of Dbp5 and Nup159 occurs through an interaction with the β-propeller domain of Nup159 and the N-terminal RecA-like domain of Dbp5, which occludes RNA binding to Dbp5 (11–15). Nup214 binds DDX19 with the highest affinity in the absence of nucleotide (11) and therefore may act to dynamically increase the local concentration of Dbp5. Recent works have provided further insight into the context of these protein interactions at NPCs that addresses positioning of Dbp5/DDX19 via Nup159/Nup214 with respect to the transport channel (16–18).
Gle1 activates the intrinsic steady-state Dbp5 ATPase and is required for Dbp5-mediated mRNA nuclear transport (4,10) which is mediated in part by the endogenous small molecule inositol hexakisphosphate (InsP6) (19,20). Structural studies have shown that Gle1 engages both RecA-like domains of Dbp5 in the presence of ADP to orient the domains in an open conformation that is incompatible with RNA binding (12). A conformation that may organize the two RecA-like domains to facilitate ATP loading and/or release of inorganic phosphate (Pi) and ADP. In vitro assays indicate that Gle1 accelerates the maximum Dbp5 ATPase cycling rate constant (kcat) and lowers the Michaelis constant (KM) for ATP (12,19–21). Binding studies of an ATPase deficient Dbp5 mutant further suggest that Gle1 may promote both the binding of ATP by Dbp5 and release of a bound RNA substrate from Dbp5 (12,21).
In this work, transient kinetic analyses were used to elucidate the basis of Dbp5 ATPase activation by Gle1in the presence of InsP6. By measuring the effects of Gle1 on nucleotide binding, hydrolysis, and product release from Dbp5, it is demonstrated that Gle1 maximally activates the Dbp5 ATPase (kcat) by accelerating Pi product release and promoting ATP binding by slowing ATP dissociation. These results provide a kinetic scheme for the regulation of the Dbp5 ATPase by Gle1. Importantly, this work establishes a framework for developing a mechanistic description and functional understanding of Dbp5 regulation via Gle1-InsP6, Nup159 and other nucleoporins in the presence of RNA during nuclear export.
MATERIALS AND METHODS
Reagents: All reagents were of the highest purity commercially available. ATP (Sigma, A7699) concentrations were determined by absorbance using ϵ259 = 15,400 M−1cm−1. mantADP (Jena Biosciences, NU-201) concentrations were determined by absorbance using ϵ255 = 23,300 M−1cm−1. Inositol hexakisphosphate (phytic acid) was purchased from SantaCruz Biotechnology (SC-253276). Full length Dbp5 and a soluble truncated Gle1(a.a. 244–538) were purified as described (12) (Supplementary Figure S4). Note that InsP6 is included in all experiments at an equimolar concentration with Gle1. The affinity is high between Gle1-InsP6 (∼120 nM (20)), as such it is treated as a single species in this study and referred to as just Gle1 throughout the text. Gle1 (i.e. without Dbp5) did not demonstrate any significant ATPase activity. Buffers were made with either DEPC treated water (American Bio, AB021028) or Millipore MilliQ® distilled deionized water (ddH2O) that had been filtered through a 0.2-μm filter. Experiments were performed at 25°C in assay buffer: 30 mM HEPES (pH 7.5), 100 mM KCl and 2 mM DTT, 2 mM MgCl2. Equimolar magnesium (MgCl2) was added to all nucleotide stocks unless otherwise noted. The solution ionic strength changes <2-fold throughout the range of [ATP] examined (0–15 mM) without affecting the results (Supplementary Information, section S5).
Transient kinetic assays: Transient kinetic measurements were performed on an Applied Photophysics SX20 stopped-flow instrument thermostatted at 25 ± 0.1°C. mant-nucleotide binding to Dbp5 was monitored by FRET between excited tryptophans (λex = 280 nm) in Dbp5 and the bound mant-labeled nucleotide. Fluorescence intensity was measured at 90° relative to excitation light after passing through a 400-nm long-pass colored glass filter. Inner filter effects are minimal in the mant-labeled nucleotide concentration range employed (22,23). Time courses shown are averages of at least two traces. Fitting was performed by nonlinear least-squares regression, and uncertainties of quantities determined from fits are given as standard errors in the fits. All assays utilizing Gle1 include equimolar InsP6 in solution (19,20). Dbp5 and Gle1 were incubated for at least 2 h at room temperature in all assays utilizing preformed Gle1–Dbp5 complex.
mantADP dissociation kinetics: Irreversible dissociation of mantADP bound Dbp5 was achieved by mixing with a large excess of unlabeled ADP to prevent mantADP rebinding. mantADP dissociation from Dbp5 was measured as a function of Gle1 concentration ([Gle1]) by incubating 2 μM Dbp5, 40 μM mantADP, and various concentrations of Gle1 (0, 2, 4, 6, 10, 20, 60, 88 μM) for at least 2 h at room temperature in assay buffer before mixing with 20 mM ADP. Final concentrations after mixing are 1 μM Dbp5, 20 μM mantADP, 10 mM ADP and 0, 1, 2, 3, 5, 10, 30, 44 μM Gle1. Time courses of dissociation were fitted to single (0 μM Gle1) or double (2, 4, 6, 10, 20, 60, 88 μM Gle1) exponential functions. The [Gle1]-dependance of both the slow and fast phase observed rate constants were fitted to Equations (1) and (2), respectively.
mantADP binding kinetics: mantADP binding to Dbp5 in the presence varying concentration of [Gle1] was measured by incubating 2 μM Dbp5 with various concentrations of Gle1 (0, 2, 6, 10, 20 μM) for at least 2 hours at room temperature and subsequently mixed with 200 μM mantADP. Final concentrations after mixing are 1 μM Dbp5, 100 μM mantADP, and 0, 1, 3, 5 and 10 μM Gle1. The [Dbp5] and [Gle1] employed in this experiment (Figure 3) precludes an analytical solution to Scheme 1 with reasonable approximations. Therefore, unknown rate constants were not determined analytically. Rather, an upper limit of Gle1 binding affinity for Dbp5 was estimated from a trend in the data (see Results). The [Gle1]-dependence of the slow phase kobs was overlaid with a rectangular hyperbola to aid visualization.
mantADP binding to preformed Gle1–Dbp5 complex was measured as a function of [mantADP] by incubating 1 μM Dbp5 with 20 μM Gle1 for at least 2 hours at room temperature and subsequently mixed with various concentrations of mantADP (10, 20, 30, 40, 60, 80, 100, 140 μM). Final concentrations after mixing are 0.5 μM Dbp5, 10 μM Gle1 and 5, 10, 15, 20, 30, 40, 50, 70 μM mantADP. Time courses of mantADP binding were fitted to double exponential functions and the [mantADP]-dependance of the observed rate constants were globally fitted to the analytical solution of a two-step binding model (24).
Competition of mantADP and unlabeled ATP: Binding of ATP to Gle1–Dbp5 complex was measured as a function of [ATP] by incubating 2 μM Dbp5 with 20 μM Gle1 for at least 2 hours at room temperature and subsequently mixed with 40 μM mantADP with various concentrations of ATP (0, 20, 40, 60, 100, 140, 200, 600 μM). Final concentrations after mixing are 1 μM Dbp5, 10 μM Gle1, 20 μM mantADP, and 0, 10, 20, 30, 50, 80, 100, 300 μM ATP. Time courses of FRET signal change from mantADP binding to Dbp5 in the presence of varying amount of unlabeled nucleotide were fitted to a MATLAB simulation of Scheme 2. Time courses of mantADP binding were also fitted to a sum of two or three exponential functions. The resulting observed rate constants were globally fitted to Equations (S2.18, S2.19 and S2.24) with equilibrium and fundamental rate constants corresponding to Scheme 2 shared across λ1, λ2 and λ3. mantADP binding rate constants were fixed to values determined previously (Figures 2–4).
Quench flow: ATP hydrolysis by Gle1–Dbp5 complex was measured as a function of time at two different ATP concentrations by incubating 36 μM Dbp5 with 120 μM Gle1 for at least 2 h at room temperature and subsequently mixing with 76 or 340 μM 32P labeled ATP, aging for various times, and quenching with 5 M formic acid. Samples were spotted (0.5 μl) onto Cellulose F TLC plates (EMD Millipore, Billerica, MA) and resolved in 0.6 M KH2PO4 (pH 3.4) for 30 min. Plates were exposed to phosphor screen, read using Amersham™ Typhoon™ imager (GE Healthcare), and quantitated using Fiji (25) software. Time courses of hydrolyzed Pi (free and enzyme bound) were fitted to Equation (5) combined with Equations (6) and (7) (26). ATP binding and dissociation rate constants (k47 and k74, respectively) were constrained to 0.2 μM−1 s−1 and 4.1 s−1 as determined from kinetic competition of ATP and mantADP binding (Figure 4).
Phosphate binding protein: Pi release by Gle1–Dbp5 complex was measured from the 8-fold increase in fluorescence (λex = 436 nm, 463 nm long pass emission filter) of MDCC-labeled PiBP upon binding phosphate. PiBP binds Pi rapidly and with a tight affinity (Kd = 0.1 μM) providing real time detection of transient and steady-state Pi release (27). 1 μM Dbp5 was incubated with 20 μM Gle1 for at least 2 h at room temperature and subsequently mixing with 0, 10, 20, 30, 40, 100, 200 μM ATP with 6 μM MDCC labeled PiBP for single mixing experiments. Contaminating Pi was removed from the buffers and instrument with a ‘Pi mop’ consisting of 0.5 mM 7-methylguanosine and 0.01 U/ml purine nucleoside phosphorylase. PiBP fluorescence was converted to [Pi] using a phosphate standard calibration curve. PiBP fluorescence was converted to [Pi] using a Pi standard curve.
MATLAB fitting: Global fits to the mantADP binding time courses (Figures 3A and 4A) were carried out using a custom MATLAB program in which the concentrations of all species in Scheme 1 were solved for at each time step using the relevant differential equations and a built-in ordinary differential equation solver (ode45 or ode15s). Global parameter optimization was achieved by minimizing the total sum of squares for all experiments, i.e. simulated time courses of mantADP binding (Figures 3A and 4A) and dissociation (Figure 2A) using a built-in, non-linear least squares solver (lsqcurvefit). Briefly, residuals between the experimental and simulated data were calculated during each fitting iteration and trust-region-reflective algorithm was employed to modify open parameters until the total sum of squares is below the default cut-off value. Quench flow data was fitted similarly to an ATP hydrolysis reaction scheme (26) where the sum of [Pi] and [EP] were used in the fit.
Figure 3.
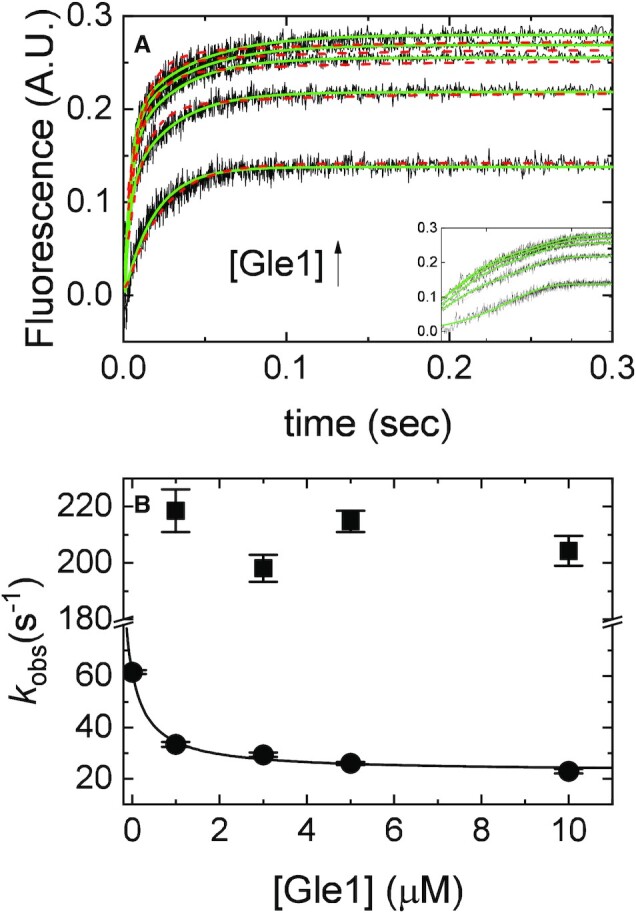
[Gle1]-dependence of mantADP binding to Dbp5. (A) Time courses of FRET signal change in pre-equilibrated solution of 2 μM Dbp5 (1 μM after mixing) with various concentrations of Gle1 ([Gle1] = 1–10 μM after mixing) upon rapid mixing with and equal volume of 200 μM mantADP (100 μM after mixing). Continuous lines through the data represent the best fits to exponential functions (solid lines) or the best global fits (with the data from Figures 2 and 4) to a kinetic simulation of Scheme 1 (dashed lines). The inset is a log-scale version of the same time courses in (A) (B). [Gle1]-dependence of the observed rate constants of mantADP binding to pre-formed Gle1–Dbp5 complex. The continuous line through the data represents the slow phase kobs (closed circles) overlaid on top with a rectangular hyperbola to aid visualization. Uncertainty bars represent standard error in the fits and are contained within the data points. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Scheme 1.
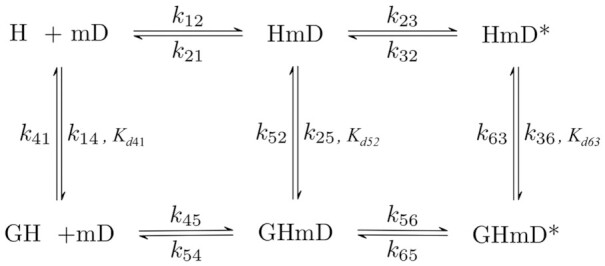
Two-step reaction mechanism for mantADP binding to Dbp5 and Gle1–Dbp5. H = Dbp5, G = Gle1- InsP6, mD = mantADP.
Scheme 2.
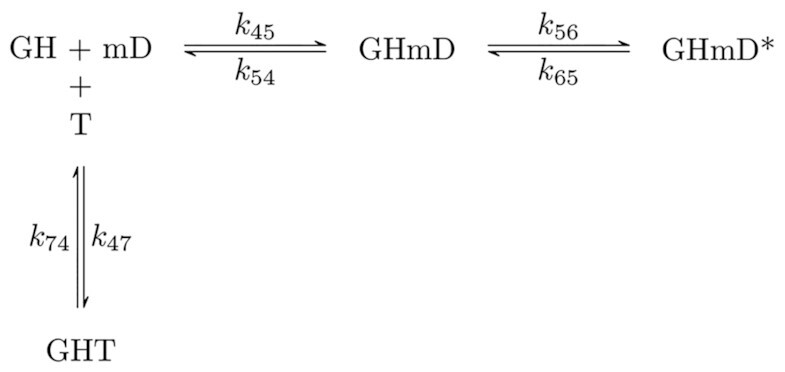
Minimal reaction scheme for kinetic competition between mantADP and ATP binding by Gle1–Dbp5. H = Dbp5, G = Gle1-InsP6, mD = mantADP, T = ATP.
Figure 2.
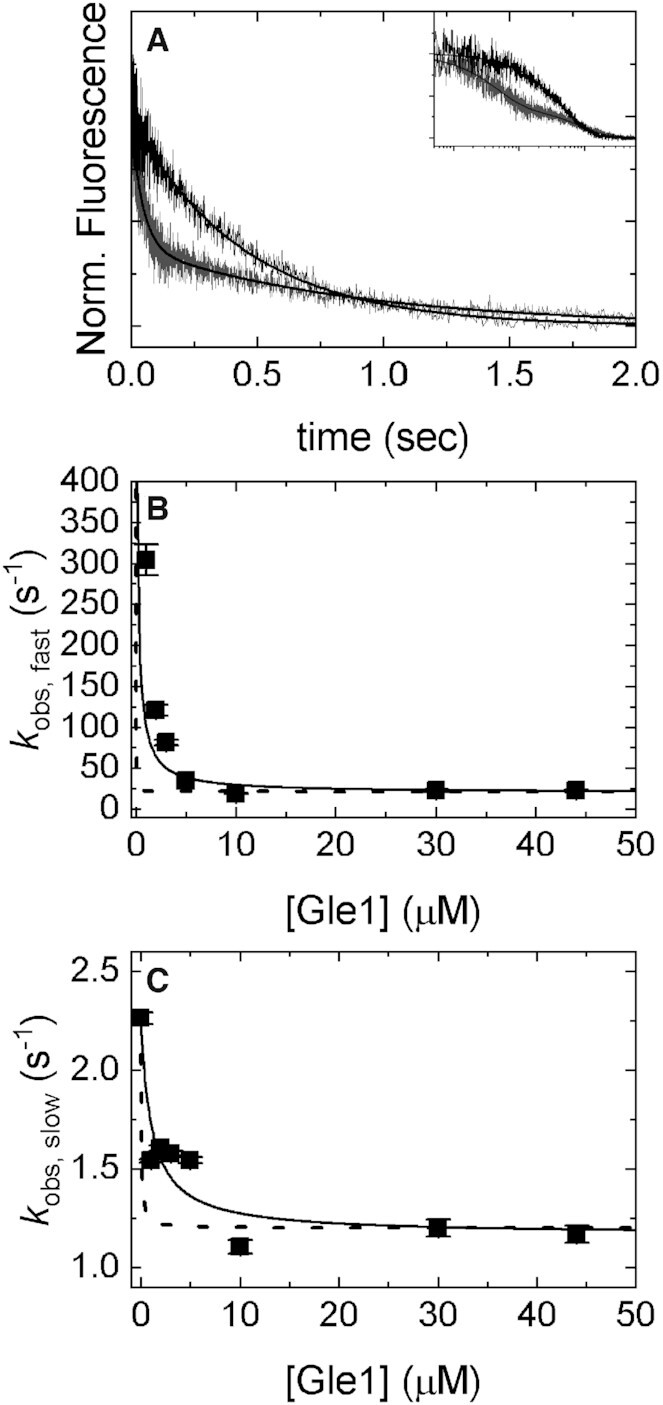
[Gle1]-dependence of mantADP dissociation from Dbp5. (A) Time courses of FRET signal changes after mixing a pre-equilibrated solution of Dbp5 (1 μM after mixing), 40 μM mantADP (20 μM after mixing), and various concentrations of Gle1 (0, 1, 2, 3, 5, 10, 30, 44 μM after mixing) with an equal volume of 20 mM ADP (10 mM after mixing). Continuous lines through the data represent the best fits to either single (0 μM Gle1) or double ([Gle1] > 0 μM) exponentials. The inset is a log-scale depiction of the same time courses in (A). (B) [Gle1]-dependence of the fast phase observed rate constants of mantADP dissociating from pre-formed Gle1–Dbp5(mant)ADP complex. Continuous lines through the data represent the best globally fits to a kinetic simulation of Scheme 1 with the data from Figures 3 and 4 (dashed lines) or the best fits to Equation (1) (continuous lines), which yields Kd52= 0.2 ± 0.1 μM. (C) [Gle1]-dependence of the slow phase observed rate constants of mantADP dissociating from pre-formed Gle1–Dbp5(mant)ADP complex. Continuous lines through the data represent the best globally fits to a kinetic simulation of Scheme 1 with the data from Figures 3 and 4 (dashed lines) or the best fits to Equation (2) (continuous lines) yielding Kd63 = 1.1 ± 0.6 μM. Fundamental rate constants garnered from the global fits are: k45 = 1 ± 0.4 s–1, k54 = 13 ± 6 s–1, k56 = 8 ± 7 s–1, k65 = 2 ± 1 s–1, Kd52 < 0.1 μM, Kd63 < 1 μM, Kd41 < 1 μM. Uncertainty bars represent standard error in the fits and are contained within the data points. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Figure 4.
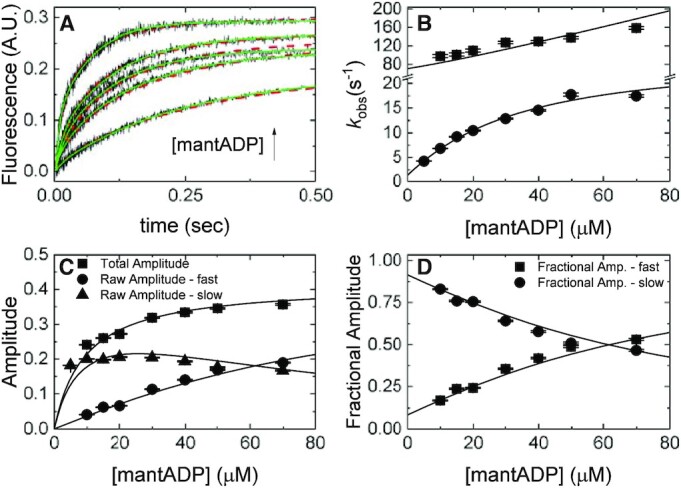
mantADP binding to Gle1–Dbp5. (A) Time courses of FRET signal changes in pre-equilibrated solution of 1 μM Dbp5 (0.5 μM after mixing) with 20 μM Gle1 (10 μM after mixing) upon rapidly mixing with an equal volume of various concentrations of mantADP (5–70 μM after mixing). Continuous lines through the data are the best fits to either double exponentials (solid lines) or global fits (with data from Figures 2B and 3) to a kinetic simulation of Scheme 1 (dashed lines). (B) [mantADP]-dependence of the observed rate constants for mantADP binding pre-formed Gle1–Dbp5 complex. Continuous lines through the data represent the best global fits to a two-step binding model (24). Rate constants resulting from this analysis are: k45 = 1.8 ± 0.14 μM–1 s–1, k54 = 47 ± 4.3 s–1, k56 = 24 ± 1.7 s–1, k65 = 2.1 ± 0.3 s–1. (C) [mantADP]-dependence of the fast and slow phase raw amplitudes and total amplitude for mantADP binding pre-formed Gle1–Dbp5 complex. (D) [mantADP]-dependence of the fast and slow phase fractional amplitudes for mantADP binding pre-formed Gle1–Dbp5 complex. Continuous lines through the data in (C) and (D) are simulated amplitudes using rate constants from fits in B. We include the amplitude data to demonstrate consistency with a two-step binding model. Uncertainty bars represent standard error in the fits and are contained within the data points. InsP6 is included in all experiments at an equimolar concentration with Gle1.
RESULTS
Previous work from others has shown that Dbp5 has a low intrinsic ATPase rate in the absence of RNA or other regulatory factors (12,19,20,28) (kcat ∼ 0.03 s−1, KM,ATP ∼ 2 mM) and Gle1 stimulates the maximum Dbp5 ATPase rate 3–5-fold (12,19,20) with an ‘apparent KM’ (KGle1; the [Gle1] needed for half maximum activation) of ∼0.4 μM (20). To validate the purified protein components used and experimental conditions that vary from previous work with respect to solution conditions, these parameters were again measured here. Consistent with previous determinations (12,19,20), Gle1 activated Dbp5 ATPase activity 5-fold from 0.03 to 0.16 s−1 with a KGle1 of 0.3 ± 0.1 μM in the presence of saturating ATP (Figure 1).
Figure 1.
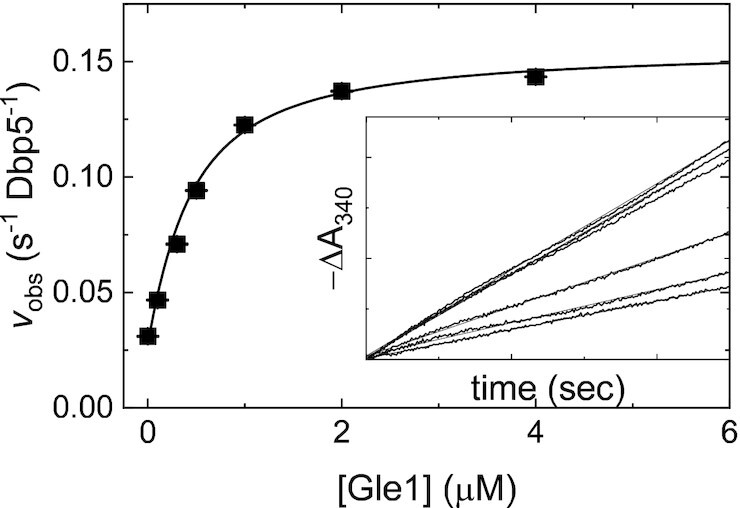
Gle1-stimulated steady-state ATPase activity of Dbp5. [Gle1]-dependance of the Gle1-stimulated Dbp5 steady-state ATPase rate (vobs) per enzyme. The continuous line through the data represents the best fit to Equation (2.9) in (31) yielding the maximum observed velocity vobs per enzyme (kcat= 0.16 ± 0.01 s–1 Dbp5–1) from the amplitude and KGle1 (apparent KM= 0.3 ± 0.1 μM) from the [Gle1] at half-maximum velocity (Table 1). Uncertainty bars represent standard errors in the fits and are contained within the data points. Inset: Time courses of absorbance change at 340 nm assayed with the NADH-coupled assay after mixing 200 nM Dbp5 (100 nM after mixing) and 30 mM ATP (15 mM after mixing) with various [Gle1] (0–4 μM after mixing). The continuous lines through the data represent the best fits to linear functions, yielding the steady-state ATPase rate from the slopes. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Assays for measuring nucleotide binding to Dbp5
Within the Dbp5 ATPase cycle, Gle1 is proposed to regulate ATP binding and RNA release (12,21), the latter being an activity that is directly influenced by changes in nucleotide binding status. Unfortunately, binding of unlabeled nucleotides (ADP or ATP) to Dbp5 yields no detectible spectroscopic signal, so nucleotide binding must be measured in kinetic competition with the fluorescent nucleotide, mantADP (23,29–31). The kinetics of mantADP binding to Dbp5 are significantly different from those of ADP, mostly due to the additional hydrophobic interaction provided by the mant moiety (28). However, since mantADP is only used as a signal source to assess Gle1 binding Dbp5 or ATP binding Gle1–Dbp5 any difference in binding kinetics caused by the mant moiety are trivial. The minimum reaction scheme considered for Dbp5, Gle1, and (mant)ADP binding involves six biochemical intermediates of Dbp5 with transitions defined by seven equilibrium and fourteen rate constants (Scheme 1). In the absence of Gle1 (top pathway of Scheme 1), mantADP binds Dbp5 following a two-step mechanism with the initial binding step in rapid equilibrium (k12 [mantADP] + k21 > 1000 s−1) with a Dbp5–(mant)ADP complex (HmD) that isomerizes (HmD*) (28). Gle1 binding to Dbp5 (32) and Dbp5–(mant)ADP (33) is then accounted for in the bottom pathway of Scheme 1.
Gle1 binds Dbp5-mantADP with an affinity ∼1 μM
The first part of Scheme 1 measured was the Gle1 affinity for Dbp5–(mant)ADP, which was estimated from the effect of Gle1 on (irreversible) mantADP release. A pre-equilibrated sample of 20 μM mantADP, 1 μM Dbp5, and a range of Gle1 concentrations ([Gle1] = 0, 1, 2, 3, 5, 10, 30, 44 μM) was rapidly mixed with 10 mM ADP. Time courses of fluorescence change, corresponding to mantADP dissociation, followed single exponentials in the absence of Gle1, while they followed double exponentials in the presence of Gle1 (Figure 2A). The effects of Gle1 on mantADP release are saturated at [Gle1] ≥10 μM (Figure 2B and C). Note that this analysis assumes that Gle1 binds Dbp5–(mant)ADP in a rapid equilibrium (i.e. Gle1 binding equilibrates faster than nucleotide is released). If this condition were not fulfilled, (at least) three exponentials would be observed in time courses of irreversible mantADP dissociation at sub-saturating [Gle1] ([Gle1] < Kd52, Kd63; Scheme 1, Figure 2A). The observed biphasic dissociation time courses are consistent with Gle1 binding of Dbp5–(mant)ADP in rapid equilibrium.
The fast and slow phases of mantADP dissociation are well separated in time, such that the rapid exponential decay is completed before the slower exponential decay appears. mantADP also dissociates more slowly when [Gle1] is saturating. Under these conditions, the two decays differ by an order of magnitude (fast observed rate constant is ∼25 s−1 and the slow observed rate constant is ∼2.2 s−1), providing temporal separation of these events and allowing independent analysis of the two phases. The fast phase of the dissociation time courses corresponds to mantADP dissociation from GHmD (Gle1–Dbp5–(mant)ADP) and HmD (Scheme 1). The slow phase originates from isomerization of GHmD* and HmD* to GHmD and HmD prior to release. The [Gle1]-dependence of the fast and slow observed mantADP dissociation rate constants (kobs, fast and kobs, slow) were fitted to the following equations accounting for the weighted population average of the parallel dissociation pathways (Scheme 1) (33):
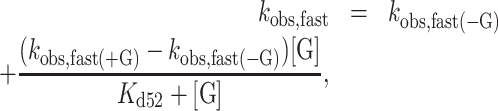 |
(1) |
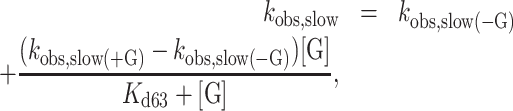 |
(2) |
where kobs, fast(−G) is the fast observed rate constant in the absence of Gle1, kobs, fast(+G) is the fast observed rate constant at saturating Gle1, [G] is the Gle1 concentration, Kd52 is the affinity of Gle1 for HmD, and Kd63 is the affinity of Gle1 for HmD*. The fast observed rate constant in the absence of Gle1 (kobs, fast(−G)) completed in the 1.2 msec instrument dead-time and was therefore constrained to 500 s−1 in fits to Equation 1. The best fits to Equations (1) and (2) yield kobs, fast(+G) = 19.8 ± 6.0 s−1, Kd52 = 0.2 ± 0.1 μM, kobs, slow(−G) = 2.2 ± 0.2 s−1, kobs, slow(+G) = 1.3 ± 0.1 s−1 and Kd63 = 1.1 ± 0.6 μM (Table 1), indicating that Gle1 binds Dbp5–(mant)ADP with an affinity ∼1 μM. Therefore at [Gle1] ≥ 10 μM, HmD and HmD* are bound to Gle1 and mantADP dissociation occurs exclusively through the bottom pathway of Scheme 1. The observed rate constants of mantADP dissociation are related to the fundamental (mant)ADP dissociation (k54) and isomerization (k56, k65) rate constants according to the following equations (24,34):
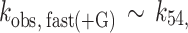 |
(3) |
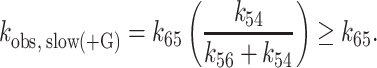 |
(4) |
Table 1.
Summary of rate and equilibrium constants for Dbp5 and Gle1–Dbp5 ATPase
| Parameter | Reaction | Value | Assay |
|---|---|---|---|
| Gle1 stimulated Dbp5 steady-state ATPase | |||
| kcat | Maximum Gle1 stimulated steady-state turnover rate | 0.15 (± 0.03) s−1 Dbp5−1 | NADH assay, Figure 7 |
| 0.16 (± 0.01) s−1 Dbp5−1 | NADH assay, Figure 1 | ||
| 0.12 (± 0.01) s−1 Dbp5−1 | PiBP, Figure 7 | ||
| 0.12 (± 0.07) s−1 Dbp5−1 | Predicted from individually measured rate and equilibrium constants, Equation (9) | ||
| KM,ATP | [ATP] at half maximal steady-state velocity | 20 (± 3) μM | NADH assay, Figure 7 |
| 26 (± 6) μM | PiBP, Figure 7 | ||
| 4 (±3) μM | Predicted from individually measured rate and equilibrium constants, Equation (11) | ||
| 15 (±13) μM | |||
| K Gle1 | [Gle1] at half maximal steady-state velocity | 0.3 (± 0.1) μM | NADH assay, Figure 1 |
| 0.24 (± 0.1) μM | Predicted from individually measured rate and equilibrium constants, Equation (10) | ||
| mantADP binding to Dbp5 | |||
| K d21 | Equilibrium constant for initial binding | 102 (± 21) μM | mantADP (28) |
| k 23 | Forward isomerization rate constant | 98 (± 15) s−1 | |
| k 32 | Reverse isomerization rate constant | 2.6 (± 0.003) s−1 | |
| K d32 | Isomerization equilibrium constant | 0.02 (± 0.003) | |
| K mD,overall | Overall mantADP binding affinity, ([HmD] + [HmD*]) | 2 (± 0.07) μM |
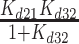
|
| mantADP binding to Gle1–Dbp5 | |||
| k 45 | Association rate constant | 1.8 (± 0.14) μM−1 s−1 | mantADP, Figure 4 |
| k 54 | Dissociation rate constant | 47 (± 4.3) s−1 | |
| 19.8 (± 6) s−1 | mantADP, Figure 2 | ||
| Kd 54 | Equilibrium constant for initial binding | 26 (± 3.2) μM |
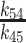
|
| 11 (± 3.4) μM | |||
| k 56 | Forward isomerization rate constant | 24 (± 1.7) s−1 | mantADP, Figure 4 |
| k 65 | Reverse isomerization rate constant | 2.1 (± 0.3) s−1 | |
| ≥1.3 (± 0.1) s−1 | mantADP, Figure 2 | ||
| 2 (± 1) s−1 | mantADP, Global fit of Figures 2–4 | ||
| K d65 | Isomerization equilibrium constant | 0.09 (± 0.01) |
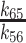
|
| ≥0.07 (± 0.006) | |||
| KmD, overall | Overall mantADP binding affinity, ([GHmD] + [GHmD*]) | 2.2 (± 0.3) μM |
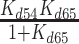
|
| ATP binding to Dbp5 | |||
| K d101 | ATP equilibrium binding affinity | 3 (± 0.4) mM | Kinetic competition of ATP and mantADP, (28) |
| ATP binding to Gle1–Dbp5 | |||
| k 47 | Association rate constant | 0.2 (± 0.1) μM−1 s−1 | Kinetic competition of ATP and mantADP, global MATLAB fit (dashed lines), Figure 5A |
| 0.6 (± 0.1) μM−1 s−1 | Kinetic competition of ATP and mantADP, Equations S2.18, S2.19 and S2.24, Figure 5B | ||
| k74 | Dissociation rate constant | 4.1 (± 2.5) s−1 | Kinetic competition of ATP and mantADP, global MATLAB fits (dashed lines), Figure 5A |
| 3.3 (± 1.1) s−1 | Kinetic competition of ATP and mantADP, Equations (S2.18), (S2.19) and (S2.24), Figure 5B | ||
| K d74 | ATP equilibrium binding affinity | 20 (± 16) μM | Ratio of rate constants from global MATLAB fits |
| 5.5 (± 2) μM | Ratio of rate constants from Equations (S2.18), (S2.19) and (S2.24) | ||
| ATP Hydrolysis by Dbp5 | |||
| k 1011 | ATP hydrolysis rate constant | 0.16 (± 5 × 10−4) s−1 | KinTek steady-state simulation, (28) |
| 2.2 (± 0.4) s−1 | PiBP, reference (28) | ||
| k 1110 | ATP resynthesis rate constant | 6 × 10−4 (± 8 × 10−7) s−1 | KinTek steady-state simulation, (28) |
| 2 × 10−4 (± 5 × 10−5) s−1 | PiBP and isotope exchange, (28) | ||
| K d1110 | Equilibrium constant for ATP hydrolysis | 0.004 (± 5 × 10−4) |
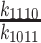
|
| 10−4 (± 3 × 10−5) | |||
| ATP Hydrolysis by Gle1–Dbp5 | |||
| k 78 | ATP hydrolysis rate constant | 0.6 (± 0.2) s−1 | Quench-flow, Figure 6 |
| k 87 | ATP resynthesis rate constant | 1.0 (± 0.9) s−1 | |
| K d87 | Equilibrium constant for ATP hydrolysis | 1.7 (± 1.6) |
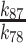
|
| Pi release from Dbp5-ADP-Pi | |||
| k 1112 | Rate constant for Pi release from HDPi | 0.02 (± 0.1) s−1 | PiBP, (28) |
| K d1211 | Equilibrium constant for Pi binding HD | >10 mM | Steady-state Pi inhibition, (28) |
| Pi release from Gle1–Dbp5-ADP-Pi | |||
| k89 | Rate constant for Pi release from GHDPi | 0.4 (± 0.1) s−1 | Quench-flow, Figure 6 |
| K d89 | Equilibrium constant for Pi binding GHD | >10 mM | Supplemental Information, section S4 |
| ADP binding Dbp5 | |||
| K d121 | Equilibrium constant for ADP binding H | 360 (± 50) μM | Kinetic competition of ADP and mantADP, (28) |
| ADP binding Gle1–Dbp5 | |||
| K d94 | Equilibrium constant for ADP binding GH | 240 (± 15) μM | Kinetic competition of ADP and mantADP, Supplementary Figure S3 |
| Gle1 binding Dbp5-ATP | |||
| K d107 | Equilibrium constant of Gle1 binding to HT | 1.5 (± 0.8) nM | Detailed balance of Scheme 3, Supplemental Information section S6 |
| Gle1 binding Dbp5-ADP-Pi | |||
| K d118 | Equilibrium constant for Gle1 binding to HDPi | 0.6 (± 0.2) μM | Detailed balance of Scheme 3, Supplemental Information section S6 |
| Gle1 binding Dbp5-ADP | |||
| K d129 | Equilibrium constant for Gle1 binding HD | 0.5 (± 0.2) μM | Detailed balance of Scheme 3, Supplemental Information section S6 |
| Gle1 binding to Dbp5 | |||
| K d41 | Equilibrium constant for G binding H | 0.8 (± 0.3) μM | mantADP, Detailed balance of Scheme 1, Figure 3 |
| Gle1 Dbp5-mantADP complexes | |||
| K d52 | Equilibrium constant for G binding HmD | 0.2 (± 0.1) μM | mantADP, Figure 2 |
| K d63 | Equilibrium constant for G binding HmD* | 1.1 (± 0.6) μM | |
| K D,overall | Equilibrium constant for G binding Dbp5-mantADP | 0.17 (± 0.15) μM |
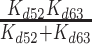
|
the values of k54 (19.8 ± 6.0 s−1) and k65 (≥ 1.3 ± 0.1 s−1) approximated in Equations (3) and (4), respectively, are consistent with more accurate determinations obtained from mantADP binding to Gle1–Dbp5 (discussed below, Figure 4; Table 1).
Gle1 binds Dbp5 (no nucleotide) with an affinity <1 μM
To measure the binding affinity of Gle1 for the nucleotide free form of Dbp5, the effect of Gle1 on mantADP binding to Dbp5 was used. A solution of 1 μM Dbp5 was equilibrated with a range of [Gle1] (0, 1, 3, 5, 10 μM) and rapidly mixed with 100 μM mantADP in a stopped flow apparatus and the time course of fluorescence change monitored. Time courses of mantADP binding under these pseudo first-order conditions ([mantADP] >> [Dbp5] and [Gle1–Dbp5]) followed a single exponential in the absence of Gle1 and double exponentials in the presence of Gle1 (Figure 3A).
In the absence of Gle1, mantADP binds Dbp5 following a two-step mechanism with the first step rapidly equilibrating within the dead time of the stopped flow instrument such that time courses of mantADP binding followed single exponentials (28). Gle1 slowed mantADP binding to Dbp5, which allows both binding phases to be observed (Figure 3). Due to the temporal separation of the two observed mantADP binding phases, it is assumed the first step for mantADP binding Gle1–Dbp5 equilibrates before the second step occurs. Therefore, the slow mantADP binding phase was analyzed independent of the fast phase. The [Dbp5] and [Gle1] employed preclude an analytical solution to Scheme 1 with reasonable approximations; moreover, little change in the [Gle1]-dependent kobs of both fast and slow phases occurs at >3–5 μM Gle1 (Figure 3B). Dbp5–(mant)ADP is saturated when [Gle1] ≥10 μM (Figure 2) indicating the affinity of Gle1 for Dbp5 in the absence of nucleotides (Kd41) is ≤1 μM (Table 1). Consequently, at [Gle1] ≥10 μM mantADP binding proceed exclusively through the bottom pathway in Scheme 1, defining experimental conditions under which nucleotide binding to the Gle1–Dbp5 complex could be measured.
mantADP binds Gle1–Dbp5 following a two-step binding mechanism
The preceding section established that Gle1 binds Dbp5 and Dbp5–(mant)ADP with affinities ≤ 1 μM, defining experimental conditions under which nucleotide binding to the Gle1–Dbp5 complex could be measured, specifically when maintaining [Gle1] ≥ 10 μM. Using this condition, the kinetics of mantADP binding to the Gle1–Dbp5 complex (bottom pathway of Scheme 1) was measured by rapidly mixing mantADP (5–70 μM) with an equilibrated mixture of 0.5 μM Dbp5 and 10 μM Gle1. Time courses of mantADP binding were best fitted by a sum of double exponential functions (Figure 4A, solid lines).
The [mantADP]-dependence of the observed rate constants (kobs) were globally fitted to a two-step binding model (24) (Figure 4B), yielding the rate and equilibrium constants for mantADP binding to Gle1–Dbp5 (k45 = 1.8 ± 0.14 μM−1 s−1, k54 = 47 ± 4.3 s−1, Kd54 = 26 ± 3.2 μM, Table 1) and subsequent isomerization of the Gle1–Dbp5–(mant)ADP complex (k56 = 24 ± 1.7 s−1, k65 = 2.1 ± 0.3 s−1, Kd65 = 0.09 ± 0.01, Table 1). The overall mantADP affinity for Dbp5 is ∼2 μM (28) and ∼2.2 μM for Gle1–Dbp5 (Table 1), indicating that Gle1 has a modest effect on the (mant)ADP binding affinity. Measurements with unlabeled ADP yield a similar conclusion (discussed below; Supplementary Figure S3).
Globally fitting the mantADP binding and release data (Figures 2-4) to the reaction matrix outlined in Scheme 1 via a custom MATLAB program yielded a shared parameter set consistent with those determined from independent analysis of the individual experimental data sets (Supplemental Information, section S1; Supplementary Figure S1).
Gle1 promotes ATP binding to Dbp5
Unlabeled ATP binding to Gle1–Dbp5 complex was then measured by kinetic competition between mantADP and ATP. A solution of 1 μM Dbp5 was incubated with 10 μM Gle1 and rapidly mixed with 20 μM mantADP and various concentrations of ATP (0–300 μM). Time courses of mantADP binding in the presence of ATP were best fitted by a sum of three exponentials. Since mantADP binds rapidly and equilibrates before any significant ATP hydrolysis occurs (see below), ATP hydrolysis was uncoupled from binding and the time courses reflect only competitive binding of the two nucleotides (Scheme 2). Kinetic competition time courses were fitted to Scheme 2 with numerical analysis using a custom MATLAB program (Figure 5A, dashed lines; Supplementary Figure S2).
Figure 5.
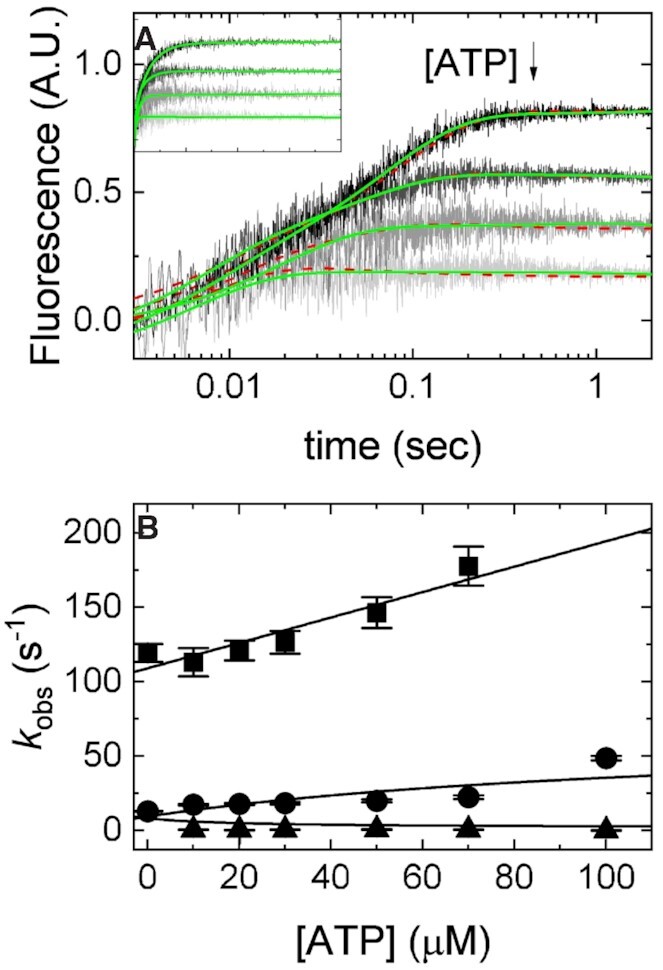
Kinetic competition between mantADP and ATP. (A) Time courses of FRET signal changes in pre-equilibrated solution of 2 μM Dbp5 (1 μM after mixing) with 20 μM Gle1 (10 μM after mixing) upon rapid mixing with an equal volume of 40 μM mantADP (20 μM after mixing) with various concentrations of ATP (from 0 to 300 μM after mixing). Continuous lines through the data are either the best fits to double or triple exponential functions (solid lines) or the best fits to a kinetic simulation of Scheme 2 (dashed lines). (B) [ATP]-dependence of the observed rate constants from exponential fits in (A) (solid lines) for mantADP binding pre-formed Gle1–Dbp5 complex. Continuous lines through the data are the best global fits to Supplemental Equations (S2.18), (S2.19) and (S2.24). Uncertainty bars represent standard error in the fits and are contained within the data points. For both fits, rate constants for mantADP binding were held to values determined from fits in Figure 4B. Fundamental rate constants resulting from the fits to a kinetic simulation of Scheme 2 are k47 = 0.2 ± 0.1 μM–1 s–1 and k74 = 4.1 ± 2.5 s–1, while fits to Equations S2.18, S2.19 and S2.24 yield k47 = 0.6 ± 0.1 μM–1 s–1 and k74 = 3.3 ± 1.1 s–1. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Best fits of the data yield association (k47) and dissociation (k74) rate constants of 0.2 ± 0.1 μM−1 s−1 and 4.1 ± 2.5 s−1 for ATP binding to the Gle1–Dbp5 complex (Table 1). Similar values for ATP binding and dissociation to Gle1–Dbp5 were obtained from an approximate analytical solution of Scheme 2 (Supplemental Information, section S2-2). Together these data suggest that the presence of Gle1 in complex with Dbp5 slows both the dissociation and binding of ATP. However, the much larger effect on ATP dissociation from Dbp5 by nearly 3-orders of magnitude, results in Gle1 effectively increasing the ATP affinity of Dbp5 ∼150-fold.
Gle1 accelerates ATPase activity of Dbp5 by increasing the Pi release rate constant
The increased ATP binding affinity of the Gle1–Dbp5 complex allowed for the direct measurement of both the rate and equilibrium constants for ATP hydrolysis by Gle1–Dbp5 using chemical quench-flow (Figure 6). The weak ATP affinity (∼3 mM (28)) of Dbp5 alone previously precluded such measurements (28). An equilibrated solution of 18 μM Dbp5 and saturating (60 μM) Gle1 was rapidly mixed with either 38 or 170 μM ATP containing trace amounts of radioactive 32P-labeled ATP, aged for various times, and quenched with formic acid. Time courses of Pi (enzyme bound and free) production by Gle1–Dbp5 reveal a rapid burst followed by a linear phase (Figure 6). The burst represents the first turnover of ATP hydrolysis, and the linear phase represents steady-state hydrolysis. Neither 20 μM Dbp5 nor 60 μM Gle1 alone generated detectable hydrolysis of ATP (50 μM) within this time range examined. All hydrolyzed ATP can therefore be attributed to the Gle1–Dbp5 complex. Time courses of Pi production were fitted to the following equation (26,35) (Figure 6, continuous lines), which accounts for reversible ATP binding and hydrolysis, and irreversible Pi release from Gle1–Dbp5 (bottom pathway of Scheme 3):
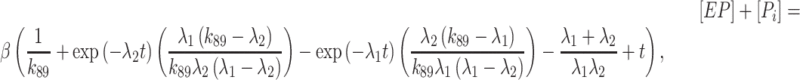 |
(5) |
where formation and loss of Gle1–Dbp5–ADP-Pi state are given by two observed exponential terms (eigenvalues λ1 and λ2, respectively):
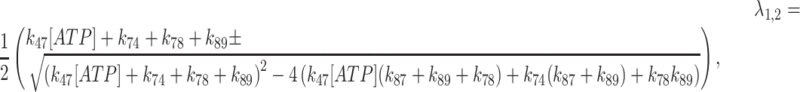 |
(6) |
and the steady-state ATP hydrolysis (slope of linear phase) is given by:
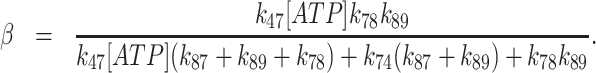 |
(7) |
Figure 6.
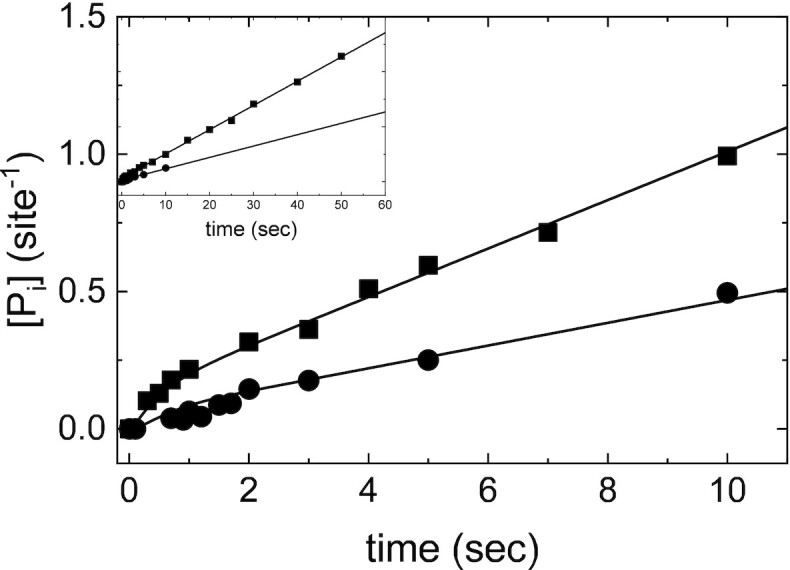
Direct measurement of ATP hydrolysis by Gle1–Dbp5 via quench flow. Time courses of phosphate production in pre-equilibrated solution of 36 μM Dbp5 (18 μM after mixing) with 120 μM Gle1 (60 μM after mixing) upon rapid mixing with either 76 μM (circles, 38 μM after mixing) or 340 μM (squares, 170 μM after mixing) ATP containing a trace of P32-ATP. Continuous lines through the data are the best fits to either the analytical solution for an ATP hydrolysis reaction scheme (Equations 5–7) (26) or the best fits to a custom MATLAB program simulating ATP hydrolysis. The best fits from each method overlap. When all parameters are left unconstrained, the best fit to the analytical solution yields: k47 = 0.09 ± 0.5 μM–1 s–1, k74 = 9.7 ± 55 s–1, k78 = 0.6 ± 0.2 s–1, k87 = 1.0 ± 0.9 s–1, k89 = 0.4 ± 0.1 s–1. Alternatively, fixing k47 and k74 to values determined from kinetic competition (Figure 5) results in a less than two-fold change in ATP hydrolysis and phosphate release rate constants: k78 = 0.6 ± 0.2 s–1, k87 = 1.9 ± 1.3 s–1, k89 = 0.6 ± 0.1 s–1. Lastly, the best fit MATLAB simulation yields: k78 = 0.4 ± 0.2 s–1, k87 = 1.7 ± 0.4 s–1, and k89 = 0.6 ± 0.1 s–1. Neither 20 μM Dbp5 nor 60 μM Gle1 alone generated detectable ATP hydrolysis with up to 10 second incubation with 50 μM ATP. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Scheme 3.

Minimal reaction scheme for Dbp5 steady-state ATPase (±Gle1). H = Dbp5, G = Gle1-InsP6, T = ATP, D = ADP, Pi= PO4. Rate and equilibrium constants for Dbp5 ATPase (top pathway) have been published elsewhere (28).
The best fit of the data (Figure 6) with unconstrained ATP binding (k47, k74), hydrolysis (k78), resynthesis (k87), and Pi release (k89) parameters indicate that Gle1 promotes ATP resynthesis (k87) and accelerates Pi release (k89) from Dbp5 ∼20-fold (k47 = 0.09 ± 0.5 μM−1 s−1, k74 = 9.7 ± 55 s−1, k78 = 0.6 ± 0.2 s−1, k87 = 1.0 ± 0.9 s−1, k89 = 0.4 ± 0.1 s−1, and the steady-state ATPase kcat = 0.1–0.2 ATP s−1 and KM,ATP = 30–50 μM; Table 1). The best fit rate constants for ATP binding are consistent with those determined from kinetic competition (Figure 5) and the steady-state ATPase consistent with those determined from NADH assay (Figures 1B and 7B) and Pi release (Figure 7B). Constraining k47 and k74 to values from kinetic competition results in a less than two-fold change in ATP hydrolysis and phosphate release rate constants: k78 = 0.6 ± 0.2 s−1, k87 = 1.9 ± 1.3 s−1, k89 = 0.6 ± 0.1 s−1 (Table 1). Note that fits to quench-flow time courses assume ADP release from Gle1–Dbp5 is more rapid than steady-state cycling and that phosphate release is irreversible, which hold in this system (Supplementary Figure S3, Supplemental Information, sections S3 and S4). Similar rate constant values were obtained from fitting the ATP hydrolysis reaction mechanism by Gle1–Dbp5 (bottom pathway of Scheme 3) with numerical analysis using a custom MATLAB program.
Figure 7.
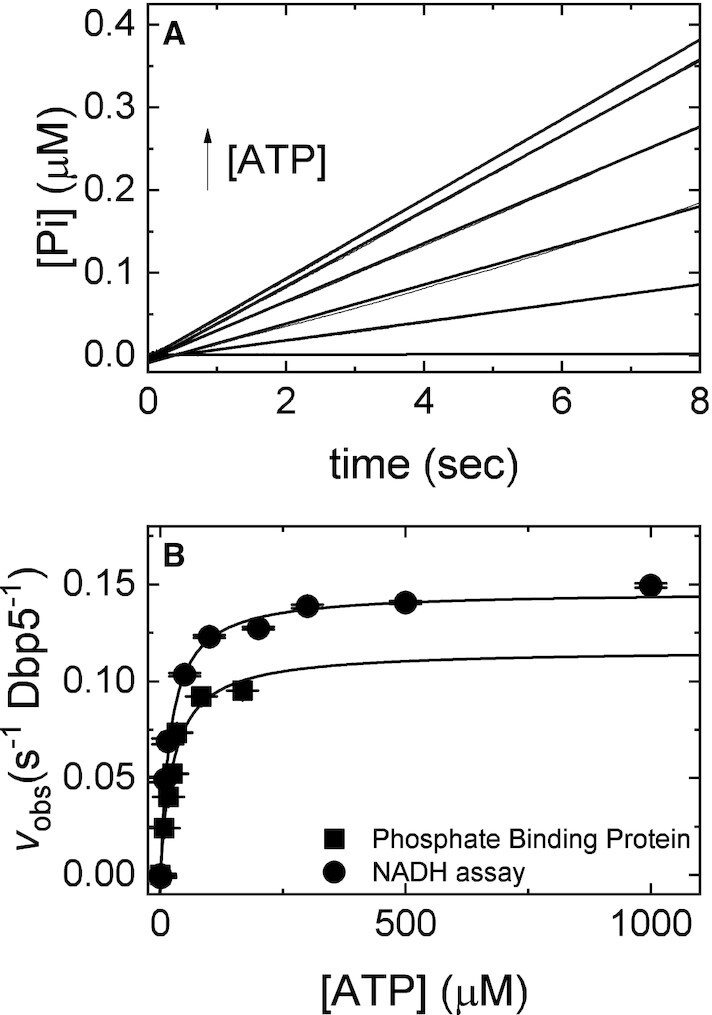
Direct measurement of Pi release from Gle1–Dbp5 via PiBP. (A) Time courses of phosphate release in a pre-equilibrated mixture of 1 μM Dbp5 (0.5 μM after mixing) and 20 μM Gle1 (10 μM after mixing) upon rapid mixing with various [ATP] (0, 5, 10, 15, 20, 50, 100 μM after mixing) containing 6 μM PiBP (3 μM after mixing). Continuous lines through the data are the best fits to a linear equation. (B) [ATP]-dependance of the observed steady-state Pi release in A (solid squares) or steady-state ATP hydrolysis in the presence of the NADH regenerating system (26,31,49). Continuous lines through the data are the best fits to a rectangular hyperbola yielding the maximum velocity per enzyme (kcat= 0.15 ± 0.02 s–1 Dbp5–1, circles; kcat= 0.12 ± 0.01 s–1 Dbp5–1, squares) from the amplitude and KM (20 ± 3 μM, circles; 26 ± 6 μM, squares) from the [ATP] at half-maximum velocity (Table 1). Uncertainty bars represent standard errors in the fits and are contained within the data points. InsP6 is included in all experiments at an equimolar concentration with Gle1.
Prior work has shown that Pi release (k1112 = 0.02 (±0.1) s−1, Table 1), is a critical transition that limits Dbp5 steady-state ATP hydrolysis in the absence or presence of RNA (28). Note that uncertainty in the measurement of k1112 is determined from the intercept of the best linear fit of the [ATP]-dependence of observed lag phase rate constants, but because the intercept is close to the origin, it is subject to significant experimental uncertainty (28). The quench-flow data presented above (Figure 6) shows that Gle1 accelerates Pi release. To directly measure the effect of Gle1 on Pi release from Dbp5, phosphate binding protein (PiBP) read out was used (27). An equilibrated solution of 0.5 μM Dbp5 and 10 μM Gle1 was rapidly mixed with a solution containing various [ATP] (0, 5, 10, 15, 20, 50, 100 μM) and 3 μM fluorescently labeled phosphate binding protein. Time courses of Pi release from Gle1–Dbp5 were linear without a detectable burst or lag phase (Figure 7A), yielding steady-state ATPase cycling velocities that are comparable to those found using the NADH coupled assay (Figure 7B, Table 1) and quench flow (Figure 6, Table 1).
In these reactions, if ATP hydrolysis and Pi dissociation were rate limiting, time courses of Pi release would display a lag phase. Alternatively, if ADP release was rate limiting, time courses would show a burst of liberated Pi (36,37). The observation that time courses of Pi release are linear with [Gle1]-dependent rates indicates that Pi release solely limits the Gle1-stimulated ATPase of Dbp5 and that Gle1 accelerates Pi release from Dbp5.
A complete thermodynamic scheme of the Gle1 activated Dbp5 ATPase cycle
The majority of the rate and equilibrium constants defining the Gle1-stimulated Dbp5 ATPase (Scheme 3) cycle have now been determined, as presented in the preceding sections (ATPase activity of Gle1–Dbp5, bottom pathway of Scheme 3) and in previous studies (ATPase activity of Dbp5 alone, top pathway of Scheme 3) (28,33). In those cases where equilibrium constants have not been measured (Kd107, Kd118 and Kd129), values can be calculated from the principles of detailed balance given that the other constants defining the cycle are known (38).
As an example, consider the cycle formed by initial mantADP binding to Dbp5 and Gle1–Dbp5 as shown in Scheme 1 accounting for a part of multistep mantADP binding to Gle1-Dbp5. In this cyclic scheme the product of equilibrium constants Kd52 × 1/Kd54 × Kd21 × 1/Kd41 must equal 1. Therefore:
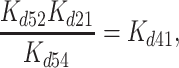 |
(8) |
allowing determination of the affinity of Gle1 binding affinity for nucleotide-free Dbp5 (Kd41) from the experimentally determined equilibrium constants for mantADP binding (Kd54 and Kd21) and Gle1 binding to Dbp5–(mant)ADP (Kd52). The affinity of Gle1 binding nucleotide-free Dbp5 (Kd41) calculated this way is 0.8 ± 0.3 μM, consistent with the value of Kd41 of ≤ 1 μM estimated from mantADP binding (Figure 3B).
Furthermore, the product of the experimentally determined equilibrium constants for mantADP isomerization (Kd32 and Kd65) and Gle1 binding to the two Dbp5–(mant)ADP states (Kd52 and Kd63) calculated from Kd52 × 1/Kd32 × Kd65 × 1/Kd63 is ∼0.82, close to the expected value of unity and indicating the experimentally determined values for mantADP isomerization (Kd32 and Kd65) and Gle1 binding to the two Dbp5–(mant)ADP states (Kd52 and Kd63) are thermodynamically consistent.
Similarly, the affinity of Gle1 for Dbp5 bound to unlabeled ATP (Kd107; Scheme 3) calculated from Kd41, Kd74, and Kd101 (the affinity of ATP for Dbp5 (28)) is 2 nM; the affinity of Gle1 for Dbp5-ADP-Pi (Kd118, Scheme 3) calculated from Kd107, Kd87 and Kd1110 (the dissociation constant for ATP hydrolysis by Dbp5 alone (28); ∼0.004) is 0.8 μM, suggesting that ATP hydrolysis is coupled to conformational rearrangement of Dbp5. The affinity of Gle1 for Dbp5-ADP (Kd129) calculated from Kd41, Kd121 (the affinity of Dbp5 for ADP(28)), and Kd94 (the affinity of Gle1–Dbp5 for ADP; Supplementary Figure S3) is 0.5 μM (Scheme 3; Table 1), consistent with the affinity of Gle1 for Dbp5–(mant)ADP (Kd52, Kd63) measured by mantADP binding (Figures 2 and 3; Table 1) and the combined affinity of the two mantADP states (Kd52Kd63/(Kd52 + Kd63) ∼ 0.17 μM).
Note that some of these calculations were carried out with parameters determined with mantADP and not unlabeled ADP. Despite the mant moiety significantly affecting ADP binding to Dbp5, the atomic-resolution structures of Dbp5-ADP and Dbp5–(mant)ADP indicate that the structure of Dbp5 is identical with both nucleotides and that the altered binding interactions with the nucleotide arise from hydrophobic contacts with the mant moiety (28). Therefore, the measured affinity of Gle1 for Dbp5–(mant)ADP is likely representative of the equilibrium constant for Gle1 binding Dbp5 bound to unlabeled ADP, which is supported by detailed balance calculations.
With these values and a complete scheme, predictions of steady-state cycling outputs can be made and compared to measured rates to validate the model. First, the measured maximum steady-state ATPase activity of Dbp5 is activated 5-fold from an intrinsic kcat ∼0.03 s−1 to a Gle1-activated kcat ∼0.16 s−1 (Figures 1 and 7; Table 1). The value of the kcat predicted from the values of the Gle1–Dbp5 ATP hydrolysis rate constant (k78), Gle1–Dbp5 ATP resynthesis rate constant (k87), and the Pi release rate constant of Gle1–Dbp5–ADP-Pi (k89) determined here and Equation (9) (26):
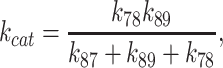 |
(9) |
yield a kcat value of 0.12 s−1 comparable to the kcat of ∼0.15 s−1 measured in steady-state ATPase assays (Figures 1 and 7; Table 1). Second, the ‘apparent KM’ of Gle1 (KGle1; [Gle1] for half-maximal Dbp5 steady-state ATPase) measured experimentally is 300 nM (Figure 1; Table 1). The value of the KGle1 predicted from the values of the Gle1 affinity for Dbp5-ATP (Kd107), Gle1 affinity for Dbp5–ADP-Pi (Kd118), Gle1–Dbp5 ATP hydrolysis rate constant (k78), Gle1–Dbp5 ATP resynthesis rate constant (k87), and the Pi release rate constant of Gle1–Dbp5–ADP-Pi (k89) determined here and Equation (10) (26):
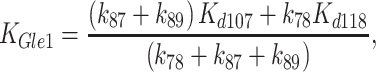 |
(10) |
yield a KGle1 value of 240 nM, comparable to the KGle1 of 300 nM measured in steady-state ATPase assays (Figure 1; Table 1). Similarly, the KM,ATP of Gle1–Dbp5 predicted from the association rate constant of ATP binding Gle1–Dbp5 (k47), ATP release rate constant of Gle1–Dbp5–ATP (k74), Gle1–Dbp5 ATP hydrolysis rate constant (k78), Gle1–Dbp5 ATP resynthesis rate constant (k87), and the Pi release rate constant of Gle1–Dbp5-ADP-Pi (k89) determined here and Equation 11 (26):
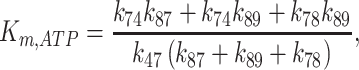 |
(11) |
yield a KM,ATP of 4–15 μM comparable to the ∼20 μM measured in steady-state ATPase assays (Figures 1 and 7; Table 1). The consistency between the predicted and measured Gle1 stimulated Dbp5 steady-state cycling parameters (kcat, KM,ATP, KGle1) indicating that the applied model and analysis are valid, and that the experimentally determined rate and equilibrium constants are consistent with the overall steady-state ATPase cycling behavior of Dbp5.
DISCUSSION
The use of transient kinetic analyses in this work has allowed for insight into the basis of Dbp5 ATPase activation by Gle1 with InsP6. Considering the effects of Gle1 on nucleotide binding, hydrolysis, and product release from Dbp5, these data provide a model of Dbp5 activation by Gle1 that includes Gle1 accelerating Pi release and promoting ATP binding. Moreover, the kinetic scheme detailing Gle1 regulation of Dbp5 supplies an important foundation for developing a complete kinetic description and functional understanding of Dbp5 regulation within the gene expression pathway via regulation by Gle1-InsP6 and Nup159.
Gle1 stimulated ATP hydrolysis by Dbp5
Previous work has shown that the intrinsic Dbp5 steady-state ATPase cycling rate constant (kcat) is slow (∼0.03 s−1) and limited by near-irreversible Pi release (28). Dbp5 also has a relatively weak affinity for ATP (KM,ATP of 1.3–1.9 mM) with RNA acting in the ATPase cycle to accelerate Pi release (28). Here, in the absence of RNA, it is demonstrated that Gle1 activates Dbp5 ATPase activity by accelerating Pi release. Although in the presence of Gle1, like RNA (28), Pi release remains the rate limiting step in the Dbp5 ATPase cycle. It is not currently known if this limitation is fully relieved when both Gle1 and RNA are present.
High resolution crystal structures indicate that Gle1 binds both RecA-like domains of Dbp5 to alter the positioning of these two domains and the nucleotide binding site (12). Gle1-induced rearrangements also include displacement of an auto-inhibitory helix in an ATP or ADP bound auto-inhibited form of DDX19 (human homolog of Dbp5) to promote an RNA-binding competent state (39). The accelerated release of phosphate reported here is consistent with these previously observed Gle1-induced rearrangements in Dbp5 that may act to weaken coordination of bound Pi and enable dissociation from Dbp5 post-hydrolysis. This Gle1activity is expected to transition Dbp5 from a high (Dbp5–ADP-Pi) to low (Dbp5–ADP) affinity RNA binding state, or resolve an auto-inhibited state, which may be critical for Dbp5 function and/or efficient recycling of Dbp5 after an ATP hydrolysis event.
A second finding of this work is that Gle1 promotes ATP binding ∼150-fold and lowers the KM,ATP 75-fold from ∼1.5 mM to ∼20 μM (Figure 7, Table 1) during Gle1 stimulated steady-state Dbp5 ATPase cycling. Previous studies reported a KM,ATP for Gle1 stimulated Dbp5 ATPase of about 100 μM (20), considerably weaker than the 20 μM reported here. The [Dbp5] and [Gle1] utilized in those measurements were not saturating, so the measured KM,ATP reflects a population weighted average of free and Gle1 bound Dbp5 steady-state turnover, yielding a significantly weaker KM,ATP.
The ability of Gle1 to affect phosphate release and ATP binding (this work), and to promote RNA-release from Dbp5 and bind a Dbp5-ADP complex (12), suggests that Gle1 engages Dbp5 in multiple configurations. Note that the full N-terminus of Dbp5/DDX19 has not been modeled by structural data with Gle1 bound, which may be engaged by Gle1 to regulate Dbp5/DDX19 activity since it harbors an autoinhibitory function (12) (39). Overall, it is envisioned that Gle1 may act within the same ATPase cycle to engage Dbp5 to promote cycling by multiple modes of action based on nucleotide state. For example, following Gle1 stimulated phosphate release, the continued binding and organization of both RecA-like domains of ADP bound Dbp5 by Gle1 may subsequently promote ATP binding by limiting the conformational flexibility of the two domains (12). In this way, enzyme recycling would be mediated by Gle1 linking ADP, Pi, and RNA release to binding of ATP. Alternatively, only a single function of Gle1 may occur in each Dbp5 ATPase cycle based on context, including the binding of other regulators, the presence and type of RNA substrate (e.g. mRNA, pre-rRNA or tRNA) and process being directed (e.g. RNA export versus translation regulation).
Implications for mRNA export models
Biochemical, structural, and genetic data strongly support a role for Gle1 in regulating Dbp5 activity to mediate mRNA export through NPCs. Several models have been presented (40–43) for individual aspects of the Dbp5 ATPase cycle given the available data. The kinetic and equilibrium analysis presented here allows for further refinement and alignment of these models.
One proposed mRNA export model suggests that Gle1 promotes ATP binding to Dbp5 based on a combination of genetic, cell, and biochemical data (21). Another model proposes that Gle1 binding would facilitate RNA-release from Dbp5 and enzyme recycling based on structural and biochemical data (12). The kinetic analyses in this work are consistent with both proposed Gle1 activities. Specifically, Gle1 promotes ATP loading via a ∼150-fold increase in ATP affinity that is achieved by slowing ATP dissociation (Table 1). In addition, Gle1 promotes Pi release and ATP resynthesis ∼20-fold, which is expected to promote release of RNA following hydrolysis, and in turn Dbp5 recycling. In combination, the kinetic effects of Gle1 on Dbp5 would favor the Dbp5-ATP state, which has a high affinity for RNA compared to other nucleotide states (1). The net effect of Gle1 on Dbp5 would be to shift the steady-state distribution of populated intermediates from weak RNA binding states to strong RNA binding states (i.e. Dbp5–ADP and nucleotide free Dbp5 to Dbp5-ATP; Figure 8) and to facilitate RNA-release and enzyme recycling (RNA–Dbp5–ADP-Pi to Dbp5–ADP), which in likely involves resolving auto-inhibited Dbp5 conformations bound to both ATP and ADP (12) (39).
Figure 8.
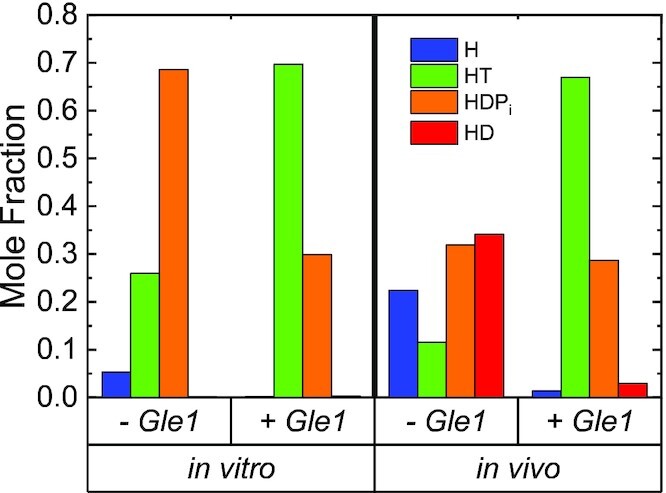
Steady-state distribution of Dbp5(±Gle1) ATPase cycle intermediates. In vitro conditions are 20 mM ATP, 0 mM ADP and 0 mM Pi. In vivo conditions are 2.1 mM ATP (50), 470 μM ADP (50) and 2.5 mM Pi (51). InsP6 is included in all experiments at an equimolar concentration with Gle1.
At NPCs, Nup159 facilitates Dbp5 localization and activity, including reports that Nup159 weakens Gle1 binding to Dbp5-ADP (33). These data, combined with the observations that Gle1 slows both ATP binding and release from Dbp5, suggest that Nup159 may play a key role at this point in the ATPase cycle to promote a weakened interaction between Gle1 and Dbp5 and enhance enzyme turnover. These activities involving Gle1 and Nup159 likely act to ensure there is an available pool of ATP bound Dbp5 at NPCs to engage RNA and facilitate export, which can maintain RNA flux from the nucleus and the kinetics of RNA export that occurs on the millisecond time scale (44–47).
Notably, Dbp5 has been linked to pre-ribosomal complex and tRNA export, as well as translation, which is suggested to involve unique enzyme activities (e.g. ATP hydrolysis appears dispensable for pre-ribosomal export but is required for mRNA export) and functions within discrete cellular compartments (7–9,48). This raises questions about the functionality of Dbp5 in these other roles that may be independent of Gle1 and/or dependent on different Dbp5 activities (e.g. stable RNA binding in the nucleus versus RNA–protein remodeling at NPCs involving ATP hydrolysis). These questions involving Dbp5 functions and differential regulation of the ATPase cycle to effect different outcomes must be addressed in the context of a fully reconstituted system that involves Nup159, Gle1-InsP6 and RNA, which is a goal of future work.
DATA AVAILABILITY
All data is available upon request.
Supplementary Material
Contributor Information
Shawn Gray, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520-8114, USA.
Wenxiang Cao, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520-8114, USA.
Ben Montpetit, Department of Viticulture and Enology, University of California, Davis, Davis, CA 95616, USA; Department of Food Science and Technology, University of California, Davis, Davis, CA 95616, USA.
Enrique M De La Cruz, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520-8114, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM136656 to E.M.D.L.C. and R01GM124120 to B.M.]; S.G. is supported by R35GM136656-S1. Funding for open access charge: National Institutes of Health [R01GM124120 to B.M.]; S.G. is supported by R35GM136656-S1.
Conflict of interest statement. None declared.
REFERENCES
- 1. Henn A., Bradley M.J., De La Cruz E.M.. ATP utilization and RNA conformational rearrangement by DEAD-box proteins. Annu. Rev. Biophys. 2012; 41:247–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao W., Coman M.M., Ding S., Henn A., Middleton E.R., Bradley M.J., Rhoades E., Hackney D.D., Pyle A.M., De La Cruz E.M.. Mechanism of mss116 ATPase reveals functional diversity of DEAD-Box proteins. J. Mol. Biol. 2011; 409:399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmitt C., von Kobbe C., Bachi A., Pante N., Rodrigues J.P., Boscheron C., Rigaut G., Wilm M., Seraphin B., Carmo-Fonseca M.et al.. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 1999; 18:4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strahm Y., Fahrenkrog B., Zenklusen D., Rychner E., Kantor J., Rosbach M., Stutz F.. The RNA export factor gle1p is located on the cytoplasmic fibrils of the NPC and physically interacts with the FG-nucleoporin rip1p, the DEAD-box protein rat8p/dbp5p and a new protein ymr 255p. EMBO J. 1999; 18:5761–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tseng S.S., Weaver P.L., Liu Y., Hitomi M., Tartakoff A.M., Chang T.H.. Dbp5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export. EMBO J. 1998; 17:2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snay-Hodge C.A., Colot H.V., Goldstein A.L., Cole C.N.. Dbp5p/Rat8p is a yeast nuclear pore-associated DEAD-box protein essential for RNA export. EMBO J. 1998; 17:2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gross T., Siepmann A., Sturm D., Windgassen M., Scarcelli J.J., Seedorf M., Cole C.N., Krebber H.. The DEAD-box RNA helicase dbp5 functions in translation termination. Science. 2007; 315:646–649. [DOI] [PubMed] [Google Scholar]
- 8. Lari A., Arul Nambi Rajan A., Sandhu R., Reiter T., Montpetit R., Young B.P., Loewen C.J., Montpetit B.. A nuclear role for the DEAD-box protein dbp5 in tRNA export. Elife. 2019; 8:e48410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neumann B., Wu H., Hackmann A., Krebber H.. Nuclear export of pre-ribosomal subunits requires dbp5, but not as an RNA-Helicase as for mRNA export. PLoS One. 2016; 11:e0149571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hodge C.A., Colot H.V., Stafford P., Cole C.N.. Rat8p/Dbp5p is a shuttling transport factor that interacts with rat7p/nup159p and gle1p and suppresses the mRNA export defect of xpo1-1 cells. EMBO J. 1999; 18:5778–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. von Moeller H., Basquin C., Conti E.. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat. Struct. Mol. Biol. 2009; 16:247–254. [DOI] [PubMed] [Google Scholar]
- 12. Montpetit B., Thomsen N.D., Helmke K.J., Seeliger M.A., Berger J.M., Weis K.. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and insp6 in mRNA export. Nature. 2011; 472:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Napetschnig J., Kassube S.A., Debler E.W., Wong R.W., Blobel G., Hoelz A.. Structural and functional analysis of the interaction between the nucleoporin nup214 and the DEAD-box helicase ddx19. Proc. Nat. Acad. Sci. U.S.A. 2009; 106:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Napetschnig J., Blobel G., Hoelz A.. Crystal structure of the N-terminal domain of the human protooncogene Nup214/CAN. Proc. Nat. Acad. Sci. U.S.A. 2007; 104:1783–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weirich C.S., Erzberger J.P., Berger J.M., Weis K.. The N-terminal domain of nup159 forms a beta-propeller that functions in mRNA export by tethering the helicase dbp5 to the nuclear pore. Mol. Cell. 2004; 16:749–760. [DOI] [PubMed] [Google Scholar]
- 16. Xie Y., Ren Y.. Mechanisms of nuclear mRNA export: a structural perspective. Traffic. 2019; 20:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin D.H., Hoelz A.. The structure of the nuclear pore complex (An update). Annu. Rev. Biochem. 2019; 88:725–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart M. Polyadenylation and nuclear export of mRNAs. J. Biol. Chem. 2019; 294:2977–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alcazar-Roman A.R., Tran E.J., Guo S., Wente S.R.. Inositol hexakisphosphate and gle1 activate the DEAD-box protein dbp5 for nuclear mRNA export. Nat. Cell Biol. 2006; 8:711–716. [DOI] [PubMed] [Google Scholar]
- 20. Weirich C.S., Erzberger J.P., Flick J.S., Berger J.M., Thorner J., Weis K.. Activation of the DExD/H-box protein dbp5 by the nuclear-pore protein gle1 and its coactivator insp6 is required for mRNA export. Nat. Cell Biol. 2006; 8:668–676. [DOI] [PubMed] [Google Scholar]
- 21. Noble K.N., Tran E.J., Alcazar-Roman A.R., Hodge C.A., Cole C.N., Wente S.R.. The dbp5 cycle at the nuclear pore complex during mRNA export II: nucleotide cycling and mRNP remodeling by dbp5 are controlled by nup159 and gle1. Genes Dev. 2011; 25:1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Worthylake D.K., Rossman K.L., Sondek J.. Crystal structure of rac1 in complex with the guanine nucleotide exchange region of tiam1. Nature. 2000; 408:682–688. [DOI] [PubMed] [Google Scholar]
- 23. Talavera M.A., De La Cruz E.M.. Equilibrium and kinetic analysis of nucleotide binding to the DEAD-box RNA helicase dbpA. Biochemistry. 2005; 44:959–970. [DOI] [PubMed] [Google Scholar]
- 24. Hannemann D.E., Cao W., Olivares A.O., Robblee J.P., De La Cruz E.M.. Magnesium, ADP, and actin binding linkage of myosin V: evidence for multiple myosin V-ADP and actomyosin V-ADP states. Biochemistry. 2005; 44:8826–8840. [DOI] [PubMed] [Google Scholar]
- 25. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B.et al.. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henn A., Cao W., Hackney D.D., De La Cruz E.M.. The ATPase cycle mechanism of the DEAD-box rRNA helicase, dbpA. J. Mol. Biol. 2008; 377:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brune M., Hunter J.L., Corrie J.E., Webb M.R.. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994; 33:8262–8271. [DOI] [PubMed] [Google Scholar]
- 28. Wong E.V., Cao W., Voros J., Merchant M., Modis Y., Hackney D.D., Montpetit B., De La Cruz E.M.. P(I) release limits the intrinsic and RNA-Stimulated ATPase cycles of DEAD-Box protein 5 (Dbp5). J. Mol. Biol. 2016; 428:492–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore K.J., Lohman T.M.. Kinetic mechanism of adenine nucleotide binding to and hydrolysis by the escherichia coli rep monomer. 2. Application of a kinetic competition approach. Biochemistry. 1994; 33:14565–14578. [DOI] [PubMed] [Google Scholar]
- 30. Moore K.J., Lohman T.M.. Kinetic mechanism of adenine nucleotide binding to and hydrolysis by the escherichia coli rep monomer. 1. Use of fluorescent nucleotide analogues. Biochemistry. 1994; 33:14550–14564. [DOI] [PubMed] [Google Scholar]
- 31. Bradley M.J., De La Cruz E.M.. Eckhard J. Methods in Enzymology. 2012; 511:Academic Press; 29–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alcazar-Roman A.R., Bolger T.A., Wente S.R.. Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with gle1. J. Biol. Chem. 2010; 285:16683–16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong E.V., Gray S., Cao W., Montpetit R., Montpetit B., De La Cruz E.M.. Nup159 weakens gle1 binding to dbp5 but does not accelerate ADP release. J. Mol. Biol. 2018; 430:2080–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henn A., De La Cruz E.M.. Vertebrate myosin VIIb is a high duty ratio motor adapted for generating and maintaining tension. J. Biol. Chem. 2005; 280:39665–39676. [DOI] [PubMed] [Google Scholar]
- 35. Henn A., Cao W., Licciardello N., Heitkamp S.E., Hackney D.D., De La Cruz E.M.. Pathway of ATP utilization and duplex rRNA unwinding by the DEAD-box helicase, dbpA. Proc. Nat. Acad. Sci. U.S.A. 2010; 107:4046–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De La Cruz E.M., Wells A.L., Sweeney H.L., Ostap E.M.. Actin and light chain isoform dependence of myosin v kinetics†. Biochemistry. 2000; 39:14196–14202. [DOI] [PubMed] [Google Scholar]
- 37. De La Cruz E.M., Ostap E.M., Sweeney H.L.. Kinetic mechanism and regulation of myosin vI. J. Biol. Chem. 2001; 276:32373–32381. [DOI] [PubMed] [Google Scholar]
- 38. De La Cruz E.M., Pollard T.D.. Nucleotide-Free actin: stabilization by sucrose and nucleotide binding kinetics. Biochemistry. 1995; 34:5452–5461. [DOI] [PubMed] [Google Scholar]
- 39. Lin D.H., Correia A.R., Cai S.W., Huber F.M., Jette C.A., Hoelz A.. Structural and functional analysis of mRNA export regulation by the nuclear pore complex. Nat. Commun. 2018; 9:2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arul Nambi Rajan A., Montpetit B.. Emerging molecular functions and novel roles for the DEAD-box protein dbp5/ddx19 in gene expression. Cell. Mol. Life Sci. 2020; 78:2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Folkmann A.W., Noble K.N., Cole C.N., Wente S.R.. Dbp5, gle1-ip6 and nup159: a working model for mRNP export. Nucleus (Austin, Tex.). 2011; 2:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tieg B., Krebber H.. Dbp5 - from nuclear export to translation. Biochim. Biophys. Acta. 2013; 1829:791–798. [DOI] [PubMed] [Google Scholar]
- 43. Stewart M. Ratcheting mRNA out of the nucleus. Mol. Cell. 2007; 25:327–330. [DOI] [PubMed] [Google Scholar]
- 44. Grunwald D., Singer R.H.. In vivo imaging of labelled endogenous [bgr]-actin mRNA during nucleocytoplasmic transport. Nature. 2010; 467:604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mor A., Suliman S., Ben-Yishay R., Yunger S., Brody Y., Shav-Tal Y.. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat. Cell Biol. 2010; 12:543–552. [DOI] [PubMed] [Google Scholar]
- 46. Siebrasse J.P., Kaminski T., Kubitscheck U.. Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy. Proc. Nat. Acad. Sci. U.S.A. 2012; 109:9426–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith C., Lari A., Derrer C.P., Ouwehand A., Rossouw A., Huisman M., Dange T., Hopman M., Joseph A., Zenklusen D.et al.. In vivo single-particle imaging of nuclear mRNA export in budding yeast demonstrates an essential role for mex67p. J. Cell Biol. 2015; 211:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Estruch F., Cole C.N.. An early function during transcription for the yeast mRNA export factor dbp5p/rat8p suggested by its genetic and physical interactions with transcription factor IIH components. Mol. Biol. Cell. 2003; 14:1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De La Cruz E.M., Sweeney H.L., Ostap E.M.. ADP inhibition of myosin ATPase activity. Biophys. J. 2000; 79:1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Theobald U., Mailinger W., Baltes M., Rizzi M., Reuss M.. In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae. I. Experimental observations. Biotechnol. Bioeng. 1997; 55:305–316. [DOI] [PubMed] [Google Scholar]
- 51. Lagunas R., Gancedo C.. Role of phosphate in the regulation of the Pasteur effect in Saccharomyces cerevisiae. Eur. J. Biochem. 1983; 137:479–483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available upon request.