

Abstract
Major progress in the understanding and treatment of cancer have tremendously improved our knowledge of this complex disease and improved the length and quality of patients’ lives. Still, major challenges remain, in particular with respect to cancer metastasis which still escapes effective treatment and remains responsible for 90% of cancer related deaths. In recent years, the advances in cancer cell biology, oncology and tissue engineering converged into the engineered human tissue models of cancer that are increasingly recapitulating many aspects of cancer progression and response to drugs, in a patient-specific context. The complexity and biological fidelity of these models, as well as the specific questions they aim to investigate, vary in a very broad range. When selecting and designing these experimental models, the fundamental question is “how simple is complex enough” to accomplish a specific goal of cancer research. Here we review the state of the art in developing and using the human tissue models in cancer research and developmental drug screening. We describe the main classes of models providing different levels of biological fidelity and complexity, discuss their advantages and limitations, and propose a framework for designing an appropriate model for a given study. We close by outlining some of the current needs, opportunities and challenges in this rapidly evolving field.
Graphical Abstract

1. Introduction
Cancer continues to claim millions of lives each year [1]. Cancer metastasis, responsible for 90% of cancer-related mortalities [2], remains poorly treatable. Current cancer therapeutic approaches are based on either killing all rapidly dividing cells or targeting a specific tumor mutation, and neither approach is sufficiently effective if the patient has metastasis.
Decades of biological research in cell culture and animal models have tremendously advanced our understanding of cancer. However, these models recapitulate human cancers to only limited extent (only about 5% of cancer therapeutics that passed screening show efficacy in clinical trials [3]) and fail to recapitulate metastatic disease. In addition, cancer cells and their metastatic potential and target organs are highly heterogeneous, both among the patients and within a single tumor. Together, these factors have contributed to the current lack of treatment options. While mice (i) reproduce and mature quickly, (ii) can be genetically defined, (iii) contain biological complexity, (iv) present a neoplastic development similar to that in humans, and (v) support the growth of implanted human cancer cells, they fail to mimic key aspects of human physiology. Notably, mice lack human immune components, fail to display metastasis to expected organ sites, and differ from humans in their genetic drift and clonal expansion of tumor cells [4].
Clearly, there is a need for human tissue models that would more accurately predict the progression of cancer and responses to treatment, ideally in a patient-specific context. Beyond genetically engineered mouse models, which spontaneously form cancers, researchers have developed patient-derived xenograft (PDX) mouse models by injecting patient’s tumor cells into immunodeficient mice. While PDX models offer patient-specific insights, they also require significant time and effort to establish, do not display the immune system of the patient, and have been shown to induce rapid mutational drifting in the transplanted cells [5]. More recently, human tissue models of cancer, in form of organoids and organs-on-a-chip (OOC), have emerged as patient specific mimics for studies of cancer progression. Notably, these models can be tailored to capture the individual aspects of cancer, while allowing a range of complexity, depending on the question asked. This feature is particularly valuable for studies of late-stage and metastatic cancers, where currently available models fail to capture cancer progression and response (or resistance) to treatment. A number of human tissue models have been developed to mimic various types and stages of cancer, from primary tumors and their niches, to intravasation and extravasation of circulating tumor cells, systemic crosstalk, metastatic preconditioning, and metastasis [12, 19, 29, 33, 45, 50, 52, 60, 64, 66, 70–75].
In this review, we focus on engineered human tissue models of cancer, and how they can contribute to cancer research and developmental drug screening, by overcoming the limitations of animal models. These models offer testing of patient-specific cells ex-vivo, overcome the biological and logistical limitations of modeling metastasis in animal models, contain human immune system components, and are more suitable for screening therapeutic modalities (biologics, vaccines, cell therapy). After describing the main classes of models providing different levels of biological fidelity and complexity, we propose a framework for selecting and designing an appropriate model and provide examples to illustrate specific opportunities in biological research, regenerative medicine, and drug development. We close by discussing some of the current needs, opportunities, and challenges in this rapidly evolving field.
2. Framework for designing an appropriate engineered model of cancer
To engineer cancer models that are suitable for clinical translation, it is necessary to create a suite of high-fidelity models of cancer progression with a complexity that increases with the biological requirements characterizing each stage (Figure 1). To determine the required level of complexity when engineering a specific model, it can be useful to work backwards starting from the intended use, and define which variables should be included to recapitulate the conditions needed to answer a specific question in the most straightforward way.
Figure 1 -. Complexity driven design of human OOC models.
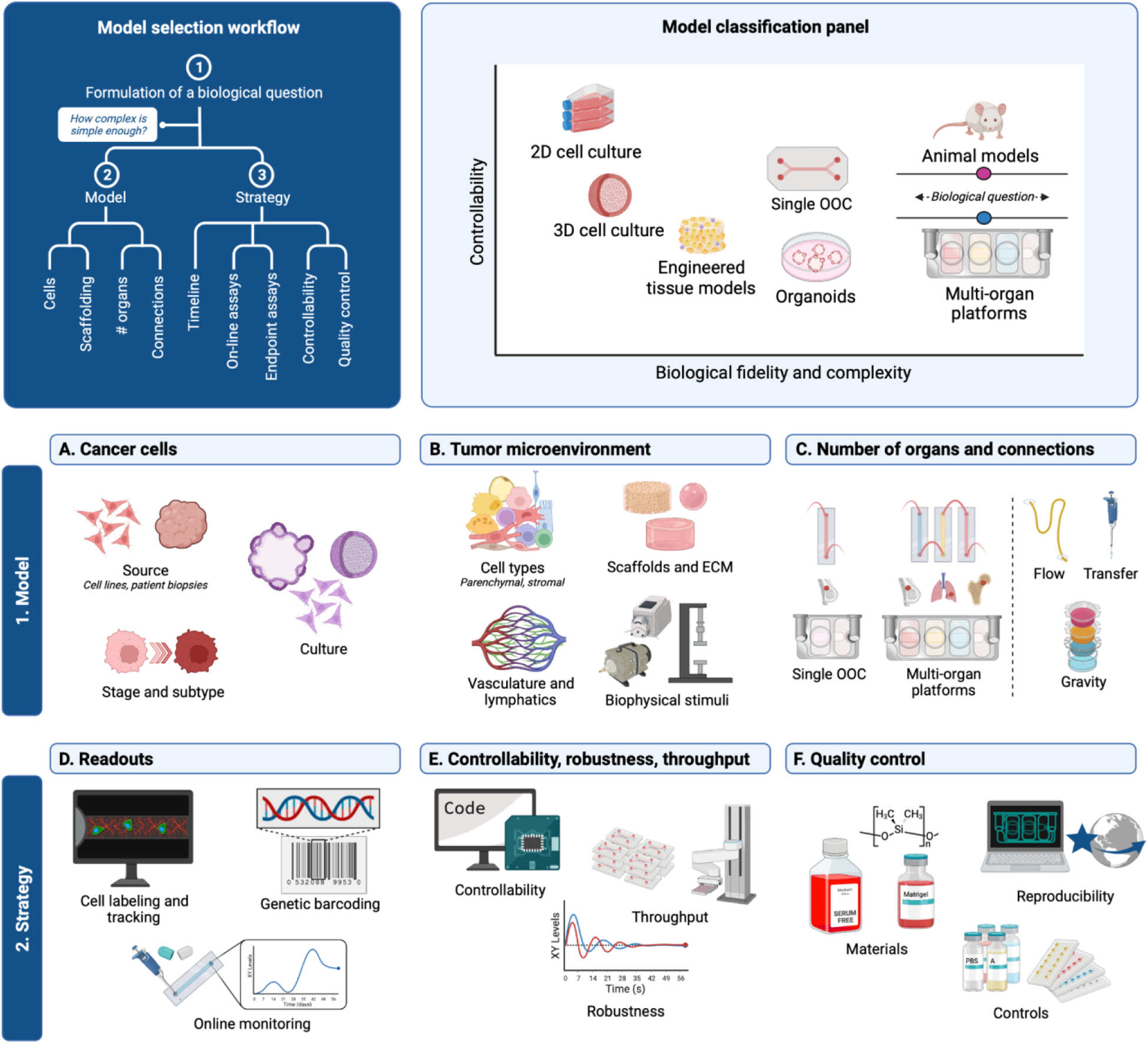
Complex questions call for complex models, with the goal to select the simplest model enabling investigation of a given question. In all cases, physiological relevance and compatibility with imaging, on-line and end-point assays are a must. Created with BioRender.com.
The case-specific input variables should be modular, to enable building a suitable model for each application in a plug-and-play manner. To demonstrate this in more detail, we will briefly introduce how cells and biologically inspired tissue engineering merge to create an array of models of increasing complexity that are capable of decoupling biological mechanisms, recapitulating organ level functions, and predicting human systems level responses. As these models can incorporate patient-specific cells and gene editing approaches, they are providing opportunities for precision medicine and characterization of patient-specific disease mechanisms in an unprecedented manner. The highly complex biology of metastatic progression is well-poised to benefit from multi-organ OOC systems, where patient tumor cells circulate within the microfluidic vascular networks and selectively home to distant organs.
The framework for designing in vitro models of cancer is based on two components: the model system, engineered to replicate defined biological functions related to the in vivo condition, and the strategy, designed to extract meaningful information from the model and establish its predictive capacity. Overall, the model should be tailored to match the simplest physiological context necessary to answer a desired biological question. The simpler the model the better, as higher complexity introduces more variability, to the point of providing diminishing returns in exchange for the increased workload.
The main parameters of interest when determining the model are the input cancer cells, tumor microenvironment (TME), and choice between a systemic multi-tissue model and an isolated single organ model, depending on the biological question. The strategy should complement these parameters with functional readouts that can be used to validate the model’s predictive capacity when defined perturbations are applied. One option is to use both positive and negative control drugs to elicit a set of baseline responses that can be correlated with the clinical responses. Furthermore, the strategy should be designed with intentional controllability to engineer models of high reproducibility that create a robust framework for consistently extracting meaningful and rapid readouts from the model system.
2.1. Cancer cells.
The input cancer cell source is an important variable that greatly influences the overall physiological relevance of the engineered model. A particularly exciting aspect of engineered models of human cancer is their increasing ability to be patient-specific and thereby enable deployment of precision medicine approaches. It is well known that cancer cells are highly heterogeneous from one patient to the next, and even within the same tumor. By using patient-derived cells, we can start understanding the innate nature of patient-specific differences at the cell, tissue, and organ levels. However, culturing patient-derived cancer cells with the preservation of their phenotypes remains an unmet challenge. The type of cancer, cancer location, procurement strategy, purity, and cell culture conditions directly influence the utility of patient’s cancer cells in downstream assays. While various types of cancer have been shown to yield more reliable results of in vitro culture, they rarely have more than a 30% success rate. The cancer sample type (core biopsy, tumor resection, aspirations of ascites or pleural effusions) also determines the subsequent cell culture strategy. The use of samples from either primary or metastatic sites should match that of the biological question intended by the experimental model. Liver and lung biopsies are more successful than lymph node biopsies, while bone biopsies have been largely unsuccessful. Samples obtained from pleural effusions or ascites show enhanced ability to successfully establish in vitro cultures, perhaps due to their adaptation to fluid-based environments and more aggressive phenotype [10].
Increasing the effectiveness of in vitro culture of cancer cell will be of great utility. Current efforts include culturing cells with rock inhibitor, removing stromal cells to enhance cancer cell purity, and using irradiated fibroblast feeder layers in a co-culture model [10,34,36,44]. Organoids can also offer a way to continuously culture patient-specific cancer cells in a way that maintains their heterogeneity, using defined signals and supporting stromal cells and extracellular matrix (ECM) components favoring selection of cancer cell phenotypes. In addition, organoids provide a means for preserving cancer cells over longer periods of time. However, both the standard cell culture and organoids show limited success and often fail due to an overgrowth of healthy stromal cells. Cell overgrowth is usually driven by fibroblasts and can be reduced by removing fibroblasts from the initial explanted sample by magnetic or fluorescent sorting. Different substrate attachment rates of fibroblasts can also be used to remove them from cancer cells during routine subculture.
Overall, there are many considerations to keep in mind when choosing the source of cancer cells for an engineered model. Primary cells offer mature functionality but have a limited lifespan and are not easy to culture ex vivo without genetic drift. Immortalized human cancer cells often represent only one of many phenotypes within the original tumor and tend to lose their physiological relevance with each passage. PDX models allow bulk tumor material to be kept alive and grow ex vivo, using mouse as a bioreactor [6]. While PDX provide a patient-specific 3D model that is multicellular and systemic, they often take 3–12 months to establish [7, 8], a time that is too long for a large portion of patients. Obtaining primary cells by dissociation of tumors following resections and the use of these cells without additional processing is in most cases most practical and time-efficient. The availability and amounts of patient samples can also greatly influence the selection of cancer cell source.
2.2. Tumor microenvironment.
While genetically engineered mouse models and PDXs have been widely used for cancer research, their misrepresentation of human tumor biology, immune system contributions, and drug responses have limited their translational impact [4, 9]. Similarly, in vitro culture of human cancer cells fails to provide the surrounding tumor microenvironment and supporting cells, limiting translational utility [10]. To overcome these limitations, stem cell biology and tissue engineering have converged into modeling the complexity of the human tumor microenvironment, using cancer cells within bioengineered human tissue niches to recapitulate in vivo conditions.
Microenvironmental factors are crucial for recapitulating both the primary tumor and secondary metastatic sites, to promote tumor progression via matrix remodeling, angiogenesis, and myeloid-derived suppressor cell maturation, among other biological events. In vivo, cancer cells are found in highly organized and complex microenvironments, that should inform the design of engineered human 3D tumor models recapitulating and maintaining biological fidelity in vitro, in terms of more physiological gene expression [11] and drug responses [12–14]. Cancer cells exhibit increased proliferation and begin to rapidly divide and take over the local microenvironment. The surrounding ECM experiences increased interstitial pressure and remodels to become mechanically stiffer by increasing collagen synthesis and matrix metalloproteinases [15, 16]. The highly proliferative cancer cells outcompete neighboring healthy cells, creating a hypoxic, acidic environment [17] and altering the local vasculature so that it becomes increasingly leaky and unable to adequately deliver oxygen and remove nutrients [18]. These changes are further influenced by the immune compartment, with innate and adaptive immune cells controlling tumor development. Subsequently, neoplastic cancer cells evolve to avoid immune mediated destruction. This shift in immune cells can be characterized as initially cold, lacking cytotoxic T cells, and then hot, as an inflammatory response is initiated and cytotoxic T cells begin to infiltrate the tumor. However, immune evasion and secreted factors often outcompete the local immune microenvironment to reverse a hot tumor into a tumor-supportive niche [19–23]. Clearly, only a few aspects of this complex and dynamic in vivo environment can be recapitulated in vitro, even using our best bioengineering tools.
Incorporation of the essential cell types and their environmental cues are imperative for tissue engineering. Most tissues are composed of different cell populations, with the tissue-specific parenchymal cells accompanied by supporting cells such as fibroblasts, pericytes, and endothelial cells that compose the stromal environment and help dictate tissue functionality [24]. Inclusion of stromal cells and their proper microenvironment plays major roles in disease progression as stromal cells provide important cell signaling and structural support via modification and deposition of ECM that can influence whether a drug treatment will succeed or fail [21, 25–29]. Such components can be incorporated into tissue systems using recent advances in tissue engineering. For example, Yu et al [30] utilized a reconfigurable microfluidic cell culture system to model tumor progression in vitro with the flexibility to add or remove multiple cell types at precise times. This approach allowed monitoring and control of spatiotemporal interactions and control over the complexity of the cell microenvironment.
While primary cells have a limited lifetime, induced pluripotent stem cells (iPS) can serve as an unlimited self-renewing source of undifferentiated cells [31] and enable derivation of patient-specific cell types for inclusion in the TME and auxiliary organ models. Recent developments are starting to enable generation of patient-specific models, typically combining tissues derived from blood derived iPS cells with the patient’s primary cells (e.g., cancer cells, immune cells, accessible tissue cells). The use of these cells to make isogenic tissue sites, especially in mimicking the primary tumor, may provide an ideal genetically identical backbone for promoting a tumor in a healthy microenvironmental niche. These models are starting to provide the tools to study patient diversity in cancer pathophysiology, progression, drug efficacy, and the development of drug resistance [31, 32]. However, the use of iPS cells to engineer patient-matched secondary tissue sites is limited by their immaturity and the long timelines (several months) needed to derive and characterize cell lines for each patient.
2.2.1. Biophysical stimuli.
Cell populations are regulated by tissue specific molecular and physical signals generated by the surrounding cells, ECM and the environment. For most tissues, biomechanical stimuli need to be incorporated into engineered models using scaffolds that recapitulate relevant biochemical and structural cues and induce assembly of more complex 3D cell structures [21]. Likewise, scaffolds can be used to engineer tumor vascularity. Lai et al. demonstrated the use of 3D stamping techniques to create a bioscaffold that provides mechanical stability for seeded endothelium to generate a perfusable luminal space and self-assembly of a vascular microenvironment [33]. With the advancement of biomanufacturing techniques, there are endless combinations of methods to recapitulate the specific aspects in vivo tumor physiology desired to study in vitro. Cancer cells can be cultured in aggregates (spheroids) [34–36], encapsulated in or printed on hydrogels (which form organoids when additional chemical signals are included) [37, 38], or cultured in biomaterial scaffolds [39]. The aggregates and organoids promote cell-cell interactions while culture in hydrogels or scaffolds introduces cell-ECM interactions and guides physical signals and spatial tissue arrangement [26, 40]. These models can be grouped into organoids, engineered tissue models, and organ-on-a-chip (OOC) models.
2.2.2. Organoid models.
Organoids are multi-cellular, 3D systems that naturally form many of the cell types within an organ from a single progenitor cell, creating multi-cellular structures through innate tissue regeneration. These multi-cellular structures may thus provide more developmentally faithful models [41, 42], although the lack of imposed biomimetic cues results in highly variable organoids that miss control or functional responses. Organoids provide a rapid approach to studying biological mechanisms independent of their phenotypic functions and for studying multicellular responses over long time periods [41, 43]. Patient-derived organoids demonstrated, as early as in 1999 [44], that two basic factors are needed for their development: adult organ specific stem cells and intercellular communication through gap junctions [41, 45, 46]. Current efforts have enabled organoids to be developed for the most common cancers by culturing patient derived cells in Matrigel and using soluble factors to select for cancer stem cells that can recapitulate the development of heterogeneous cancers in vitro [41]. Organoid models show great utility for screening drugs in a patient specific manner [47], and for creating models of blood cancers that are not well recapitulated in animals. Notably, organoid models of multiple myeloma were established using patient derived bone marrow aspirates [13]. These models are advantageous as they naturally contain most of the relevant cell types within a target tissue and can subsequently be exploited as an optimal source of cells for tissue engineering. While this approach has not been fully realized, the merging of organoids and tissue engineering shows promise for enhancing the cellular complexity to further increase physiological relevance [48].
2.2.3. Tissue engineered models.
To engineer tissues with the functionality of their in vivo counterparts, we must fully understand the cellular mechanisms, genetic circuitry, extracellular matrix and signaling that drive tissue development and homeostasis. Biological insights are then used to guide tissue formation and functionality in a controlled manner, using engineering techniques such as 3D printing, to directly seed cells and ECM in an organized way and subsequently expose them to appropriate biomimetic stimuli as needed (ie physical, chemical, electrical cues) [13, 14, 19, 49–52]. The focus is on controlling the cells in 3D to directly result in the desired tissue state in a reproducible, and sometimes accelerated, manner [29, 53]. The culture of cancer cells on scaffolds is the most widely used approach, enabling ECM specific features. In this setting, cancer models better mimic cell-ECM interactions and the development of drug resistance for many cancer types [54–56].
2.2.4. Organs-on-a-chip models.
The utility of tissue engineering approaches to model their human counterparts is currently being developed through “organs-on-a-chip” (OOC) [57–59]. OOCs combine microfluidics, 3D cell culture, and precise control of environmental conditions to mimic in vivo conditions [60]. The development of OOCs provide user-friendly approaches to study human responses to drugs and environmental factors [61–63]. By coupling multiple OOCs together through vasculature, perfusion of a shared blood substitute, or supernatant exchange, one can study organ-organ interactions and systemic diseases [19, 64–66]. Further efforts to introduce vascular components, including endothelial cells and flow, have resulted in OOC platforms. By introducing biophysically relevant fluid flow, engineered OOC models show increased similarity in gene expression and biomechanical regulation pathways associated with cancer progression [21, 28, 33, 56, 67–71].
2.3. Isolated or systemic organ models.
The individual tissue units representing specific organs can be used on their own or linked together to create human cancer models of varying complexity that mimic human cancer physiology and drug responses [48, 72–75]. It may be ideal to focus on a bioengineered model of a single tumor when looking at its initial development, growth profile, tumor niche remodeling, drug treatment and resistance, and premetastatic niches priming. However, cell lines and organoids may also be appropriate when looking into the molecular and cellular events, if the goal is to measure responses at a cellular level instead of within a tissue niche. With this said, some events (e.g., those involving tissue-cross-talk and immune factors) are only recapitulated within physiological tissue and multi-tissue models capturing some systemic factors of the in vivo environment. The current direction in engineered tissue and OOC models is to provide biological complexity while keeping the models user-friendly [59, 76].
Stromal cells and ECM are critical players in premetastatic tumor remodeling and the subsequent intravasation. Thus, engineered tissues and OOCs would be more appropriate models. When examining intravasation or extravasation, it is ideal to use OOC systems that include vascular and lymphatic flow and barriers, to evaluate tumor cell migration into or out of the vasculature [20, 47, 77, 78]. As studies of metastasis and advanced cancer are becoming increasingly more systemic in nature [79], they require the use of multi-organ platforms where different engineered tissues/organs are linked together to facilitate inter-organ communication [19]. Similarly, the timeline of the biological mechanism or drug treatment should also be considered when choosing the model. Engineered tissues, particularly those supported by perfusion, can support culture timelines from weeks to months to enable physiologically relevant studies [80–84].
2.4. Readouts.
Engineered cancer models can only be useful if they are able to provide physiologically relevant information over time. The use of fluorescent or bioluminescent labeled cells enables imaging-based tracking of where the cells are at any given point in time, enabling studies of cancer cell growth and spread while also tracking drug efficiency and immune mediated cancer responses. Genetic barcoding of cancer cells now enables longitudinal tracking and characterization of the clonal expansion of tumors within engineered models. Just as patient blood sampling provides biomarkers indicative of the patient’s disease status, supernatant sampling from the model systems enables similar analysis of biomarkers and secreted factors. High-content endpoint data can be similarly extracted from the system by single-cell sequencing, phenotypic analysis, and multiplexed immunohistochemistry/immunofluorescence. Circulating immune and cancer cells can be sampled from the platform and evaluated by single-cell sequencing and flow cytometry. New approaches include characterizing the extracellular vesicles, both in the supernatant and within engineered tissues. These assays provide results that can be compared to clinical results as they share the same methodologies.
2.5. Controllability, robustness, and throughput.
To facilitate reliable use of engineered cancer models, they must be continuously improved with increasing automation and standardized tissue fabrication methods that enable reproducible formation of comparable tissue models. Currently the formation of tissue models is laborious and manual, unintentionally bringing variability and user bias into the system, that must be eliminated for their broad utility. Model systems that are compatible with automated liquid handling and standard analytics (i.e. well plate formats) would greatly maximize their controllability, robustness, and throughput.
2.6. Quality control.
On-line readouts enable evaluation of the same tissue over and over, and thus provide longitudinal data for tissue functionality that can serve as a measure of tissue quality. These parameters can be fed into a digital model of manufacturing process, known as a “digital twin”, to track the fidelity of the generated tissues over time. This approach can be furthered by using a set of reference compounds, for which there are validated tissue responses, that can be used to elicit both positive and negative responses, serving as a quality control or each batch of tissues. Assessing the variability of these known compounds within the system, and comparing them to the average response for a given batch, enables the user to quantitatively determine how much confidence to have when using the system for drugs or perturbations with unknown responses.
Decreasing the variability of these engineered systems depends on eliminating the ill-defined cell culture supplements and ECM components, such as serum and Matrigel, particularly when using these models for regulatory approvals. Similarly, the materials used to create the engineered model environment can introduce variability and should be manufactured using quality control guidelines and evaluated for parameters of interest with each batch. Commonly used fabrication materials, such as Polydimethylsiloxane (PDMS), are known to absorb drugs and can show differing stiffness depending on the detailed conditions and timing for mixing and curing. Users can create their own suite of quality control indices that define the acceptability of the device for the intended use.
3. Bioengineering complexity into cancer models
Bioengineered models coupled with microfluidic devices enable temporal control of dose-dependent signals that can help recapitulate these complex dynamics (Figure 2). Tumor cells are characterized by peculiar adaptability and are prone to phenotypic changes. These phenomena involve complex and often redundant time-dependent molecular pathways, with the interplay between the ECM, immune system, local vasculature, and primary tumor regulating spatial and temporal gene transcription [35]. The adaptive strategies of cancer cells enable them to shift between highly proliferating to quiescent states, in response to temporal changes in the local environments such as varying blood flow, transient hypoxia and ECM remodeling [34, 36]. Although the formation of metastatic lesions can take months to years, transient cellular properties such as the epithelial-to-mesenchymal transition (EMT) and its reverse mesenchymal-to-epithelial transition (MET) could be captured at shorter time frames [85].
Figure 2. Bioengineering models of human tumors.
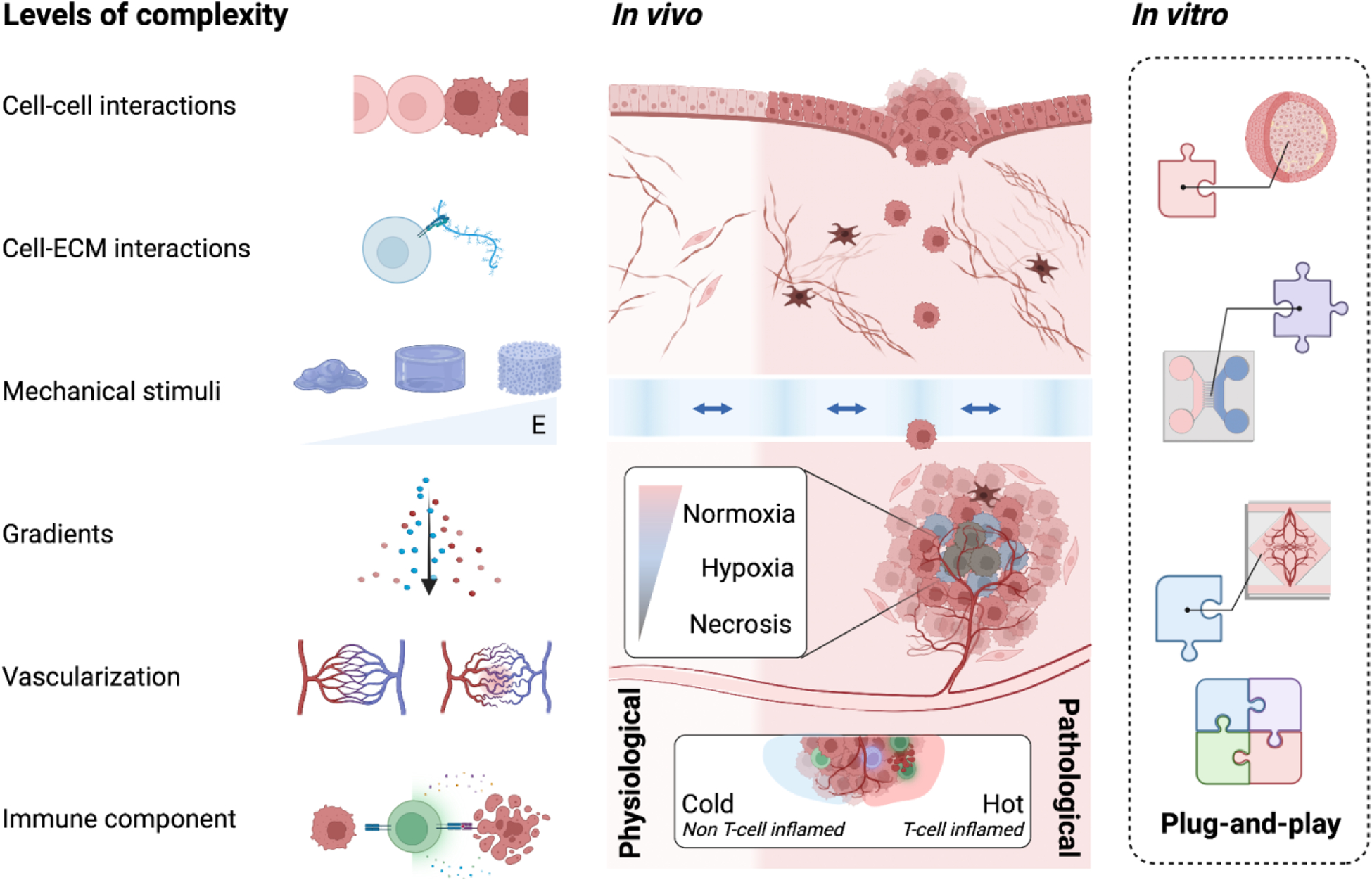
Our knowledge of the in vivo tissue conditions of human cancers guides the design of in vitro tumor models. Varying levels of complexity can be engineered by incorporating parameter inputs that replicate specific components of the TME. Such inputs include but are not limited to (1) cell-cell interactions, (2) cell-ECM interactions, (3) mechanical stimuli, (4) molecular gradients (chemical, hypoxic, metabolic), (5) vascular integrity, and (6) incorporation of immune components. Created with BioRender.com.
3.1. Primary tumor models.
Primary tumors are complex, heterogeneous organ systems consisting of multiple cell types and dynamic extracellular matrix. Over time, the interplay of these components creates mechanical and nutrient gradients, intricate vascular networks, and differing immune landscapes that comprise the complex ecosystem of the tumor microenvironment. In the TME, cancer cells influence the local environment to support the pathological shifts in the stromal cells, ECM, and signaling molecules [71, 86]. These shifts are outcompeting the surrounding healthy cells to favor the needs of the rapidly growing tumor [28, 87]. Thus, recapitulation of the multi-dimensional structure, organization, and communication of the TME is essential for bioengineering physiologically relevant models of human tumors. These efforts rely on our understanding of the in vivo conditions that maintain physiological tissue homeostasis and drive disease progression (Figure 2).
In addition, dormancy and reawakening are regulated by complex interactions between disseminating tumor cells and the homing niches. Residual disease can be associated with dormancy of disseminated tumor cells (DTCs), characterized by reversible cell-cycle arrest, or to tumor mass dormancy, featuring steady-state proliferation balanced by cell death due to either angiogenic impairment or immune pruning [88]. Relapse occurs once tumor cells reactivate causing overt lesions. Thus, it is not surprising that in vitro primary tumor models have been beneficial to the mechanistic identification of factors inducing dormancy and reactivation. Foundational studies by Ghajar et al. of bioengineered organotypic models of the perivascular niche demonstrated the role of endothelial-derived thrombospondin-1 in the induction of dormancy. Stable vasculature that secretes this factor was identified as a dormant niche, while sprouting neovasculature where this cue is suppressed was shown to be a proliferative niche [56]. More recently, it was found that lung epithelial cells interact with breast cancer cells regulating their indolence through induced deposition of fibronectin fibrils [89].
Interestingly, ECM remodeling was also identified as a key regulator of dormancy through cell culture on finely tuned substrates [90]. While not necessarily related to disseminated tumor cells, the role of mechanical stimuli in the modulation of dormancy and proliferation of tumor cells emerged in a cancer-on-a-chip model of non-small-cell lung cancer. In particular, indolence and therapeutic resistance were found to be sustained by breathing motion [30]. As recently reviewed by Montagner et al. [89], similar models can be highly informative, however bona fide models of dormancy including the immune components are still lacking. Since dormant DTCs are immune evasive and subject to culling by cytotoxic immune cells in some scenarios of tumor mass dormancy [41], it is likely that the field will greatly benefit from this further level of complexity.
3.2. Cancer metastasis models.
A human tissue engineered cancer model that would accurately model key aspects of metastatic progression would be transformative to cancer research and therapeutic discovery. As mentioned above, OOC platforms may provide utility for modeling metastasis by offering controlled models of the key metastatic mechanisms, from intravasation to extravasation [19] (Figure 3). Multi-organ OOCs may further enable systemic crosstalk to evaluate premetastatic niche conditioning and tissue tropism of metastatic sites specific to each cancer and patient subtype.
Figure 3. Microfluidic models of tumor metastasis.
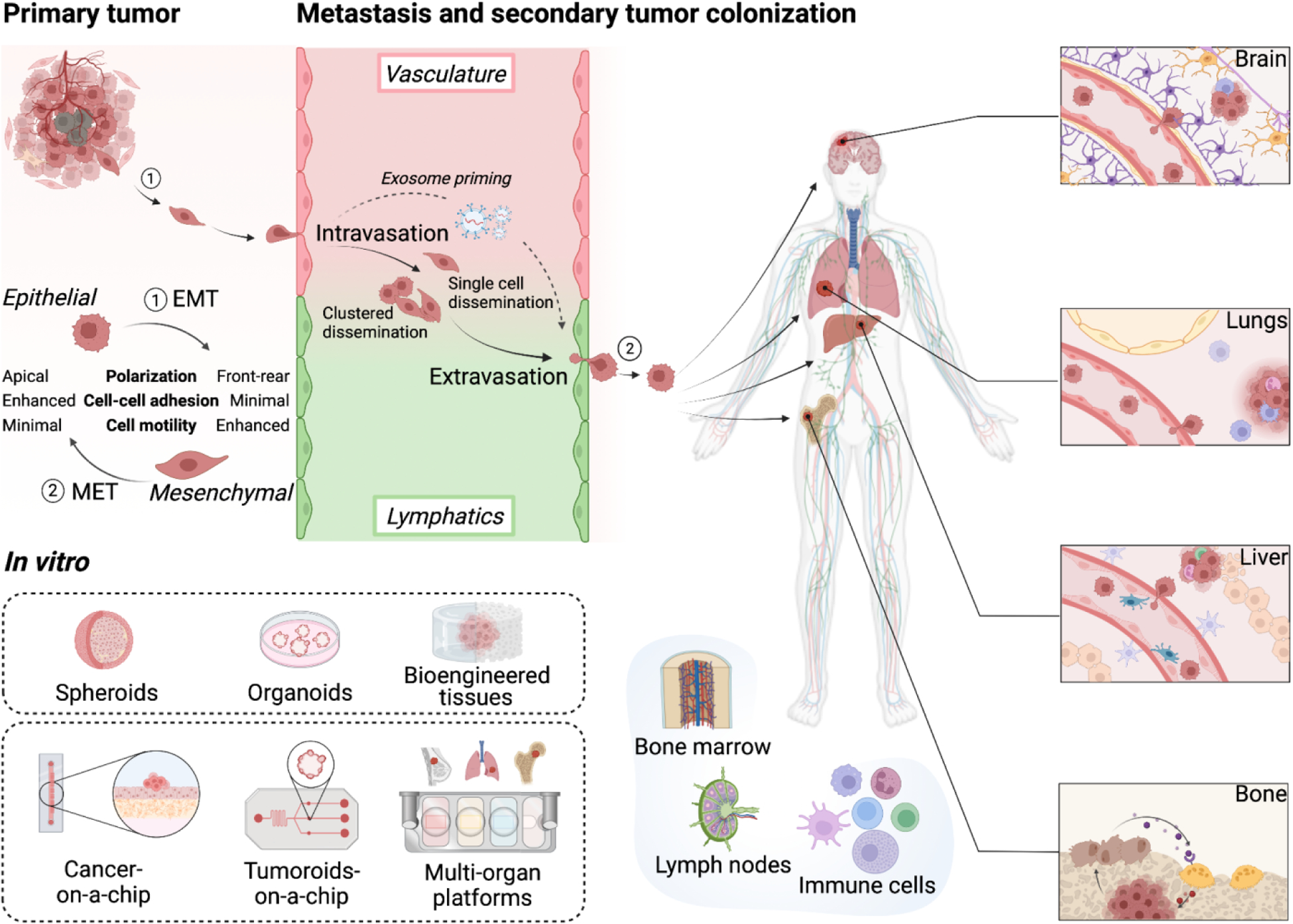
Different models of primary tumors, vasculature and engineered tissues that are common targets for invasion of circulating tumor cells can be utilized alone and in combinations, to model the key steps of metastasis (premetastatic niche, intravasation, dissemination, extravasation, metastatic colonization, formation of secondary tumors). Created with BioRender.com.
3.2.1. Premetastatic niche formation.
The formation of the premetastatic niche, locus of homing for cancer cells evading the primary tumor, is a complex and fundamental piece of the metastatic dissemination. It is now known that the preparation of premetastatic niche involves tumor secreted factors such as extracellular vesicles (EVs), capable of traveling from the primary tumor to a secondary site [91, 92]. The abundance, cargo, surface presentation and overall behavior of EVs are dependent on the cells of origin and microenvironmental cues such as oxygen tension, ECM composition and mechanical properties of the TME [93, 94].
Signaling to secondary locations promotes vascular remodeling towards a leakier phenotype, enabling tumor cells to more readily escape the vasculature and colonize tissues [95, 96]. These signals further prime the metastatic site to enhance cancer cell attachment, survival and stemness, and provide immune surveillance [97]. The bidirectional signaling between the primary and secondary tumor sites enable the premetastatic niche to actively contribute to tumor cell extravasation and colonization of target tissues [98]. This time-dependent chain of events, impossible to reproduce using standard in vitro models, could be emulated by multi-component OOCs, capable of better reproducing each compartment and its characteristic factors [12, 99–109]. The presence of fluid flow not only serves as a connection between the individual compartments and a shuttle for the delivery of the secreted factors, but is also fundamental in adding physiological mechanical stimuli [67]. In preparation of premetastatic niche, the target site undergoes EVs-mediated changes such as ECM remodeling, activation and cell recruitment, both locally and in the surrounding areas, and modification to the vascular compartment such as increased angiogenesis and permeabilization [110].
3.2.2. Intravasation.
Intravasation occurs when cancer cells leave the primary tumor microenvironment by crossing the endothelial barrier and entering the bloodstream where they can subsequently migrate to secondary sites [111]. Cancer cells acquire mesenchymal like characteristics, where they dedifferentiate to be better able to leave the primary tumor, cross the vascular membrane, and invade downstream tissues [112]. The visualization of cancer cell morphology and vimentin staining can be used to determine where a cell is in the EMT process [113]. Engineered models that include both the primary tumor and vasculature would be ideal to model this stage of metastatic progression. In vivo, blood vessels become leaky during cancer development, exposing the underlying cancer cells to blood flow, which can cause them to dislodge and enter circulation. Current OOC models with microfluidic flow and imaging are well suited for these studies [70, 114–125]. For example, a 3D tissue engineered model with co-culture of primary tumor organoids and perfusable microvessels was able to depict tumor-vascular interactions of mosaic vessel formation, organoid entrapment of microvessels, and organoid microvessel pulling, all of which are known to drive intravasation [51].
3.2.3. Dissemination
Once cancer cells have entered the bloodstream, they must survive in circulation if they are to colonize a secondary organ site. Thus, studies of circulating cancer cells (CTCs) are of high importance. It has been shown that cancer cells that survive in circulation are able to exploit the biomechanics of blood flow to their advantage towards supporting metastasis [126, 127]. Microfluidic OOC platforms offer unprecedented insights into how single versus clusters of CTCs survive in circulation and interact with the surrounding microvasculature [128–134]. While it has been demonstrated that CTCs in clumps better survive the shear stresses induced by blood flow than single cells, it is currently unknown whether this advantage also translates to enhanced extravasation [135]. Microfluidic models enable control over parameters of blood flow shear, CTCs cell cluster size, vessel architecture and size, and lymphatic versus circulatory endothelial cells.
3.2.4. Extravasation
CTCs that attach to the endothelium or arrest in microvasculature subsequently extravasate into the surrounding tissue. The arrest of CTCs is driven by the low shear forces and flow rates, and geometrical constraints, ultimately supporting extravasation [136]. OOC models with varying microvascular flow and specific geometrical features can be deployed to decipher the parameters influencing CTC extravasation. Similarly, these platforms enable drug discovery and screening of anti-metastatic compounds that would prevent CTC extravasation.
3.2.5. Metastatic colonization and organ tropism
Extravasated cells begin to proliferate and remodel the tissue environment, co-opting it to support secondary tumor growth. This is known as metastatic colonization. The mechanisms driving organ specific tropism in metastatic colonization of secondary sites are currently unknown. Suggested theories include Paget’s “seed and soil” hypothesis, where CTCs preferentially seed in secondary sites that are more favorable to cancer cell growth by providing a more fertile “soil” [137], and Ewing’s “flow and filter” hypothesis [138], which posits that CTCs preferentially metastasize based on vascular flow patterns governed by flow path, capillary bed size, and adhesion. Both theories are widely accepted, each partially explaining aspects of metastatic colonization and may coexist [139].
Tissue engineered cancer models can allow further deciphering of the mechanisms behind metastatic colonization via microfluidic flow [140, 141]. The inclusion of secondary tissues, through the use of multi-tissue OOC platforms where the tumor module is fluidically connected to the tissue modules representing the potential metastatic targets, enables studies into organ specific tropism. Of note, this model is suitable for reductionist approaches, facilitating translation.
3.3. Secondary tumors.
The liver is the main site of secondary metastasis, while primary liver tumors are rarely seen [142]. The liver is thought to be a favorable secondary tumor site because of its dual blood supply, high vascular to tissue ratio, high total volume of blood flow, slow blood flow rates, and leaky blood vessel structure that stems from the innate high permeability of liver sinusoidal endothelial cells (LSECs). A successful in vitro OOC model of liver metastasis recreated the sinusoidal architecture on a two-channel device, enabling both control over blood flow and tissue geometry [143]. This model was able to reveal the role of cancer-secreted extracellular vesicles in remodeling the secondary tumor site, primarily by increasing CTC binding to the LSECs, promoting secondary tumor colonization.
Bone marrow microenvironment also offers a rich “soil” for secondary tumor colonization, with minerals and growth factors readily available for cancer cells to utilize for their growth, to the detriment of bone marrow [144–146]. It also has a ripe “flow and filter” setting characterized by dense vasculature, high blood flow, and permeable blood vessels that work together to facilitate CTC extravasation.
Metastatic bone lesions are primarily osteolytic, where tumor cells activate osteoclast breakdown of the bone to secrete growth factors favorable to the secondary tumor [147]. Some cancers exhibit an osteoblastic phenotype where osteoblasts are activated instead, leading to enhanced bone nodule formation. Breast and prostate cancers routinely show secondary metastasis to the bone, with breast cancer showing an osteolytic phenotype and prostate cancer showing a mixed or osteoblastic phenotype [147]. These mechanisms remain highly unknown and the studies are hindered by the lack of clinical bone and bone marrow samples. Thus, 3D engineered bone tissue models can be used to study cancer cell growth, recapitulating proper tumor markers only when cultured in 3D bone matrices [11], and enabled predictive evaluation of anticancer drugs [12].
Brain metastasis is similarly difficult to access in vivo, and the development of engineered models of brain metastasis is of great significance. Brain organoids and OOC cultures have provided high-fidelity models of human brain regions [148] and can be further leveraged to model the devastating pathological consequences of brain metastasis. Brain metastasis occurs in 10–30% of cancers and is characterized by dense vasculature and high blood flow, providing a highly favorable flow path for CTCs. Critically, CTCs must pass through the restrictive blood-brain-barrier (BBB). Microfluidic OOC models have been leveraged to model both the healthy BBB and the cancer modified BBB, showing how lung cancer cells must co-opt their ability to attach to the BBB to support extravasation into the brain parenchyma [149].
While the lung was the first OOC developed [69], in vitro models of lung metastasis remain limited. The use of OOC models that contain the relevant cell sources, mechanical strain induced by breathing, and the air-liquid interface characteristic of the lung will be critical for modeling lung metastasis in vitro.
4. Integration of immune components
Until recently, the immune system has been largely neglected in OOC development. In the adult body, the immune system is responsible for maintaining tissue homeostasis and largely involved in the development of disease [150]. Further, the immune system directly contributes to cancer cell survival and disease progression [151], making it a critical component of modeling a patient that cannot be overlooked moving forward. Importantly, the immune system is critical in maintaining the primary tumor microenvironment, aiding in malignant metastasis, and responding to therapeutic measures; the extent of immune system involvement in cancer is summarized in other thorough reviews [12,180,213,214, 224].
The challenges in deriving and using immune cells include the lack of protocols widely available to attain these cells via differentiation from adult stem cells, and their limited life span in vitro. To that end, iPSC-derived protocols are often laborious and have limitations in modeling both myeloid and lymphoid progenitor maturation in vitro [152–163]. Immune organs have been studied extensively in murine models, while their human equivalents are markedly less developed. As OOCs are expanding, the emergence of immune OOCs is of high interest to drug developers, as many new cancer therapeutics take advantage of the human body’s immune system [12, 68, 115, 121, 151,163, 177, 180, 213,214].
4.1. Incorporating the immune system into OOC platforms
The innate immune system consists of myeloid cells which are responsible for killing intruders and releasing signals triggering the adaptive immune response[164, 165]. The adaptive immune system consists of cells from the lymphoid lineage and is responsible for generating antigen-presenting cells, B and T cells, and memory cells capable of re-recognizing the same foreign entity to trigger a stronger response upon re-encounter [166–171]. To that end, engineering models of cancer not only need to incorporate the CTCs involved in systemic responses, like in metastasis or drug treatment, but also must consider the tissue-resident immune cells, which have major roles in promoting protumorigenic microenvironment for primary tumor growth and dissemination. CTCs are now being used in OOC systems [172–177], in parallel to the establishment of immune organs that can serve as a source of blood and immune cells [68, 176, 178–182]. As engineered systems are being developed, new technologies to model immune organs are becoming more relevant for building complex and systemic multi-scale cancer models (Figure 4).
Figure 4. Incorporation of immune components into organs-on-a-chip models.
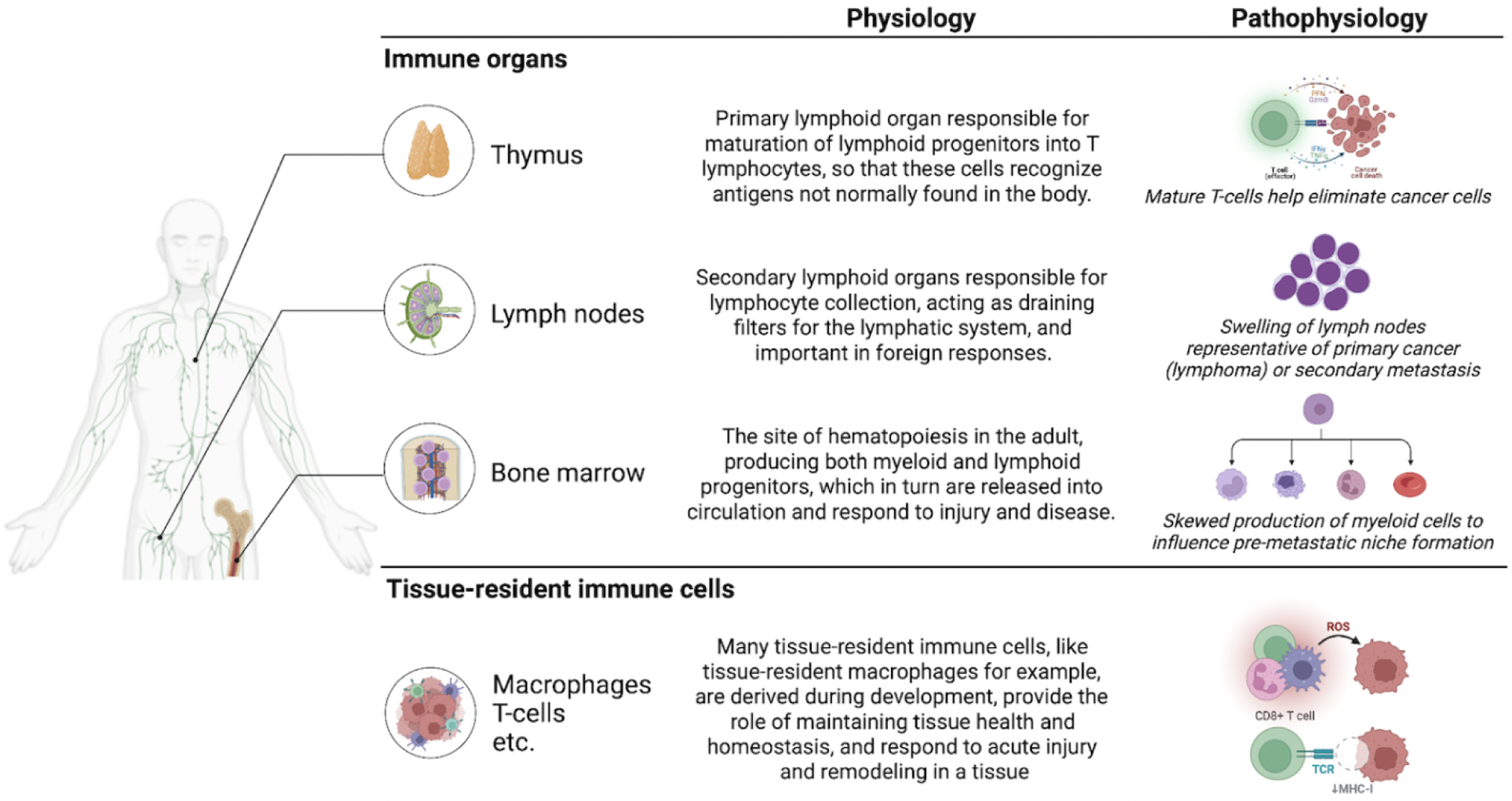
Overview of the human immune organs that are of interest for in vitro models of cancer, with their patho/physiological roles in the body. Created with BioRender.com.
4.2. Bioengineered bone marrow
In the human body, the bone marrow is the site of adult hematopoiesis, where hematopoietic stem cells (HSCs) reside and produce myeloid and lymphoid progenitors in homeostasis and in response to injury or disease [183]. Over the past few years, a number of groups have developed physiological models of human bone marrow, creating a unique environment to study marrow toxicity in chemotherapeutic drug delivery, as well as to study cancer metastasis and colonization [184]. Chou et al. established one of the first human, multicellular microfluidic models of bone marrow, as a platform for studying radiation and chemotherapeutic toxicity (5-FU), and disease modeling in the scope of blood disorder Shwachman-Diamond Syndrome [185]. More recently, George and colleagues developed a model of the human bone marrow for specific studies of cancer cell migration, and in this case, the migration of triple negative breast cancer within the bone marrow niche as compared to acellular controls (MDA-MB-231) [132].
4.3. Bioengineered thymus
Only few studies have attempted to engineer the human thymus, another critical immune organ regulating T lymphocyte development and maturation. In ex vivo maturation of T lymphocytes from hematopoietic progenitors, many groups have demonstrated the ability of scaffolds and stromal cells presenting delta-like-1 notch ligand to mature T lymphocytes from human or murine origin [186–188]. However, few models exist to recapitulate the thymus stromal cells (e.g. thymic epithelial cells, cytoreticular cells, fibroblasts), extracellular matrix components, and reoccurring ability to mature T lymphocytes outside the body [189].
4.4. Bioengineered lymph node
Similarly, there are a few emerging models to study human lymph node tissue, including microfluidic platforms with for culture of lymph node slices ex vivo, as well as lymph-node-on-a-chip platforms for studying antigen presenting cell – T lymphocyte interactions in vitro [20, 78]. Platforms like these enable development of new therapeutics by eliciting antibody production from germinal center-like lymphoid tissues, though these studies have not yet been extended to mechanistic studies of solid tumor interactions. Future work towards integrating lymphoid immune components is crucial to mimicking the systemic dissemination of cancer cells during metastasis of the lymph nodes [77].
5. Framework for engineered models of cancer based on biological question
Current preclinical studies of cancer rely primarily on animal models or cancer cell lines cultured in monolayers. Specifically, while animal models are inherently complex and enable systemic studies of cancer formation and spread over time, their nonhuman physiology hinders clinical translation. This is particularly true when using animal models to predict human immune related responses and metastatic spread [190]. Similarly, the predictive utility of cancer cell lines is hindered by their loss of tumor cell heterogeneity and significant transcriptional drift in culture. Despite these limitations, these models have provided highly useful tools for the field of cancer research, enabling mechanistic study of cancer formation, insight into the drivers of cancer progression, and providing preclinical models for therapeutic development.
Subsequently, engineered human cancer models were primarily designed to serve as a complementary missing piece to these existing preclinical studies that enable enhanced insight into cancer disease, rather than being designed to replace animal models. These engineered models are designed to serve as tools for answering specific questions related to cancer physiology and therapeutic development that current preclinical methods are not well suited to address. They are well suited for applications requiring a humanized setting, such as immunocompetent personalized cancer modeling, and for characterizing metastatic progression, a challenge both in the clinical setting and the existing preclinical models. Overall, these engineered models are finding utility in cancer research, including mechanistic studies of cancer biology, drug development, and precision medicine. Here we detail these use cases in more detail and subsequently provide an example framework used to design each engineered model of cancer in the context of a specific biological question (Figure 5).
Figure 5. Framework for designing engineered models of cancer.
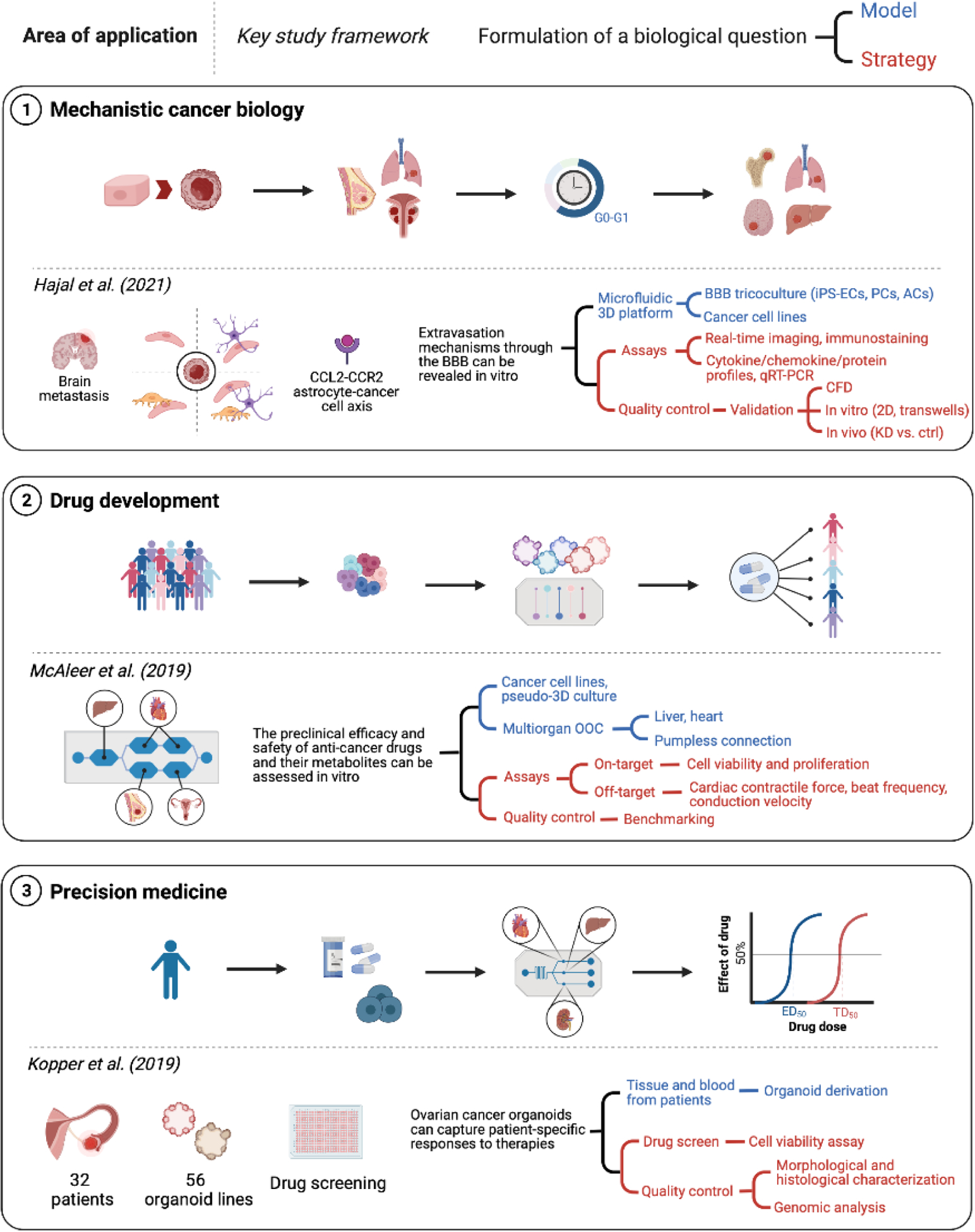
Mechanistic cancer biology, drug development, and precision medicine are three major areas of application of engineered cancer models. The workflow for key studies in each field of research can be summarized identifying the biological question of interest to the investigators and the fundamental elements of the model and strategy adopted to answer it. Created with BioRender.com
5.1. Mechanistic cancer biology.
The advent of iPSCs and gene editing technologies provided opportunities to mechanistically study specific gene mutations and their functional consequences [191–196]. The information gleaned from these experimental studies is invaluable in structuring biologically relevant computational models which in turn drive the development of algorithms that can make biological predictions of enhanced accuracy. While the advances in technology have greatly reduced the cost of genetic sequencing and allowed patients to obtain their genomic data, we still do not fully understand how genetic and epigenetic changes lead to acquired diseases and patient specific responses to environmental changes and therapeutic interventions. Engineered tissue models enable investigations of stromal populations and their roles in the tumor microenvironment [197, 198]. Engineered model systems have been used in conjunction with new technologies, like CRISPR/Cas9 gene editing, to run multiplexed genetic screens of known tumor suppressor pathways and identify checkpoints and therapeutic targets [199, 200]. For example, sequential gene editing of iPSCs with known mutations in leukemia helped identify early targets of myeloid leukemia progression in vitro [201]. Parsing out the contributions of malignant cells and their microenvironment is instrumental for mechanistic cancer-on-a-chip studies, identifying potential new biomarkers of metastatic progression [202, 203].
The first step in designing a framework for using engineered cancer models for mechanistic cancer biology is to think about the model and strategy in the context of the specific biological question intended. The model can then be simplified to focus on the key aspects of the biological question, where input variables can be directly evaluated and interrogated, through either their direct removal/addition or genetic under/overexpression, thereby providing direct evidence for the importance of each input variable on the resulting biological response. The strategy should include assays that provide insight into these biological responses and allow meaningful data extraction. The inclusion of quality control measures, such as using a defined cell response to a known perturbation, serves to validate the platform and provides a baseline for subsequent perturbations which are less well understood. To detail the framework for evaluating mechanistic cancer biology through engineered cancer models, we will use the work of Hajal et al [204]. The drivers of brain metastasis are largely unknown, in part because of the inaccessibility of the brain-blood-barrier (BBB) for in vivo studies. To overcome this, an engineered cancer model of the BBB was developed with the specific goal of enabling evaluation of extravasation mechanisms in vitro. The model system therefore needed to focus on recapitulating the BBB and subsequent cancer cell migration. This was accomplished by using a BBB coculture model of the relevant cell types in a barrier system. The strategy relied on real time tracking of cancer cells as they migrate across the barrier, and characterization of the TME through immunostaining and supernatant assays. The experimental data were analyzed using computational models and validated by comparing to the corresponding in vivo response. Overall, this framework provided support for the CCL2-CCR2 astrocyte-cancer cell axis as a key driver of brain metastases [204].
5.2. Drug development.
The development of a new drug often costs over $1 billion, takes an average of 10 years, and still results in only one out of every nine drugs that have passed preclinical studies making it to market [205]. Among the drugs that have advanced through Phase III into patients, the top 10 drugs only work in 4–25% of the patient population [206]. The high costs and risks associated with drug development are partly due to the use of inadequate models to predict drug behavior in humans [207–209]. The drug fialuridine (FIAU) is an example of how animal testing can lead to disastrous effects in humans, with no toxicities during animal screening but five deaths due to liver failure when the drug was administered to patients [210]. It was later shown that these deaths occurred due to species-specific drug induced mitochondrial toxicity, that would only be identified by the use of preclinical human models. Subsequently, it has been shown that human in vitro liver models were capable of detecting FIAU induced liver toxicity [211], further supporting the need to include human models in drug development. Similarly, the drug TGN1412, a CD28 superantagonist antibody that was safe in all animal studies, caused life-threatening side effects in the first six healthy volunteers enrolled in the clinical trial [212].
To bridge this large unmet need in drug development and predict how a drug will work in human patient populations, there is a need for drug testing in models that are human, systemic, incorporating an immune component, and capturing the heterogeneity of the patient responses [197, 213]. Engineered human cancer models will further enable preclinical evaluation of potential therapeutics towards selecting those with the highest likelihood of clinical success [27, 45].
Human mimics of metastasis are most critically needed. A paradigm shift towards human tissue models of metastatic cancer would tilt the cancer therapeutic pipeline towards treating cancer metastasis, from the current focus on primary tumors. Bioengineered cancer tissues are beginning to serve as the missing link in developing human therapeutics for cancer treatment. By providing human data at early time points during drug development, the engineered cancer models would decrease the time and costs of getting a drug to market, while also ensuring only safe drugs to get to patients.
To provide an example framework for designing engineered cancer models for drug development, we will use the work by McAleer [214]. The biological question they sought to model was the preclinical efficacy and safety of anti-cancer drugs, where there is a specific need for a human liver module to actively metabolize the drugs and capture the subsequent effects of both the parent drug and its metabolites. The model system they chose included the use of cancer cell lines with known drug sensitive and resistant phenotypes, to enable direct evaluation of the ability to replicate on-target anti-cancer drug responses, and a simple psuedo-3D culture of the cells to recreate a TME that enables tracking cancer cell proliferation and viability. The systemic nature of anti-cancer drugs, acting on both the intended cancer cells and being both metabolized by the liver and causing off-target toxicity in the liver and heart, necessitated the inclusion of multiple organs using a pumpless multi-organ-on-a-chip system with shared recirculating medium. This simple model and strategy enabled efficient preclinical drug testing in a human setting, replicating both the on-target efficacy and off-target toxicity of anti-cancer drugs and their metabolites.
When designing the framework for drug development applications it is of utmost importance to include quality control measures into the strategy. Here they accomplished this by using cancer cell lines with known drug responsiveness, enabling the researchers to validate their model by replicating these known responses in the model. As these model systems become more developed, it will be of increasing importance to establish a standard panel of negative and positive control drugs and recapitulate the known tissue responses, as a basis for predictive screening of new drugs. Further, adapting these engineered systems for studying primary tumor samples will be crucial for assessing personalized approaches to cancer therapeutics.
5.3. Precision medicine.
As prevention and therapy continue to progress, we live longer and better than ever, prompting the healthcare to enter the era of precision medicine. Rather than reactive medicine aimed at treating patient symptoms as they arise, precision medicine (focused on a specific cohort of patients) is becoming personalized (focused on a specific patient). The evolving personalized medicine approaches seek to individualize patient healthcare so that diseases can be predicted, prevented when possible, and treated in a personalized manner that involves participation of the patient [215]. This forward-thinking approach will greatly benefit from tissue engineering, where patient cells and engineered tissues can serve as patient specific in vitro models of disease progression and treatment.
For large patient populations, tissue engineering provides models that help test preventative medicine modalities for reducing the burden of diseases known to affect specific cohorts. For each patient, a specific disease could be modeled using the patient’s own cells so that the treatment regimen can be customized. It is for the first time that we can go beyond population studies into determining the effects of sex, race, age and conducting “patient on a chip” studies of drugs. This approach can inform the design of clinical trials by determining patient populations that will most benefit from the drug and those that may be at risk. Overall, the long-term impact of engineered human cancer models directly relates to their patient specificity, enabling the optimization of the therapeutic treatment regimen for each individual.
Focus on patient specific models of drug resistance and metastasis puts the emphasis on the patients most vulnerable to disease, a piece missing in the current paradigm. The proposed synergy between clinical, bioengineering, and computational expertise is expected to enable the optimization of drug dosage, combination therapy, and drug development in a way that prioritizes the individual therapeutic outcome for each patient. Patient-derived organoids have been used to predict patient specific response to cancer treatments by directly screening cell responses to drug panels [46]. While many patients have specific mutations for which drug prioritization is straightforward, more than half of patients are ineligible for targeted treatments [216]. However, by leveraging patient-specific engineered cancer models, clinicians could determine the best treatment for each patient even when traditional genomic biomarkers are absent.
The model system for precision medicine studies should (i) ensure efficiency to effectively impact treatment regimens, and (ii) be based on patient-derived cells in order to inform personalized responses. The strategy should rely on a robust, controlled approach where multiple model replicates can be screened by the full library of available cancer treatments and have a meaningful readout of drug response, such as cell viability. Organoid models are well poised for this framework as they are patient derived and can be engineered to create reproducible model arrays in a well-plate format that is compatible with existing drug screening approaches. It is particularly important to include quality control measures when working within a precision medicine framework, as these efforts have the potential to directly influence patient outcomes and therefore require direct benchmarking to evaluate whether the patient-derived cells change in culture and whether their drug responses mimic that of the patient in the clinical setting.
To provide an example framework for designing an engineered cancer model for precision medicine, we will use the work by Kopper et al [217]. They created patient-derived organoid models from 32 patients, to yield 56 successful organoid lines. These organoids were used for high throughput screening where on-target drug efficacy responses were evaluated using a cell viability assay. Quality control measures were included by performing genomic analysis of the organoids over time. The results demonstrated that organoids maintained the diverse molecular and genetic traits of the original tumor, further supporting the use of organoids as a suitable engineered cancer model for precision medicine. This is in stark contrast to what is seen with standard patient cell line development, which rapidly lose the molecular and genetic characteristics of the original tumor due to in vitro selection during 2D culture [218]. Overall, patient-derived organoid models, which capture patient-specific responses and maintain tumor heterogeneity, and high throughput screening strategies, coupled with quality control measures, provide an exciting engineered model of cancer for personalized medicine.
6. Future directions
6.1. Increased complexity.
With respect to biological complexity of tissue models of cancer, a major limitation is the current lack of methods to form tumor vasculature from appropriate endothelial cells and to attain high permeability comparable to that in native tumors, an important component for studies of cancer progression and responses to drugs. Biological value of engineered tissues, such as those being targets for metastasis (liver, lung, brain, bone) would markedly increase from establishing perfusable vasculature, innervation, tissue interfaces, and microbiome. The immune and endocrine systems are particularly important, both for their direct relevance to cancer research and the inability to model cancer-immune-endocrine interactions in animal models. Bioengineered models of lymphatics and immune organs are just beginning to be developed, and they are important for having sustained production of immune cells and lymphatic flow. While progress with the model of human bone marrow with hematopoietic function is encouraging, generation of lymphoid cells matching the patient remains challenging. As both the adaptive and innate immune systems are critical to cancer progression and treatment of primary tumor tissue, there still is an intense need to recapitulate the multifaceted interactions of immune cells in vitro [219, 220]. Current tissue engineering models lack vasculature perfusable by blood, a key component to cancer disease biology and drug responses. In particular, the tumor vasculature needs to consist of not only tissue-specific ECs, but ECs that are able to adapt to tumor-inducing signals to drive high permeability of vasculature, comparable to that of a native tumor. As dysregulated angiogenesis is a hallmark of cancer, it is important that the choice of ECs reflects that of microvasculature found in individual organs, rather than commonly used macrovascular ECs such as those from the human umbilical vein. New protocols to establish iPSC-derived equivalents may help circumvent this challenge, producing high quantities of endothelial progenitors that may further adopt phenotypic changes of tumor tissue [221].
6.2. Benchmarking and validation
Benchmarking and validation of bioengineered models against retrospective and prospective clinical data and appropriate animal models will be critical to building models of primary and metastatic tumors capable of predicting clinical outcomes. It remains to determine what are the required levels of authenticity of molecular, structural and functional tissue phenotypes, and which exact readouts are relevant for validation. While the use of patient derived human iPS cells enables the development of patient specific in vitro models, their use in precision medicine is currently limited by the lack of understanding of how in vitro models correlate to their clinical patient counterparts. Future research utilizing clinical data and patient matched experimental in vitro data will be critical to building higher throughput models capable of accurately predicting clinical outcomes. By identifying commonalities and irregularities in matched clinical and in vitro model generated data, we can determine the limitations of in vitro models and subsequently optimize these models to properly match the in vivo data. The use of tissue engineered models also enables systematic investigations in the human context, where current mechanistic investigations are limited to clinical observations, animal models, or simple cell culture. The future use of engineered models to obtain patient-specific clinical predictions can facilitate identification of shared mechanisms related to disease risk, discovery of early-stage biomarkers of cancer, and optimal, patient-specific therapeutic regimens.
To validate the clinical predictive capacity of engineered models of cancer, tumor cells studies in OOC platforms should be treated with the same therapeutics and directly compared using similar metrics. Such “clinical-trial-on-a-chip” studies will be instrumental to quantify the predictive power of these models, and to identify gaps in their translation that need to be overcome or taken into account when interpreting the experimental results. Example matched data include patient-specific tumor response to drugs, maintenance of tumor phenotype within the engineered model, and demonstrated secondary tumor metastasis to sites seen within the patient. Opportunities to extract large datasets from these matched studies enable the use of artificial intelligence to correlate patient and model responses and extrapolate how engineered models can be used to decipher the complex biology of cancer in vitro. This aligns with current approaches that leverage the advances in transcriptomic and multiplexed assays to generate large patient specific datasets that can be further used to train predictive algorithms for next generation models.
6.3. Scaling.
Another critical challenge in recapitulating the physiological demands of multiple tissues integrated on a chip is matching the metabolic and organ-organ interactions seen in vivo [222]. Approximating the appropriate cell numbers and ratios between individual tissue masses remains an important area for consideration, as the number of cells may dictate the impact cell-cell interactions (i.e. circulating cells) and cell secretome signaling may have on an off-target tissue. In addition, the liver, known as the site of metabolism in the body, may have differential responses to drugs depending on the in vitro tissue size and function. Therefore, for studies of drug efficacy and personalized responses, we must attempt to try to match the functional load of the liver in a healthy individual, as to match the drug metabolism demands in vitro. In many current multi-OOC systems, tissues have been engineered individually and linked with a common perfusing channel, though there have been a number of proposed frameworks for integrating scaling into the design of multi-OOC platforms [223]. In modeling tumor burden and metastasis, future models will have to better recapitulate the metabolic demands of cancer, benchmarking and paralleling the simplicity of cancer cell migration in vitro to the metastatic cascade in vivo. Not only in scaling of solid organs, but the scaling of the complex vascular and immune networks seen in vivo will remain an important challenge in developing new human OOC platforms [19, 189].
6.4. Regulatory and stakeholder adoption.
To advance biological research and drug development, tissue culture platforms for modeling cancer will need to become broadly available, cost-effective and user friendly. The simplest models, such as organoids, are already widely used, while more complex systems (engineered tissues, OOCs) reside in laboratories where they were developed, and are just starting to be used in collaborations between scientists, engineers, clinicians and pharmaceutical companies [121]. Further progress development of modular, configurable platforms would greatly facilitate standardization and adoption of even complex systems. Longitudinal studies are invaluable for improving the rigor and consistency of experimentation, removing sample-to-sample variability and capturing the biological dynamics of cancer. To this end, noninvasive real-time measurements are emerging as one of the important technical requirements for the further development of cancer models. The adoption of engineered models beyond academic use will rely on their increased robustness and throughput, a more defined view of their predictive capacity for various use cases, and their acceptance by regulatory agencies for therapeutic development and clinical utilization. The tradeoff between model complexity, system control, and “time-to-results” continues to be further evaluated, positioning these models as a mostly academic research tool for the meantime. To drive industry adoption of these models, various regulatory agencies and government funding initiatives have established concerted efforts to standardize, qualify, and validate these models in a coherent manner [224].
6.5. Individualized approach.
Patient-specificity of bioengineered tissue models of cancer remains a major need in the field. Tissue niches for the formation of primary and metastatic tumors are typically formed using blood-derived iPSCs that are differentiated into tissue specific and supporting cells and induced to form tissues. iPSCs provide defined genetic background that is a basis for patient specificity, allow parsing out the effects of genetic and environmental factors, and enable studies of biologic diversity. Tissues derived from iPSCs are being matured in culture towards adult-like phenotypes. A major challenge in establishing patient-specific models of cancer is the introduction of the patient’s tumor cells and immune cells, matching with all tissues in the model. While myeloid cells do not have to be matched, matching of lymphoid cells is needed. Also, there is no alternative to using the patient’s cancer cells, because of the heterogeneity of tumor cell populations not only from one patient to another, but also within the same tumor. It is possible that this challenge can be addressed by generating immune-agnostic tissues and combining these with the patient’s immune cells and cancer cells.
7. Summary
Modeling integrated human physiology in vitro is a rapidly expanding area, where bioengineered models of human tissues are used to study development, regeneration, disease and responses to drugs in human physiological context. The unique feature of these models is that they consist of micro-sized human tissues, cultured alone or linked to each other, in a way that allows recapitulation of organ-level and systemic responses [120]. This technology has emerged from converging advances in tissue engineering, microfabrication and cell biology. The ability to establish patient-specific tissue models is offering unique advantages over simple cell culture and small animal models in a number of areas, and most critically in cancer where patient-to-patient differences can largely determine the progression of disease and response to drugs.
As the adoption and use of these models by the research community and pharma are increasing, we review here their state of the art, current designs and applications, advantages and limitations. We focus on the complexity of human tissue models of cancer, and how the level of complexity relates to the biological fidelity and functional outcomes of the models, and their utility in biological and pharmacological studies. We ask how simple a model can be to provide sufficient complexity for studying a specific question. We discuss a framework for designing the model of the right type and complexity for meeting a specific study goal. Thinking forward, we outline some of the research gaps and ongoing developments that may overcome the current challenges and further increase the adoption of human tissue models.
Highlights.
Current models of cancer result in low yield and high cost of drug testing
Cancer metastasis is of greatest interest and most difficult to model
New models provide patient-specific studies of cancer progression and treatment
The model complexity can be tailored to capture the specific aspects of cancer
We propose a framework for selecting and designing an appropriate model
Acknowledgments
Studies described in this review are funded by NIH (grants R01 CA249799, UH3 EB025765 and P41 EB027062 to GVN), NASA (grant NNX16AO69A to GVN), NSF (Graduate Research Fellowship DGE1644869 to DNT) and ASPR-BARDA (grant 75A50121C00017 to GVN). European Research Council (ERC StG-UERI17, to EC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no competing interests.
References
- 1.Bray F, et al. , Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 2018. 68(6): p. 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Gupta GP and Massagué J, Cancer Metastasis: Building a Framework. Cell, 2006. 127(4): p. 679–695. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, et al. , Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduction and Targeted Therapy, 2020. 5(1): p. 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mestas J and Hughes CC, Of mice and not men: differences between mouse and human immunology. J Immunol, 2004. 172(5): p. 2731–8. [DOI] [PubMed] [Google Scholar]
- 5.Ben-David U, et al. , Patient-derived xenografts undergo mouse-specific tumor evolution. Nature genetics, 2017. 49(11): p. 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X and Lewis MT, Establishment of Patient-Derived Xenograft (PDX) Models of Human Breast Cancer. Curr Protoc Mouse Biol, 2013. 3(1): p. 21–9. [DOI] [PubMed] [Google Scholar]
- 7.Lai Y, et al. , Current status and perspectives of patient-derived xenograft models in cancer research. Journal of Hematology & Oncology, 2017. 10(1): p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada S, Vaeteewoottacharn K, and Kariya R, Establishment of a Patient-Derived Tumor Xenograft Model and Application for Precision Cancer Medicine. Chem Pharm Bull (Tokyo), 2018. 66(3): p. 225–230. [DOI] [PubMed] [Google Scholar]
- 9.Day CP, Merlino G, and Van Dyke T, Preclinical mouse cancer models: a maze of opportunities and challenges. Cell, 2015. 163(1): p. 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitra A, Mishra L, and Li S, Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends in biotechnology, 2013. 31(6): p. 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villasante A, Marturano-Kruik A, and Vunjak-Novakovic G, Bioengineered human tumor within a bone niche. Biomaterials, 2014. 35(22): p. 5785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chramiec A, et al. , Integrated human organ-on-a-chip model for predictive studies of antitumor drug efficacy and cardiac safety. Lab on a Chip, 2020. 20(23): p. 4357–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de la Puente P, et al. , 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials, 2015. 73: p. 70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassell BA, et al. , Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Reports, 2017. 21(2): p. 508–516. [DOI] [PubMed] [Google Scholar]
- 15.Bonnans C, Chou J, and Werb Z, Remodelling the extracellular matrix in development and disease. Nature reviews. Molecular cell biology, 2014. 15(12): p. 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gehler S, et al. , Bi-directional signaling: extracellular matrix and integrin regulation of breast tumor progression. Crit Rev Eukaryot Gene Expr, 2013. 23(2): p. 139–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato Y, et al. , Acidic extracellular microenvironment and cancer. Cancer Cell International, 2013. 13(1): p. 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaaf MB, Garg AD, and Agostinis P, Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death & Disease, 2018. 9(2): p. 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graney PL, et al. , Engineered models of tumor metastasis with immune cell contributions. iScience, 2021. 24(3): p. 102179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moura Rosa P, et al. , The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab on a Chip, 2016. 16(19): p. 3728–3740. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues J, et al. , 3D In Vitro Model (R)evolution: Unveiling Tumor-Stroma Interactions. Trends Cancer, 2021. 7(3): p. 249–264. [DOI] [PubMed] [Google Scholar]
- 22.Villasante A, et al. , Human Serum Enhances Biomimicry of Engineered Tissue Models of Bone and Cancer. Front Bioeng Biotechnol, 2021. 9: p. 658472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler J, et al. , Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nature Communications, 2020. 11(1): p. 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tlsty TD and Gascard P, Stromal directives can control cancer. 2019. 365(6449): p. 122–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghajar CM, et al. , The perivascular niche regulates breast tumour dormancy. Nature Cell Biology, 2013. 15(7): p. 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gill BJ and West JL, Modeling the tumor extracellular matrix: Tissue engineering tools repurposed towards new frontiers in cancer biology. J Biomech, 2014. 47(9): p. 1969–78. [DOI] [PubMed] [Google Scholar]
- 27.Harris AR, Yuan JX, and Munson JM, Assessing multiparametric drug response in tissue engineered tumor microenvironment models. Methods, 2018. 134–135: p. 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mierke CT, The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep Prog Phys, 2019. 82(6): p. 064602. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, et al. , 3D cell culture models: Drug pharmacokinetics, safety assessment, and regulatory consideration. Clin Transl Sci, 2021. 14(5): p. 1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J, et al. , Reconfigurable open microfluidics for studying the spatiotemporal dynamics of paracrine signalling. Nature Biomedical Engineering, 2019. 3(10): p. 830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye L, Swingen C, and Zhang J, Induced pluripotent stem cells and their potential for basic and clinical sciences. Current cardiology reviews, 2013. 9(1): p. 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfannkuche K, et al. , Induced pluripotent stem cells: a new approach for physiological research. Cell Physiol Biochem, 2010. 26(2): p. 105–24. [DOI] [PubMed] [Google Scholar]
- 33.Lai BFL, et al. , A well plate–based multiplexed platform for incorporation of organoids into an organ-on-a-chip system with a perfusable vasculature. Nature Protocols, 2021. 16(4): p. 2158–2189. [DOI] [PubMed] [Google Scholar]
- 34.Guo X, et al. , Enrichment of cancer stem cells by agarose multi-well dishes and 3D spheroid culture. Cell Tissue Res, 2019. 375(2): p. 397–408. [DOI] [PubMed] [Google Scholar]
- 35.Ishiguro T, et al. , Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci, 2017. 108(3): p. 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee KH and Kim TH, Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors (Basel), 2021. 11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gebeyehu A, et al. , Polysaccharide hydrogel based 3D printed tumor models for chemotherapeutic drug screening. Scientific reports, 2021. 11(1): p. 372–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, et al. , Advances in 3D peptide hydrogel models in cancer research. npj Science of Food, 2021. 5(1): p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unnikrishnan K, Thomas LV, and Ram Kumar RM, Advancement of Scaffold-Based 3D Cellular Models in Cancer Tissue Engineering: An Update. 2021. 11(4468). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loessner D, et al. , Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials, 2010. 31(32): p. 8494–506. [DOI] [PubMed] [Google Scholar]
- 41.Drost J and Clevers H, Organoids in cancer research. Nature Reviews Cancer, 2018. 18(7): p. 407–418. [DOI] [PubMed] [Google Scholar]
- 42.Prince E, et al. , Microfluidic Arrays of Breast Tumor Spheroids for Drug Screening and Personalized Cancer Therapies. Adv Healthc Mater, 2021: p. e2101085. [DOI] [PubMed] [Google Scholar]
- 43.Jung JW, et al. , Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One, 2011. 6(11): p. e28068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun W, et al. , High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res, 1999. 59(24): p. 6118–23. [PubMed] [Google Scholar]
- 45.Trosko JE, A Historical Perspective for the Development of Mechanistic-Based 3D Models of Toxicology Using Human Adult Stem Cells. Toxicol Sci, 2018. 165(1): p. 6–9. [DOI] [PubMed] [Google Scholar]
- 46.Wensink GE, et al. , Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. npj Precision Oncology, 2021. 5(1): p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Driehuis E, et al. , Correction: Patient-derived oral mucosa organoids as an in vitro model for methotrexate induced toxicity in pediatric acute lymphoblastic leukemia. PLOS ONE, 2020. 15(8): p. e0237488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takebe T, Zhang B, and Radisic M.J.C.s.c., Synergistic engineering: organoids meet organs-on-a-chip. 2017. 21(3): p. 297–300. [DOI] [PubMed] [Google Scholar]
- 49.Atat OE, et al. , 3D modeling in cancer studies. Hum Cell, 2021. [DOI] [PubMed] [Google Scholar]
- 50.Infanger DW, Lynch ME, and Fischbach C, Engineered culture models for studies of tumor-microenvironment interactions. Annu Rev Biomed Eng, 2013. 15: p. 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silvestri VL, et al. , A Tissue-Engineered 3D Microvessel Model Reveals the Dynamics of Mosaic Vessel Formation in Breast Cancer. Cancer Res, 2020. 80(19): p. 4288–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan ML, Ling L, and Fischbach C, Engineering strategies to capture the biological and biophysical tumor microenvironment in vitro. Adv Drug Deliv Rev, 2021. 176: p. 113852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bregenzer ME, et al. , Integrated cancer tissue engineering models for precision medicine. PLOS ONE, 2019. 14(5): p. e0216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ananthanarayanan B, Kim Y, and Kumar S, Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials, 2011. 32(31): p. 7913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin J-W and Mooney DJ, Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. 2016. 113(43): p. 12126–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marturano-Kruik A, et al. , Human bone perivascular niche-on-a-chip for studying metastatic colonization. Proc Natl Acad Sci U S A, 2018. 115(6): p. 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Low LA and Tagle DA, Organs-on-chips: Progress, challenges, and future directions. Exp Biol Med (Maywood), 2017. 242(16): p. 1573–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watson DE, Hunziker R, and Wikswo JP, Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp Biol Med (Maywood), 2017. 242(16): p. 1559–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vunjak-Novakovic G, Ronaldson-Bouchard K, and Radisic M, Organs-on-a-chip models for biological research. Cell, 2021. 184(18): p. 4597–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sontheimer-Phelps A, Hassell BA, and Ingber DE, Modelling cancer in microfluidic human organs-on-chips. Nature Reviews Cancer, 2019. 19(2): p. 65–81. [DOI] [PubMed] [Google Scholar]
- 61.Dhiman N, et al. , On-chip anticancer drug screening - Recent progress in microfluidic platforms to address challenges in chemotherapy. Biosens Bioelectron, 2019. 137: p. 236–254. [DOI] [PubMed] [Google Scholar]
- 62.Ewart L, et al. , Navigating tissue chips from development to dissemination: A pharmaceutical industry perspective. Exp Biol Med (Maywood), 2017. 242(16): p. 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rothbauer M, et al. , Tomorrow today: organ-on-a-chip advances towards clinically relevant pharmaceutical and medical in vitro models. Curr Opin Biotechnol, 2019. 55: p. 81–86. [DOI] [PubMed] [Google Scholar]
- 64.Caballero D, et al. , Organ-on-chip models of cancer metastasis for future personalized medicine: From chip to the patient. Biomaterials, 2017. 149: p. 98–115. [DOI] [PubMed] [Google Scholar]
- 65.Hughes DJ, Kostrzewski T, and Sceats EL, Opportunities and challenges in the wider adoption of liver and interconnected microphysiological systems. Exp Biol Med (Maywood), 2017. 242(16): p. 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skardal A, Shupe T, and Atala A, Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov Today, 2016. 21(9): p. 1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Follain G, et al. , Fluids and their mechanics in tumour transit: shaping metastasis. Nature Reviews Cancer, 2020. 20(2): p. 107–124. [DOI] [PubMed] [Google Scholar]
- 68.Henderson AR, Choi H, and Lee E, Blood and Lymphatic Vasculatures On-Chip Platforms and Their Applications for Organ-Specific In Vitro Modeling. Micromachines (Basel), 2020. 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huh D, et al. , Reconstituting organ-level lung functions on a chip. Science, 2010. 328(5986): p. 1662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee E, Song HHG, and Chen CS, Biomimetic on-a-chip platforms for studying cancer metastasis. Current Opinion in Chemical Engineering, 2016. 11: p. 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mierke CT, Mechanical Cues Affect Migration and Invasion of Cells From Three Different Directions. Front Cell Dev Biol, 2020. 8: p. 583226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duzagac F, et al. , Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research. Cancers, 2021. 13(4): p. 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haque MR, et al. , Organ-Chip Models: Opportunities for Precision Medicine in Pancreatic Cancer. Cancers, 2021. 13(17): p. 4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sontheimer-Phelps A, Hassell BA, and Ingber DE, Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer, 2019. 19(2): p. 65–81. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, et al. , From cell spheroids to vascularized cancer organoids: Microfluidic tumor-on-a-chip models for preclinical drug evaluations. Biomicrofluidics, 2021. 15(6): p. 061503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ronaldson-Bouchard K and Vunjak-Novakovic G, Organs-on-a-Chip: A Fast Track for Engineered Human Tissues in Drug Development. Cell Stem Cell, 2018. 22(3): p. 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gu Y, et al. , Tumor-educated B cells selectively promote breast cancer lymph node metastasis by HSPA4-targeting IgG. Nature Medicine, 2019. 25(2): p. 312–322. [DOI] [PubMed] [Google Scholar]
- 78.Ross AE, et al. , Spatially resolved microfluidic stimulation of lymphoid tissue ex vivo. Analyst, 2017. 142(4): p. 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clark AM, Allbritton NL, and Wells A, Integrative microphysiological tissue systems of cancer metastasis to the liver. Semin Cancer Biol, 2021. 71: p. 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Faley SL, et al. , iPSC-Derived Brain Endothelium Exhibits Stable, Long-Term Barrier Function in Perfused Hydrogel Scaffolds. Stem Cell Reports, 2019. 12(3): p. 474–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sassi L, et al. , A Perfusion Bioreactor for Longitudinal Monitoring of Bioengineered Liver Constructs. Nanomaterials (Basel), 2021. 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tronolone JJ, et al. , Pumpless, modular, microphysiological systems enabling tunable perfusion for long-term cultivation of endothelialized lumens. Biomed Microdevices, 2021. 23(2): p. 25. [DOI] [PubMed] [Google Scholar]
- 83.Wang YI and Shuler ML, UniChip enables long-term recirculating unidirectional perfusion with gravity-driven flow for microphysiological systems. Lab Chip, 2018. 18(17): p. 2563–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei L, et al. , Microfluidics-enabled 96-well perfusion system for high-throughput tissue engineering and long-term all-optical electrophysiology. Lab Chip, 2020. 20(21): p. 4031–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bado IL, et al. , The bone microenvironment increases phenotypic plasticity of ER+ breast cancer cells. Developmental Cell, 2021. 56(8): p. 1100–1117.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walker C, Mojares E, and Del Río Hernández A, Role of Extracellular Matrix in Development and Cancer Progression. International journal of molecular sciences, 2018. 19(10): p. 3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mierke CT, et al. , The two faces of enhanced stroma: Stroma acts as a tumor promoter and a steric obstacle. NMR Biomed, 2018. 31(10): p. e3831. [DOI] [PubMed] [Google Scholar]
- 88.Risson E, et al. , The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nature Cancer, 2020. 1(7): p. 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Montagner M and Sahai E, In vitro Models of Breast Cancer Metastatic Dormancy. 2020. 8(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barney LE, et al. , Tumor cell–organized fibronectin maintenance of a dormant breast cancer population. 2020. 6(11): p. eaaz4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong Q, et al. , Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front Cell Dev Biol, 2021. 9: p. 733627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ghoroghi S, et al. , Tumor extracellular vesicles drive metastasis (it’s a long way from home). 2021. 3(11): p. 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bhatta B and Cooks T, Reshaping the tumor microenvironment: extracellular vesicles as messengers of cancer cells. Carcinogenesis, 2020. 41(11): p. 1461–1470. [DOI] [PubMed] [Google Scholar]
- 94.Naito Y, et al. , How cancer cells dictate their microenvironment: present roles of extracellular vesicles. Cell Mol Life Sci, 2017. 74(4): p. 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Farnsworth RH, et al. , Vascular remodeling in cancer. Oncogene, 2014. 33(27): p. 3496–3505. [DOI] [PubMed] [Google Scholar]
- 96.Hooglugt A, et al. , Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Frontiers in oncology, 2021. 10: p. 612802–612802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marar C, Starich B, and Wirtz D, Extracellular vesicles in immunomodulation and tumor progression. Nature Immunology, 2021. 22(5): p. 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hansen MT, et al. , A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene, 2015. 34(4): p. 424–35. [DOI] [PubMed] [Google Scholar]
- 99.Carvalho MR, et al. , Colorectal tumor-on-a-chip system: A 3D tool for precision onconanomedicine. 2019. 5(5): p. eaaw1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Esch EW, Bahinski A, and Huh D, Organs-on-chips at the frontiers of drug discovery. Nature Reviews Drug Discovery, 2015. 14(4): p. 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeon JS, et al. , Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. 2015. 112(1): p. 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lai BFL, et al. , InVADE: Integrated Vasculature for Assessing Dynamic Events. 2017. 27(46): p. 1703524. [Google Scholar]
- 103.Lai BFL, et al. , Recapitulating Pancreatic Tumor Microenvironment through Synergistic Use of Patient Organoids and Organ-on-a-Chip Vasculature. 2020. 30(48): p. 2000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maoz BM, et al. , A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat Biotechnol, 2018. 36(9): p. 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marx U, et al. , Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. Altex, 2016. 33(3): p. 272–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCracken KW, et al. , Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature, 2014. 516(7531): p. 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Skardal A, et al. , Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Scientific Reports, 2017. 7(1): p. 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Trapecar M, et al. , Gut-Liver Physiomimetics Reveal Paradoxical Modulation of IBD-Related Inflammation by Short-Chain Fatty Acids. Cell Syst, 2020. 10(3): p. 223–239.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao S, et al. , A microfluidic culture model of the human reproductive tract and 28-day menstrual cycle. Nat Commun, 2017. 8: p. 14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zeng Z, et al. , Cancer-derived exosomal miR-25–3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nature Communications, 2018. 9(1): p. 5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zavyalova MV, et al. , Intravasation as a Key Step in Cancer Metastasis. Biochemistry (Mosc), 2019. 84(7): p. 762–772. [DOI] [PubMed] [Google Scholar]
- 112.Thompson EW and Newgreen D.F.J.C.r., Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? 2005. 65(14): p. 5991–5995. [DOI] [PubMed] [Google Scholar]
- 113.van Zijl F, Krupitza G, and Mikulits W, Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res, 2011. 728(1–2): p. 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Agarwal P, et al. , Microfluidics Enabled Bottom-Up Engineering of 3D Vascularized Tumor for Drug Discovery. ACS Nano, 2017. 11(7): p. 6691–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bai J, et al. , Identification of drugs as single agents or in combination to prevent carcinoma dissemination in a microfluidic 3D environment. Oncotarget, 2015. 6(34): p. 36603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim S, et al. , Comparison of Cell and Organoid-Level Analysis of Patient-Derived 3D Organoids to Evaluate Tumor Cell Growth Dynamics and Drug Response. SLAS Discov, 2020. 25(7): p. 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mencattini A, et al. , High-throughput analysis of cell-cell crosstalk in ad hoc designed microfluidic chips for oncoimmunology applications. Methods Enzymol, 2020. 632: p. 479–502. [DOI] [PubMed] [Google Scholar]
- 118.Michna R, et al. , Vascularized microfluidic platforms to mimic the tumor microenvironment. Biotechnol Bioeng, 2018. 115(11): p. 2793–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pradhan S, et al. , A Microvascularized Tumor-mimetic Platform for Assessing Anti-cancer Drug Efficacy. Sci Rep, 2018. 8(1): p. 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rajasekar S, et al. , IFlowPlate-A Customized 384-Well Plate for the Culture of Perfusable Vascularized Colon Organoids. Adv Mater, 2020. 32(46): p. e2002974. [DOI] [PubMed] [Google Scholar]
- 121.Shang M, et al. , Microfluidic modelling of the tumor microenvironment for anti-cancer drug development. Lab Chip, 2019. 19(3): p. 369–386. [DOI] [PubMed] [Google Scholar]
- 122.Shirure VS, et al. , Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip, 2018. 18(23): p. 3687–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Skardal A, et al. , Liver-Tumor Hybrid Organoids for Modeling Tumor Growth and Drug Response In Vitro. Ann Biomed Eng, 2015. 43(10): p. 2361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Virumbrales-Muñoz M, et al. , Multiwell capillarity-based microfluidic device for the study of 3D tumour tissue-2D endothelium interactions and drug screening in co-culture models. Sci Rep, 2017. 7(1): p. 11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang Y, et al. , Utilizing a high-throughput microfluidic platform to study hypoxia-driven mesenchymal-mode cell migration. Integr Biol (Camb), 2015. 7(6): p. 672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martin JD, Seano G, and Jain RK, Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol, 2019. 81: p. 505–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Massagué J and Obenauf AC, Metastatic colonization by circulating tumour cells. Nature, 2016. 529(7586): p. 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bayir Garbioglu D, et al. , Determination of therapeutic agents efficiencies of microsatellite instability high colon cancer cells in post-metastatic liver biochip modeling. Faseb j, 2021. 35(9): p. e21834. [DOI] [PubMed] [Google Scholar]
- 129.Bhat SM, et al. , 3D tumor angiogenesis models: recent advances and challenges. J Cancer Res Clin Oncol, 2021. 147(12): p. 3477–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Changirwa D, Schlechte J, and McDonald B, A Multi-Modal Toolkit for Studying Neutrophils in Cancer and Beyond. Cancers (Basel), 2021. 13(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.de Haan L, et al. , A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease. Int J Mol Sci, 2021. 22(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Glaser DE, et al. , Organ-on-a-chip model of vascularized human bone marrow niches. Biomaterials, 2021: p. 121245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Monteiro MV, et al. , 3D-bioprinted cancer-on-a-chip: level-up organotypic in vitro models. Trends Biotechnol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang J, et al. , A Fully Automated and Integrated Microfluidic System for Efficient CTC Detection and Its Application in Hepatocellular Carcinoma Screening and Prognosis. ACS Appl Mater Interfaces, 2021. 13(25): p. 30174–30186. [DOI] [PubMed] [Google Scholar]
- 135.Giuliano M, et al. , Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res, 2018. 78(4): p. 845–852. [DOI] [PubMed] [Google Scholar]
- 136.Al-Mehdi AB, et al. , Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med, 2000. 6(1): p. 100–2. [DOI] [PubMed] [Google Scholar]
- 137.Paget SJTL, The distribution of secondary growths in cancer of the breast. 1889. 133(3421): p. 571–573. [PubMed] [Google Scholar]
- 138.Ewing J, Neoplastic diseases. 1919: WB Saunders. [Google Scholar]
- 139.Font-Clos F, Zapperi S, and La Porta CAM, Blood Flow Contributions to Cancer Metastasis. iScience, 2020. 23(5): p. 101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hao S, et al. , A Spontaneous 3D Bone-On-a-Chip for Bone Metastasis Study of Breast Cancer Cells. Small, 2018. 14(12): p. e1702787. [DOI] [PubMed] [Google Scholar]
- 141.Sigdel I, et al. , Biomimetic Microfluidic Platforms for the Assessment of Breast Cancer Metastasis. Front Bioeng Biotechnol, 2021. 9: p. 633671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mielgo A and Schmid M.C.J.C.S.H.p.i.m., Liver tropism in cancer: the hepatic metastatic niche. 2019: p. a037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim J, et al. , Three-Dimensional Human Liver-Chip Emulating Premetastatic Niche Formation by Breast Cancer-Derived Extracellular Vesicles. ACS Nano, 2020. 14(11): p. 14971–14988. [DOI] [PubMed] [Google Scholar]
- 144.Ban J, et al. , Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells, 2021. 10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Batoon L and McCauley LK, Cross Talk Between Macrophages and Cancer Cells in the Bone Metastatic Environment. Front Endocrinol (Lausanne), 2021. 12: p. 763846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yip RKH, et al. , Mammary tumour cells remodel the bone marrow vascular microenvironment to support metastasis. Nat Commun, 2021. 12(1): p. 6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ortiz A and Lin SH, Osteolytic and osteoblastic bone metastases: two extremes of the same spectrum? Recent Results Cancer Res, 2012. 192: p. 225–33. [DOI] [PubMed] [Google Scholar]
- 148.Marton RM and Pașca SP, Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol, 2020. 30(2): p. 133–143. [DOI] [PubMed] [Google Scholar]
- 149.Liu W, et al. , AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomaterialia, 2019. 91: p. 195–208. [DOI] [PubMed] [Google Scholar]
- 150.Sattler S, The Role of the Immune System Beyond the Fight Against Infection. Adv Exp Med Biol, 2017. 1003: p. 3–14. [DOI] [PubMed] [Google Scholar]
- 151.Shurin MR, Cancer as an immune-mediated disease. ImmunoTargets and therapy, 2012. 1: p. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Alisjahbana A, et al. , CD5 Surface Expression Marks Intravascular Human Innate Lymphoid Cells That Have a Distinct Ontogeny and Migrate to the Lung. Front Immunol, 2021. 12: p. 752104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bunis DG, et al. , Single-Cell Mapping of Progressive Fetal-to-Adult Transition in Human Naive T Cells. Cell Rep, 2021. 34(1): p. 108573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Campinoti S, et al. , Reconstitution of a functional human thymus by postnatal stromal progenitor cells and natural whole-organ scaffolds. Nat Commun, 2020. 11(1): p. 6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cichocki F, Grzywacz B, and Miller JS, Human NK Cell Development: One Road or Many? Front Immunol, 2019. 10: p. 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Euchner J, et al. , Natural Killer Cells Generated From Human Induced Pluripotent Stem Cells Mature to CD56(bright)CD16(+)NKp80(+/−) In-Vitro and Express KIR2DL2/DL3 and KIR3DL1. Front Immunol, 2021. 12: p. 640672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Galat Y, et al. , Cytokine-free directed differentiation of human pluripotent stem cells efficiently produces hemogenic endothelium with lymphoid potential. Stem Cell Res Ther, 2017. 8(1): p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Guo R, et al. , Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res, 2020. 30(1): p. 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nagasawa M, et al. , Human CD5(+) Innate Lymphoid Cells Are Functionally Immature and Their Development from CD34(+) Progenitor Cells Is Regulated by Id2. Front Immunol, 2017. 8: p. 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bonte S, et al. , T-cells with a single tumor antigen-specific T-cell receptor can be generated in vitro from clinically relevant stem cell sources. Oncoimmunology, 2020. 9(1): p. 1727078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.García-García A, et al. , Culturing patient-derived malignant hematopoietic stem cells in engineered and fully humanized 3D niches. Proc Natl Acad Sci U S A, 2021. 118(40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hansen M, et al. , Efficient production of erythroid, megakaryocytic and myeloid cells, using single cell-derived iPSC colony differentiation. Stem Cell Res, 2018. 29: p. 232–244. [DOI] [PubMed] [Google Scholar]
- 163.Volk-Draper L, et al. , Myeloid-Derived Lymphatic Endothelial Cell Progenitors Significantly Contribute to Lymphatic Metastasis in Clinical Breast Cancer. Am J Pathol, 2019. 189(11): p. 2269–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Goldstein ME and Scull MA, Modeling Innate Antiviral Immunity in Physiological Context. J Mol Biol, 2021: p. 167374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Melchjorsen J, Learning from the messengers: innate sensing of viruses and cytokine regulation of immunity - clues for treatments and vaccines. Viruses, 2013. 5(2): p. 470–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cousin N, et al. , Lymphatic PD-L1 Expression Restricts Tumor-Specific CD8(+) T-cell Responses. Cancer Res, 2021. 81(15): p. 4133–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ghosh D, et al. , New insights into B cells as antigen presenting cells. Curr Opin Immunol, 2021. 70: p. 129–137. [DOI] [PubMed] [Google Scholar]
- 168.Minute L, et al. , Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer, 2020. 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ren H, et al. , Tumor-Derived Autophagosomes (DRibbles) Activate Human B Cells to Induce Efficient Antigen-Specific Human Memory T-Cell Responses. Front Immunol, 2021. 12: p. 675822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Synn CB, et al. , Primary Tumor Suppression and Systemic Immune Activation of Macrophages through the Sting Pathway in Metastatic Skin Tumor. Yonsei Med J, 2022. 63(1): p. 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Welsh RA, Song N, and Sadegh-Nasseri S, How Does B Cell Antigen Presentation Affect Memory CD4 T Cell Differentiation and Longevity? Front Immunol, 2021. 12: p. 677036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gjorevski N, et al. , Neutrophilic infiltration in organ-on-a-chip model of tissue inflammation. Lab Chip, 2020. 20(18): p. 3365–3374. [DOI] [PubMed] [Google Scholar]
- 173.Irimia D and Wang X, Inflammation-on-a-Chip: Probing the Immune System Ex Vivo. Trends Biotechnol, 2018. 36(9): p. 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miller CP, et al. , Engineering Microphysiological Immune System Responses on Chips. Trends Biotechnol, 2020. 38(8): p. 857–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sasserath T, et al. , Differential Monocyte Actuation in a Three-Organ Functional Innate Immune System-on-a-Chip. Adv Sci (Weinh), 2020. 7(13): p. 2000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shanti A, Teo J, and Stefanini C, In Vitro Immune Organs-on-Chip for Drug Development: A Review. Pharmaceutics, 2018. 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sharifi F, et al. , A Foreign Body Response-on-a-Chip Platform. Adv Healthc Mater, 2019. 8(4): p. e1801425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hallfors N, et al. , Multi-Compartment Lymph-Node-on-a-Chip Enables Measurement of Immune Cell Motility in Response to Drugs. Bioengineering (Basel), 2021. 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nelson MR, et al. , A multi-niche microvascularized human bone marrow (hBM) on-a-chip elucidates key roles of the endosteal niche in hBM physiology. Biomaterials, 2021. 270: p. 120683. [DOI] [PubMed] [Google Scholar]
- 180.Shanti A, et al. , Lymph Nodes-On-Chip: Promising Immune Platforms for Pharmacological and Toxicological Applications. Front Pharmacol, 2021. 12: p. 711307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shanti A, et al. , Multi-Compartment 3D-Cultured Organ-on-a-Chip: Towards a Biomimetic Lymph Node for Drug Development. Pharmaceutics, 2020. 12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shim S, et al. , Two-way communication between ex vivo tissues on a microfluidic chip: application to tumor-lymph node interaction. Lab Chip, 2019. 19(6): p. 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yu VWC and Scadden DT, Hematopoietic Stem Cell and Its Bone Marrow Niche. Current topics in developmental biology, 2016. 118: p. 21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tavakol DN, et al. , Lessons from Biology: Engineering Design Considerations for Modeling Human Hematopoiesis. Current Stem Cell Reports, 2021. 7(4): p. 174–184. [Google Scholar]
- 185.Chou DB, et al. , On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nature Biomedical Engineering, 2020. 4(4): p. 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bredenkamp N, et al. , An organized and functional thymus generated from FOXN1-reprogrammed fibroblasts. Nature Cell Biology, 2014. 16(9): p. 902–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lee J and Kotov NA, Notch Ligand Presenting Acellular 3D Microenvironments for ex vivo Human Hematopoietic Stem-Cell Culture made by Layer-By-Layer Assembly. 2009. 5(9): p. 1008–1013. [DOI] [PubMed] [Google Scholar]
- 188.Shah NJ, et al. , An injectable bone marrow-like scaffold enhances T cell immunity after hematopoietic stem cell transplantation. Nat Biotechnol, 2019. 37(3): p. 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Polini A, et al. , Towards the development of human immune-system-on-a-chip platforms. Drug Discovery Today, 2019. 24(2): p. 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sharpless NE and Depinho RA, The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov, 2006. 5(9): p. 741–54. [DOI] [PubMed] [Google Scholar]
- 191.Lee JH, Oh JO, and Lee CS, Induced Pluripotent Stem Cell Modeling of Best Disease and Autosomal Recessive Bestrophinopathy. Yonsei Med J, 2020. 61(9): p. 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sayed N, Liu C, and Wu JC, Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J Am Coll Cardiol, 2016. 67(18): p. 2161–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shi Y, et al. , Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov, 2017. 16(2): p. 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sinha D, et al. , Human iPSC Modeling Reveals Mutation-Specific Responses to Gene Therapy in a Genotypically Diverse Dominant Maculopathy. Am J Hum Genet, 2020. 107(2): p. 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Turhan AG, et al. , iPSC-Derived Organoids as Therapeutic Models in Regenerative Medicine and Oncology. Front Med (Lausanne), 2021. 8: p. 728543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhou H, et al. , Gene Editing in Pluripotent Stem Cells and Their Derived Organoids. Stem Cells Int, 2021. 2021: p. 8130828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Morgan MM, et al. , Modeling chemical effects on breast cancer: the importance of the microenvironment in vitro. Integr Biol (Camb), 2020. 12(2): p. 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wu M and Swartz MA, Modeling tumor microenvironments in vitro. J Biomech Eng, 2014. 136(2): p. 021011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dong MB, et al. , Tumor immunology CRISPR screening: present, past, and future. Trends in Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sohn TA, et al. , High-throughput drug screening of the DPC4 tumor-suppressor pathway in human pancreatic cancer cells. Annals of surgery, 2001. 233(5): p. 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wang T, et al. , Sequential CRISPR gene editing in human iPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell Stem Cell, 2021. 28(6): p. 1074–1089.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mehta AK, et al. , Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nature Cancer, 2021. 2(1): p. 66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Quan Y, et al. , Organ-on-a-chip: the next generation platform for risk assessment of radiobiology. RSC Advances, 2020. 10(65): p. 39521–39530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hajal C, et al. , The CCL2-CCR2 astrocyte-cancer cell axis in tumor extravasation at the brain. Sci Adv, 2021. 7(26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Paul SM, et al. , How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Reviews Drug Discovery, 2010. 9(3): p. 203–214. [DOI] [PubMed] [Google Scholar]
- 206.Schork NJ, Personalized medicine: Time for one-person trials. Nature, 2015. 520(7549): p. 609–11. [DOI] [PubMed] [Google Scholar]
- 207.Dirven H, et al. , Performance of preclinical models in predicting drug-induced liver injury in humans: a systematic review. Scientific Reports, 2021. 11(1): p. 6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kannt A and Wieland T, Managing risks in drug discovery: reproducibility of published findings. Naunyn-Schmiedeberg’s archives of pharmacology, 2016. 389(4): p. 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Moors EHM, Cohen AF, and Schellekens H, Towards a sustainable system of drug development. Drug Discov Today, 2014. 19(11): p. 1711–1720. [DOI] [PubMed] [Google Scholar]
- 210.McKenzie R, et al. , Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med, 1995. 333(17): p. 1099–105. [DOI] [PubMed] [Google Scholar]
- 211.Jolly CE, et al. , The utility of a differentiated preclinical liver model, HepaRG cells, in investigating delayed toxicity via inhibition of mitochondrial-replication induced by fialuridine. Toxicology and applied pharmacology, 2020. 403: p. 115163–115163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Attarwala H, TGN1412: From Discovery to Disaster. Journal of young pharmacists : JYP, 2010. 2(3): p. 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kerns SJ, et al. , Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. Elife, 2021. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.McAleer CW, et al. , Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. 2019. 11(497): p. eaav1386. [DOI] [PubMed] [Google Scholar]
- 215.Hood L, Balling R, and Auffray C, Revolutionizing medicine in the 21st century through systems approaches. 2012. 7(8): p. 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Meric-Bernstam F, et al. , Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol, 2015. 33(25): p. 2753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kopper O, et al. , An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nature Medicine, 2019. 25(5): p. 838–849. [DOI] [PubMed] [Google Scholar]
- 218.Torsvik A, et al. , U-251 revisited: genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. 2014. 3(4): p. 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Binnewies M, et al. , Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine, 2018. 24(5): p. 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Whiteside TL, Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Seminars in Cancer Biology, 2006. 16(1): p. 3–15. [DOI] [PubMed] [Google Scholar]
- 221.Fleischer S, Tavakol DN, and Vunjak-Novakovic G, From Arteries to Capillaries: Approaches to Engineering Human Vasculature. Advanced Functional Materials, 2020. 30(37): p. 1910811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Low LA, et al. , Organs-on-chips: into the next decade. Nat Rev Drug Discov, 2021. 20(5): p. 345–361. [DOI] [PubMed] [Google Scholar]
- 223.Wikswo JP, et al. , Scaling and systems biology for integrating multiple organs-on-a-chip. Lab on a Chip, 2013. 13(18): p. 3496–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ingber DE, Is it Time for Reviewer 3 to Request Human Organ Chip Experiments Instead of Animal Validation Studies? 2020. 7(22): p. 2002030. [DOI] [PMC free article] [PubMed] [Google Scholar]