

ABSTRACT
Bacterial type IV secretion systems (T4SSs) are macromolecular machines that translocate effector proteins across multiple membranes into infected host cells. Loss of function mutations in genes encoding protein components of the T4SS render bacteria avirulent, highlighting the attractiveness of T4SSs as drug targets. Here, we designed an automated high-throughput screening approach for the identification of compounds that interfere with the delivery of a reporter-effector fusion protein from Legionella pneumophila into RAW264.7 mouse macrophages. Using a fluorescence resonance energy transfer (FRET)-based detection assay in a bacteria/macrophage coculture format, we screened a library of over 18,000 compounds and, upon vetting compound candidates in a variety of in vitro and cell-based secondary screens, isolated several hits that efficiently interfered with biological processes that depend on a functional T4SS, such as intracellular bacterial proliferation or lysosomal avoidance, but had no detectable effect on L. pneumophila growth in culture medium, conditions under which the T4SS is dispensable. Notably, the same hit compounds also attenuated, to varying degrees, effector delivery by the closely related T4SS from Coxiella burnetii, notably without impacting growth of this organism within synthetic media. Together, these results support the idea that interference with T4SS function is a possible therapeutic intervention strategy, and the emerging compounds provide tools to interrogate at a molecular level the regulation and dynamics of these virulence-critical translocation machines.
KEYWORDS: effector protein, small molecule library, high throughput screen, beta-lactamase reporter, Dot/Icm secretion system
INTRODUCTION
To successfully establish an infection, bacterial pathogens employ a variety of strategies to evade the host immune response and to establish conditions favorable for their own survival and growth. Host cell signaling and trafficking processes are manipulated by effector proteins and microbial toxins that are encoded by pathogens and then shuttled into the host cell using specialized delivery systems. One widespread effector translocator is the type IV secretion system (T4SS) that is present in a variety of animal and plant pathogens (1, 2). Based on their architecture, T4SSs are categorized into two major classes: (i) the type IVA secretion system (T4ASS) which is comprised of at least 12 different proteins and found in organisms like Helicobacter, Brucella, and Bartonella; and (ii) the more complex type IVB secretion system (T4BSS), present in Legionella pneumophila, Coxiella burnetii, and Rickettsiella grylli, which is assembled from twice as many components as the T4ASS (2–6). Despite their differences in composition and complexity, both classes of T4SSs are evolutionarily related to DNA conjugation systems and share a set of subassemblies that operate in a similar manner, such as an outer membrane core complex and an inner membrane complex.
Recent advances in structural biology, electron microscopy, electron cryotomography, and single-particle cryo-electron microscopy have provided much-needed insight into the three-dimensional organization of subassemblies from several T4SSs, including the VirB/VirD4 T4SS from Agrobacterium tumefaciens (7), the Helicobacter pylori Cag system (8, 9), and Escherichia coli conjugation apparatuses encoded by the R388 (10) and pKM101 plasmids (11–13). The Dot/Icm T4BSS system from L. pneumophila is one of the most-well characterized secretion system of the type IVB class (14–18). It is composed of at least ∼27 components that assemble into several architectural subcomplexes, including an outer membrane core complex composed of at least five proteins (DotC, DotD, DotH, DotK, and Lpg0657) that has a pinwheel-shaped structure with a 13-fold symmetry, a periplasmic ring with an 18-fold symmetry, and an inner membrane subcomplex consisting of six proteins (DotL, DotM, DotN, IcmS, IcmW, and LvgA) that traverses the inner membrane. A wide channel at the center of a stalk, composed mainly of DotG, connects the inner and outer membrane complexes of the Dot/Icm T4SS and likely ushers substrate proteins from the cytosol of the bacteria across their outer membrane. Other components function as cytosolic chaperones (IcmQ, IcmR) or as inner membrane-associated ATPases (DotL, DotB, and DotO) that convert chemical into mechanical energy for cargo translocation through the T4SS conduit. How the remaining proteins contribute to the structure and function of the L. pneumophila T4SS remains to be determined.
L. pneumophila relies on its Dot/Icm T4SS for colonization and proliferation within a wide range of host cells, including freshwater amoeba in the environment and alveolar macrophages during Legionnaires' pneumonia in humans (19). Throughout its intracellular replication cycle, the bacterium resides within a membrane-enclosed compartment, the Legionella-containing vacuole (LCV), that avoids fusion with destructive endosomes and lysosomes (20, 21). Instead, the bacterium acquires proteins and membranes from the early secretory pathway and other sources to establish a camouflaged replication compartment (22). L. pneumophila mutants with a non-functional T4SS fail to control trafficking of their LCV and are quickly delivered to lysosomes for degradation (23–25), thus underscoring the importance of the Dot/Icm system for Legionella pathogenesis.
Given their importance for virulence, bacterial type IV secretion systems are being increasingly recognized as putative drug targets. Because T4SSs are typically not essential for bacterial fitness outside the host, they experience low selective pressure in the environment, making it less likely for resistance mechanisms against inhibitory compounds to already exist or to easily spread throughout microbial populations. Moreover, because commensal bacteria within the microbiota of humans are not known to require a T4SS for survival, therapeutics that specifically target these translocation machines will likely affect only pathogens while leaving the healthy microbiota undisturbed, thus reducing the risk of secondary infections by creating niches for pathogens.
Recently, several groups have reported the discovery of small molecules that interfere with the activity of type III secretion systems (T3SSs) (26), another major bacterial translocation machine found in pathogens. While the exact type of reporter assays used in these studies varied, they all made use of the convenient fact that T3SS activity could be monitored during bacterial growth in broth without the need for host cells to be present. This is in stark contrast to bacterial T4SS that do not exhibit effector translocation activity during axenic growth and that are activated only upon contact of the pathogen with a target cell, which can either be another bacterium in the case of T4SS-mediated DNA conjugation, or a host cell during infection (27). The requirement for both bacteria and host cells to be present at the same time and to interact in a productive manner that allows for efficient reporter transfer to occur explains why high throughput screens for compounds that interfere with T4SS function have remained a major challenge.
To bypass these limitations, alternative approaches have been developed, for example by identifying inhibitors of individual protein components of T4SSs, such as Brucella VirB8 (28, 29) or H. pylori VirB11 (30). While these surrogate screening approaches have made some progress toward the discovery of T4SS inhibitors, they did not fully recapitulate the complex behavior of host-pathogen interactions during infection. Shuman and colleagues (31) have made a first progress toward identifying molecules that attenuate the delivery of a effectors from L. pneumophila into infected host cells. In their screening campaign, they tested a library of approximately 2,600 compounds and identified several candidates that reduced delivery of a reporter-effector fusion protein by L. pneumophila into J774 macrophages. Importantly, the vast majority of hit compounds that were detected in this way appeared to indirectly interfere with processes on the host side, most notably actin polymerization which is a dynamic process critically important for bacterial uptake via phagocytosis. By blocking phagocytosis, these compounds likely prevented the establishment of an intimate cell-cell contact between the bacterium and its host, thus reducing the efficiency of reporter protein translocation. Although valuable as tools for the experimental analysis of T4SS function, compounds that target human cell are undesirable as therapeutics against infectious diseases due to the likelihood of them causing side effects.
Here, we implemented a modified screening approach to identify novel compounds that interfere with the process of reporter translocation without affecting phagocytosis. After screening a library of more than 18,000 compounds at a wide range of concentrations, several hits emerged that showed high efficacies in a variety of biological assays that require a functional T4SS, indicating that our approach was successful in selecting potentially novel therapeutics to antagonize bacterial infections by L. pneumophila and, notably, also Coxiella burnetii whose virulence relies on a related T4BSS.
RESULTS
Development of a cell-based reporter assay for monitoring bacterial effector translocation.
To identify compounds that specifically interfere with the process of bacterial type IV secretion, we developed a screening protocol that minimized the enrichment of compounds that function by altering host cell physiology and that favored detecting compounds that block other aspects of effector translocation by the T4SS. For this purpose, several important criteria and screening parameters had to be tested and optimized, including: the type of reporter enzyme and the substrate used for its detection; the type of host cell that, upon contact with L. pneumophila, favors reporter translocation under infection conditions; the ratio of bacteria to host cells and the duration for which they will be incubated; and the concentration and incubation periods of compounds from the screening libraries.
Prior to our study, several reporter protein-based translocation monitoring assays have been successfully used in L. pneumophila. These included the reporter enzyme Cre recombinase from P1 bacteriophage (32), the adenylate cyclase CyaA from B. pertussis (33, 34), and the Enterobacteriaceae β-lactamase (βLac) (35). The CyaA and Cre recombinase assays require either extensive washing steps or a time-intensive reporter step which makes them less desirable for a high-throughput screen (36). The βLac translocation assay bypasses many of these limitations because no wash steps are required, the experimental procedure can be completed in a single day, and the time between the addition of the reporter substrate and the readout occurs within a few hours. When fused to a known translocated substrate of the Dot/Icm T4SS, the βLac reporter is shuttled into target cells where its presence can be optically detected using CCF4/AM, a membrane-permeable lipophilic ester that is easily taken up by cultured cells (Fig. 1A) (37). CCF4/AM is composed of two fluorophores (7-hydroxycoumarin and fluorescein) that form a fluorescence resonance energy transfer (FRET) pair linked by a β-lactam ring. The excitation of intact CCF4/AM with light of 409 nm wavelength results in FRET from the coumarin donor to the fluorescein acceptor, causing cells without βLac to emit light in the green spectrum (518 nm). In the presence of βLac activity, CCF4/AM cleavage occurs which separates the two fluorophores and disrupts FRET, resulting in cells that fluoresce in the blue spectrum (447 nm) of the coumarin donor (Fig. 1A). The ratio of blue-to-green light in infected cells is an approximation of the level of translocated effector-reporter fusion protein and can serve as an indicator for the activity of the L. pneumophila T4SS.
FIG 1.

Small molecule library screen using a FRET-based reporter assay. (A) Schematic depiction of the reporter translocation assay. Compounds that interfere with T4SS-mediated reporter delivery into infected cells will prevent β-Lac-mediated cleavage of the mammalian cell-permeable CCF4/AM substrate, retaining its green fluorescence, whereas reporter delivery into host cells will result in a shift in emission light from green to blue due to cleavage of the FRET pair. (B) RAW264.7 macrophages were challenged at an MOI of 20 with either Lp02 (functional T4SS) or Lp03 (defective T4SS) harboring plasmids encoding either βLac (control) or βLac-LidA. At the indicated time points (hours postinfection [hpi]), CCF4/AM was added to the cells, and fluorescence emission light was detected by epifluorescence microscopy. (C) Schematic overview of the different high-throughput screen stages. (D) Waterfall plot depicting the dose response to all compounds. Compounds that result in reduced reporter translocation (assessed by FRET) are shown in red, compounds without effect are shown in green, activators or false activators/fluorescent compounds are shown in blue. (E) Inhibition profile for representative hits from the FRET confirmation assay using an 11-point dose range. Data were globally fit using a Hill equation.
We created a genetic fusion between βLac and LidA, a known substrate of the L. pneumophila T4SS and a protein shown to efficiently shuttle βLac into host cells (38). Synthesis of equal levels of βLac-LidA in both L. pneumophila wild-type (Lp02) and the T4SS-defective L. pneumophila dotA3 (Lp03), a strain with a non-functional variant of the polytopic inner membrane protein DotA (39, 40), was confirmed by immunoblot analysis (Fig. S1). Using the FRET-based read-out, we detected efficient translocation of βLac-LidA during infection of mouse RAW264.7 macrophages, a cell type that can be easily propagated and readily takes up L. pneumophila (Fig. 1B) (36). Infection with the Lp02 strain producing βLac-LidA resulted in efficient hydrolysis of the CCF4/AM substrate within RAW264.7 macrophages, causing them to fluoresce blue, whereas no hydrolysis was detected upon macrophage challenge with either Lp03 producing βLac-LidA or Lp02 producing unconjugated βLac (Fig. 1B). Thus, reporter translocation depended on its fusion to a translocated effector and the presence of a functional Dot/Icm system.
The production of βLac or βLac_LidA was induced overnight by incubation with IPTG in both strains Lp02 and Lp03. Proteins within the bacterial lysate were separated by SDS-PAGE. After transfer to nitrocellulose membranes, the βLac-tagged proteins were immunologically detected using a β-lactamase specific antibody (top). Isocitrate dehydrogenase (ICDH) served as a loading control (bottom). Download FIG S1, PDF file, 0.6 MB (668.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
One of the critical aspects of the high-throughput library screen was the miniaturization of the FRET assay from a 96-well to a 1,536-well plate format. This increases the number of samples that can simultaneously be screened in each plate by 16-fold and, at the same time, reduces the total sample volume per well from 220 μL to 6 μL. Despite these benefits, miniaturization also is associated with a variety of challenges, most notably enhanced fluid surface tension, limited sample mixing or liquid aspiration capability, as well as liquid evaporation and an increased surface-to-volume ratio that can dramatically affect reagent absorption and stability (41). To assure assay reproducibility under these difficult conditions, the translocation assay described above (Fig. 1A) was further optimized for a 1,536-well plate format. We found that a bacteria-to-macrophage ratio of 20:1 and an infection period of 60 min produced the best results, with an average Z’ factor (42) of 0.63 ± 0.14, which exceeded the generally accepted criteria of a Z’ factor >0.5 required for high-throughput screening. These results suggest a reproducible assay that is not adversely affected by minor variabilities in experimental conditions.
High-throughput screen for compounds that attenuate reporter delivery by the L. pneumophila T4SS.
Once optimized for a 1,536-well plate format, the FRET-based translocation assay was carried out by performing qHTS against commercially available small libraries (see “Materials and Methods”) at multiple concentrations, to evaluate compounds for their ability to interfere with T4SS-mediated β-Lac-LidA translocation (Fig. 1C). A total of 18,272 compounds were initially screened at either five or six concentrations ranging from 22 nM to 46 μM (43–46). Importantly, compounds and bacteria were added to macrophage monolayers in very short succession (<15 min) as to minimize effects of the compounds on host cell physiology. A dose-response curve was generated for each compound and classified to one of four curve classes based on the shape of the curve and the quality of the fit (r2) (45) (Fig. 1D and E). Briefly, curve class 1 represents a full curve with full efficacy range and high degree of fit (high R2), curve class 2 represents curve with partial efficacy and lower degree of fit (low R2), class 3 is a dose response with a single-point trend, while class 4 is a flat, or inactive, response. We represented the screening outcome in three categories: all actives shown in red were compounds that inhibited in the assay (negative curve classes 1–3), inactives were in green (class 4), and in blue we displayed compounds that appeared to activate in the assay (positive curve classes) (Fig. 1D). Of the 18,272 compounds evaluated in the primary screen, 501 compounds were active with a maximum response (≥35% signal reduction), corresponding to a hit rate of 2.7% (Fig. 2).
FIG 2.

Compound triage. Schematic overview of the multi-step compound triage workflow. Numbers represent the quantity of compounds that emerged from each triage step.
Active primary hits were further evaluated in the same primary assay using an 11-point dose response, and 113 compounds (0.6%; Table S1) were confirmed as positive hits (Fig. 2). These compounds were further assessed in counter-screens and orthogonal assays as described next.
Data of 113 compounds selected from the qHTS reporter translocation assay Table S1, PDF file, 0.5 MB (475.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Triaging of active compounds.
Given the complexity of the bacteria-to-macrophage translocation process, several irrelevant mechanisms could have caused a reduction in the FRET-based reporter signal without directly affecting T4SS-mediated translocation. These include a potentially inhibitory effect of hit compounds on the enzymatic activity of β-Lac; compounds that cause host cell toxicity or lysis; and compounds that are highly promiscuous and thus non-specifically react with macromolecules. Each of these issues was addressed using the following validation methods.
First, we determined if any of the active compounds functioned as β-Lac inhibitors that interfered with the enzyme's ability to hydrolyze its substrate. To do so, we adapted a chromogenic assay that monitors the ability of β-Lac to hydrolyze the cephalosporin-type substrate nitrocefin in solution (47) (Fig. S2A). Upon hydrolysis of the amide bond in its β-lactam ring, nitrocefin undergoes a rapid and distinctive color change from yellow (390 nm) to orange-red (486 nm) that can easily be measured using a spectrophotometer. Sixteen of the 113 compounds blocked β-Lac enzymatic activity, of which three were known inhibitors of β-Lac (Pivoxil Sulbactam, NCGC00249610; Tazobactam sodium, NCGC00159340; and Clavulanate lithium, NCGC00180892).
(A) β-lactamase specificity assay principle based on hydrolysis of the chromogenic substrate nitrocefin, an indicator of β-Lac enzymatic activity. (B) Cell cytotoxicity assay principle based on ATP abundance, an indicator of metabolically active cells. Download FIG S2, PDF file, 0.4 MB (404.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We also explored the possibility if any of the active compounds affected mammalian cell viability, which could have prevented uptake and/or conversion of CCF4/AM during the FRET-based assay. Moreover, cytotoxicity is an undesirable trait in therapeutics as it increases the chance of side effects. To determine cell viability, RAW264.7 macrophage monolayers were incubated for 24 h with hit compounds at concentrations ranging from 22 nM to 46 μM. The cells were subsequently lysed, and ATP levels, a proxy for live cell metabolism, were measured using a luminescence-based signal that is proportional to the amount of ATP present (Fig. S2B). Of the 113 active compounds selected from the primary assay (Table. S1), 62 compounds had only a moderate or no effect on cell viability (all negative curve classes and max response more than 35) even after 24 h of incubation and were advanced to the next stage. To eliminate nonspecific inhibitors, we deployed an orthogonal GFP intracellular growth L. pneumophila assay in macrophages as described below.
Compound treatment protects macrophages from intracellular growth of L. pneumophila.
Given that effector translocation is essential for L. pneumophila intracellular survival and growth, we speculated that treatment of L. pneumophila with hit compounds that affect T4SS function should render the bacteria less virulent and protect host cells from intracellular bacterial replication (Fig. 3A). We developed a 1,536-well plate-based automated microscopy and image processing approach to monitor growth of GFP-producing L. pneumophila within host cells. After pre-treatment with compounds (22 nM to 46 μM final concentration) for 15 to 30 min, RAW264.7 macrophages were challenged with a GFP-producing L. pneumophila strain. After incubation for 14 h, which is equivalent to the time required by L. pneumophila to complete its intracellular replication cycle, macrophages were chemically fixed, host cell nuclei were labeled by staining DNA with the dye Hoechst-33342, and cells were imaged using an INCell Analyzer 2200 Imaging system to identify cell nuclei and GFP-positive bacteria (Fig. 3B and C). INCell analyzer software (GE Healthcare Life Sciences) was used to quantify cells with nuclei stain. GFP intensity was normalized to Dimethylsulfoxide(DMSO) control to identify the cells with positive-GFP. Of 62 compounds tested in our L. pneumophila intracellular growth assay in conjunction with the β-Lac assay resulted in 33 compounds that demonstrated inhibition with an efficacy greater than 60% and AC50s lower than 25 μM (Fig. S3). Of these, we manually excluded 11 compounds with known antibacterial activities and mercury containing promiscuous compounds. The remaining 22 compounds were reacquired from a commercial source, purified, and retested in 384-well format to validate previous results. Of these 22 compounds, seven compounds (NCGC-00167765, -00345082, -00015397, -00015826, -00025216, -00178913, -00165875, called C1 to C7 hereafter) were promoted for further evaluation (Fig. 3D, Table S2).
FIG 3.

L. pneumophila intracellular growth is attenuated in the presence of hit compounds. (A) Schematic overview of the intracellular growth assay. (B) Growth of Lp02ΔflaA in RAW264.7 macrophages. RAW264.7 macrophages were challenged with Lp02 in the presence of DMSO (vehicle). Images were captured using an InCell Analyzer at 14 hpi. Bacteria are shown in green and DNA (DAPI staining) in blue. (C) Growth of Lp02 in RAW264.7 macrophages is attenuated in the presence of compounds. RAW264.7 macrophages were challenged as described in (A) in the presence of the indicated compounds at 57 μM (NCGC IDs shown). Panels are merged images of GFP-producing L. pneumophila (green) and DAPI-stained host nuclei (blue). (D) Inhibition of L. pneumophila growth by select compounds is dose-dependent. In RAW264.7 macrophages, L. pneumophila growth was quantified by measuring the GFP signal strength relative to macrophages not infected by GFP- Lp02. Experiments were done in triplicate.
Legionella growth inhibition in a 1,536-well plate format. RAW264.7 macrophages were challenged with a GFP-producing L. pneumophila for 14 h in the presence of the indicated compounds, and the GFP signal (as a measure for bacterial growth) was determined using an IN Cell Analyzer 2200 Imaging system. Download FIG S3, PDF file, 0.5 MB (473.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data of 7 compounds selected following validation assays. Download Table S2, PDF file, 0.4 MB (387KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Studying the effect of hit compounds on bacterial uptake.
Efficient effector translocation requires close contact between bacteria and their host cell. This condition is strongly favored during phagocytosis where the pathogen is being engulfed by the host plasma membrane. Many of the compounds previously shown to block T4SS-mediated effector translocation indirectly did so by attenuating L. pneumophila uptake (31). Although the screening approach implemented here had been designed to reduce the enrichment of compounds that interfere with host cell processes, we nonetheless evaluated if and to what extent the hit compounds negatively impacted phagocytosis of L. pneumophila. RAW264.7 macrophages were challenged for 1 h with Lp02 in the presence of either the vehicle (DMSO), hit compounds C1 to C7, or cytochalasin D, a cell-permeable and potent inhibitor of actin polymerization, and the percentile of cells with intracellular bacteria was microscopically determined (Fig. S4). Unlike cytochalasin D, which reduced the number of intracellular bacteria by almost 95%, four of the seven hit compounds had no significant effect on L. pneumophila phagocytosis. Compound C4 and C5 still allowed 59% and 71% of bacterial uptake by macrophages, respectively, while C2 more strongly attenuated phagocytosis by −65%. Thus, of the seven hit compounds tested here, six showed moderate to low effects on bacterial phagocytosis, confirming that our screening protocol had indeed favored the identification of hit compounds that attenuate effector delivery into host cells by some other means, a notable difference to the aforementioned study by Shuman and colleagues (31).
Bacterial uptake by macrophages in the presence of compounds. RAW264.7 macrophages were treated with the indicated compounds, Cytochalasin D, or DMSO vehicle prior to infection with Lp02, and intracellular bacteria were enumerated visually. Data shown are the mean with standard deviation from six experimental replicates, with the DMSO control considered as 100%. P-values (t-test) are <0.05 (*), <0.001 (**), or <0.005 (***) relative to the control. Download FIG S4, PDF file, 0.2 MB (198KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Effect of hit compounds on Legionella growth outside of host cells.
The inhibitory effect of the seven selected hits on L. pneumophila growth in RAW264.7 macrophages might suggest that they could have attenuated effector protein translocation through the T4SS, its most important known function in host intracellular growth. Alternatively, these compounds could also exhibit bacteriostatic or bactericidal activity, comparable to the role of conventional antibiotics, which would reduce the ability of L. pneumophila to proliferate within host cells. To distinguish between these two possibilities, we measured the effect of the seven hit compounds on growth of Lp02 outside its host, conditions under which the T4SS can be considered dispensable. The bacteria were incubated in AYET media containing either DMSO (vehicle), the antibiotic chloramphenicol (5 μg/mL), or C1 to C7 (28 μM final concentration), and growth was monitored by measuring the optical density at 600 nm (OD600) (Fig. 4A). Over a period of 18 h, Lp02 grew robustly and uninhibited in the presence of the DMSO vehicle, whereas no increase in cell numbers was detectable in AYET media supplemented with chloramphenicol. Five compounds had no detectable effect on growth of Lp02 in AYET, suggesting that they did not have a bacteriostatic or bactericidal effect on L. pneumophila (Fig. 4A). Notably, two compounds C2 and C6 did cause a dramatic reduction in growth similar to the level of inhibition observed upon cultivation of the bacteria with chloramphenicol. Unexpectedly, we found that, under similar growth conditions, neither compound had an inhibitory effect on the replication of two other Gram-negative bacteria, E. coli and Pseudomonas aeruginosa (Fig. 4C and D), suggesting that these two compounds did not function as general Gram-negative antibiotics but were directed primarily against processes or components present in L. pneumophila while absent from E. coli and P. aeruginosa.
FIG 4.

Effect of hit compounds on growth of L. pneumophila in liquid media. (A) Growth of L. pneumophila Lp02 or Lp03 in the presence of hit compounds. Bacteria were inoculated in AYET media at an OD600 of 0.1 in the presence of either DMSO, the antibiotic chloramphenicol (control; 5 μg/mL), or the indicated compounds (NCGC IDs 28 μM). Growth was monitored for at least 18 h by measuring the absorbance at OD600. (B) Compounds C2 and C6 do not affect growth of E. coli MG1655 or Pseudomonas aeruginosa. (C) C2 and C6 do inhibit growth of Lp02_26Δdot/icm.
Although it is generally accepted that the L. pneumophila Dot/Icm T4SS transport system is dispensable for axenic reproduction in growth media, there are reports where a dysregulated T4SS system can affect L. pneumophila physiology and proliferation outside its host. Specifically, a functional T4SS renders L. pneumophila sensitive to high concentrations of sodium chloride within the growth media, while isolates with a non-functional T4SS, such as the Lp03, are salt-resistant (48). Although the molecular details underlying this Dot/Icm-dependent salt-sensitivity in growth media have yet to be determined, it is likely that the intact T4SS translocation pore of Legionella may allow unregulated passage of ions across the bacterial membrane, thus disturbing the bacterium’s electrochemical gradients. To determine if C2 and C6 had caused a similar disturbance by targeting the integrity of the Dot/Icm system, we tested whether growth in media could be restored by disabling the T4 translocon. Interestingly, we found no difference in sensitivity between Lp02 and Lp03 to either C2 or C6 during growth in AYET media (Fig. 4B). In fact, even the axenic growth of a L. pneumophila strain lacking the entire dot/icm gene cluster (Lp02[Δ26]) was robustly repressed in the presence of either C2 or C6 but not of the vehicle DMSO (Fig. 4E), demonstrating that the growth-inhibitory effects of these two compounds were independent of components of the Dot/Icm system.
Treatment with hit compounds increases delivery of Legionella to lysosomal compartments.
Failure to translocate effectors results in L. pneumophila to be rapidly shuttled to lysosomal compartments for degradation. To evaluate the effect of the five remaining hit compounds (C1, C3, C4, C5, C7) on intracellular trafficking, RAW264.7 cells were challenged with L. pneumophila producing mCherry, and LCVs were enumerated by indirect immunofluorescence microscopy for their colocalization with lysosomal marker protein Lamp1 (Fig. 5). While L. pneumophila showed the commonly observed basal level (∼15%) of Lamp1-positive LCVs in vehicle-treated cells, five of the six hit compounds caused a 2-fold or higher increase in the number of Lamp1-positive LCVs, with C4 having the most dramatic effect with up to 80% Lamp1-positive LCVs. Combined with the results from the intracellular growth analysis and the reporter translocation assay, these data favor the scenario where the hit compounds attenuate L. pneumophila intracellular growth and lysosomal avoidance by altering the dynamics of effector translocation through the T4SS.
FIG 5.
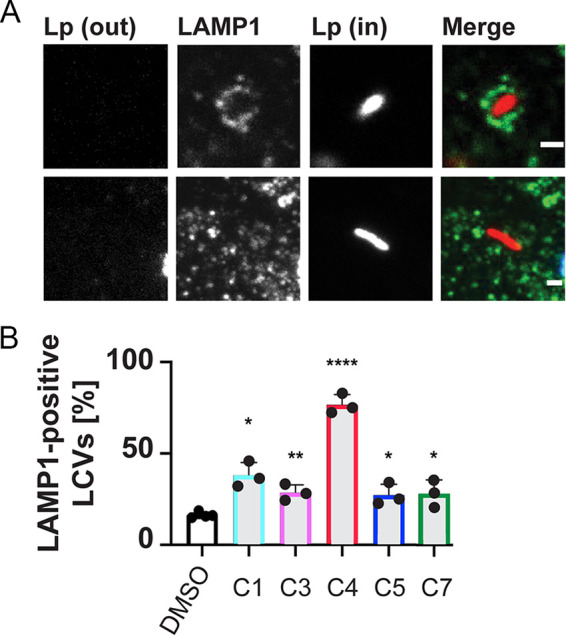
Increased delivery of L. pneumophila to Lamp1 positive compartments in the presence of compounds. RAW264.7 macrophages were infected with Lp02-mCherry and indicated compounds or DMSO vehicle for 2 hpi at an MOI of 25, then enumerated for the percent of internalized bacteria in Lamp1 positive compartments. (A) Representative image of Lamp1 (green) positive internalized L. pneumophila (red), and Lamp1 negative internalized L. pneumophila. (B) Enumeration of Lamp1 positive Legionella containing vacuoles (LCVs). Data are represented as the mean of 3 to 4 experimental replicates with standard deviation and individual replicate points shown. (*) indicates P < 0.05 compared with DMSO control.
Hit compounds interfere with the delivery of Coxiella T4SS effector-reporter fusions.
Coxiella burnetii is the causative agent of zoonotic Q fever in humans. Upon internalization by a permissive macrophage, the obligate intracellular bacterium resides within the Coxiella-containing vacuole (CCV). C. burnetii encodes a T4SS that is homologous to the Dot/Icm system from L. pneumophila that delivers bacterial effector proteins across the CCV membrane into the host cytosol (4, 49, 50). Here, we tested whether compounds that interfere with the L. pneumophila T4SS also inhibit translocation of Coxiella Dot/Icm effectors CvpA and CvpB. THP-1 macrophages were cultured for 24 h with C. burnetii expressing the Dot/Icm effector CvpA or CvpB fused to the CyaA reporter tag and then incubated an additional 24 h with 50 μM each of the five indicated compounds. Cytosolic cAMP measured within infected cell lysates was used to determine the translocation efficiency of the CyaA fusions in response to compound treatment. With the exception of C4 each of the compounds reduced the cAMP levels at least 10-fold compared to cells without inhibitor treatment, both for cells infected with C. burnetii expressing either CyaA-CvpA or CyaA-CvpB (Fig. 6). In all instances, low levels of translocated protein were still detected above the 2.5-fold cutoff for background signal. This response could be due to persistent low levels of secretion in the presence of inhibitors or active remnants of CyaA fusions that were delivered to the host cell cytosol prior to addition of the inhibitor compounds. Importantly, none of the compounds had an inhibitory effect on C. burnetii growth in axenic media (Fig. S5), further supporting the idea that they did not cause a global disturbance within the bacteria’s physiology. Together, these result support the conclusion that the inhibitor screen performed here succeeded in identifying compounds that likely interfere with cargo translocation by the bacterial T4BSS found in pathogens like L. pneumophila and C. burnetii.
FIG 6.
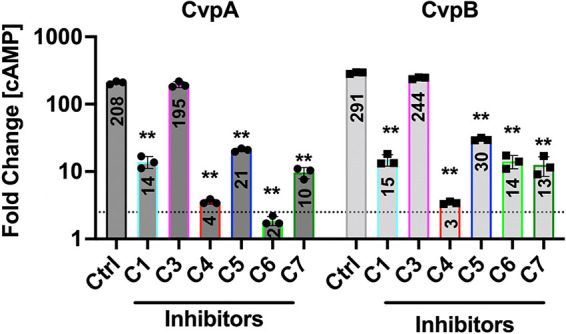
Compound treatment attenuates effector translocation by C. burnetii Dot/Icm. Histograms depict the fold change in cytosolic (cAMP) for THP-l cells infected for 48 h with C. burnetii producing CyaA-CvpA or CyaA-CvpB fusion proteins. The cutoff for positive secretion is indicated by a dotted line at 2.5-fold change (cAMP). Results are representative of three independent experiments and error bars indicate the standard deviations from triplicate samples. Asterisks indicates a statistically significant difference (P < 0.01).
In vitro replication of C. burnetii in the presence of compounds. ACCM-D broth cultures containing 5 μM of the indicated compounds or DMSO and inoculated with ∼ 1 × 105 per mL of C. burnetii were incubated at 37° C in a 2.5% O2 and 5% CO2. The plot depicts the number of C. burnetii genomes measured in ACCM-D broth cultures at the start of the experiment (day 0) and after incubation (day 6) detected by qRT-PCR (K. M. Sandoz, D. E. Sturdevant, B. Hansen, R. A. Heinzen. (2014). Developmental transitions of Coxiella burnetii grown in axenic media. J Microbiol Methods 96:104–110.). Bars indicate the mean ± SEM of combined data from three independent experiments each performed with triplicate samples (N = 9). One-way ANOVA analysis indicated differences between groups at D6 were found not significant (ns) using a 95% confidence interval. Download FIG S5, PDF file, 0.7 MB (754.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
In this study, we have identified and experimentally validated five hit compounds that interfered with processes that depend on a functional T4BSS, such as the delivery of a β-lactamase reporter into host cells (Fig. 1A), the proficiency of L. pneumophila to avoid endolysosomal trafficking (Fig. 5), and the capability to replicate within human macrophages (Fig. 3A). Importantly, none of the five compounds affected the ability of either L. pneumophila (Fig. 4A) or C. burnetii (Fig. S5) to replicate outside their host when grown within synthetic media, conditions under which a functional Dot/Icm system is dispensable. Together, these results suggest that the five hit compounds, by targeting either the bacterial T4SS or factors required for its activation or function, attenuate L. pneumophila’s ability to fully employ this translocation system to deliver cargo into recipient cells.
The T4SS inhibitors described here emerged from a high throughput small molecule library screen in which we employed a protocol that strongly favored the discovery of compounds that likely function by directly altering the regulation and/or function of the Dot/Icm system instead of compounds that indirectly affect reporter delivery, for example, by altering host cell processes such as phagocytosis, a process known to be essential for L. pneumophila uptake. This was accomplished by adding both the bacteria and each compound in rapid succession to macrophage monolayers, thus limiting the exposure time of cultured macrophages to them. Another feature of our screening protocol was that each compound was tested at a wide range of concentrations, which allowed us to generate a dose response profile for each compound (Fig. 1D and E). As a result, our screen yielded a diverse set of hits and was enriched in compounds that seemed to affect T4SS function without altering host cells processes like phagocytosis (Fig. S4) or cell viability in general (Fig. S2B).
The structures of the hit compounds and their pharmacological properties are summarized in Table S2. Briefly, among these five hits, C7 (PAP-1, muscarinic acetylcholine receptor M1 antagonist), C5 (roxindole, serotonin 1a receptor agonist), C1 (LP-44, sigma1/2 receptor agonist), and C3 (ethamivan, 5-HT7 receptor agonist) are associated with modulation of listed G protein-coupled receptor (GPCR) activities, while C4 (perphenazine) is a known disruptor of HIF-β/transforming acidic coiled coil containing protein 3 (TACC3) complex in the HIF pathway. Importantly, while each of these compounds has a known host cell target, we do not imply that engagement of those targets was responsible for the inhibitory effect on cargo translocation by the L. pneumophila T4SS. Instead, these compounds may have additional targets on either the bacterial side or the host cell side that were responsible for inhibiting effector translocation, and it will be interesting to determine the identity of those alternate targets in the future.
The fact that the hit compounds identified here show a wide range of molecular structures and chemical properties suggests that they may have different modes of inhibition, perhaps offering the opportunity to explore future dual synergistic anti-microbial actions. This is further supported by the variation in lysosomal delivery of L. pneumophila upon compound treatment, where the bacteria were trafficked with higher frequency to destructive lysososmal compartments within infected host cells (Fig. 5). One obvious possibility would be for hit compounds to sterically obstruct the T4SS channel, thereby preventing cargo from entering or passing through the translocon. A compound might also block a yet-to-be identified sensor on the bacterial surface or its cognate host cell receptor, thus preventing their engagement which would otherwise trigger T4SS activation upon bacterial contact with and engulfment by recipient cells. Membrane-permeable compounds might enter the bacterial cell and target cytosolic or integral membrane components of the Dot/Icm system, including subunits with ATPase activity, such as DotL or DotB (51, 52), which would disable the force-providing components of the T4SS. And, some of the compounds could bind to and block the function of the coupling protein complex composed of DotL, DotM, and DotN, which serve as a platform for the recruitment of effector proteins prior to translocation (53). Despite the briefness of the exposure of the bacteria to compounds prior to bacterial contact with macrophages, it cannot be excluded that the compounds functioned by inhibiting expression of dot/icm genes or by preventing proper assembly of the pre-synthesized T4SS components. This is particularly true during Coxiella infection where incubation periods with the compounds were prolonged compared to the Legionella infection assay and where effects of the compounds on the assembly of the T4SS cannot be excluded (Fig. 6).
Hit compound C4 emerged as a particularly promising candidate for interfering with T4SS-based virulence processes of Legionella. This compound robustly decreased the ability of L. pneumophila to escape lysosomal compartments (Fig. 5) and to proliferate within mouse macrophages (Fig. 3). It also dramatically reduced reporter-effector translocation by the L. pneumophila Dot/Icm system (Fig. 1E). Importantly, while C4 had no detectable effect on effector-reporter delivery by C. burnetii (Fig. 6), the four other hit compounds, C1, C3, C5, and C7, did, reducing cAMP levels by at least 10-fold compared with untreated cells (Fig. 6). Despite the high degree of homology between the T4SS components from L. pneumophila and C. burnetii, the two pathogens show notably different infection dynamics: While L. pneumophila effector translocation begins immediately upon host cell contact, Coxiella are metabolically inactive at early times postinfection and do not secrete proteins until 24 to 48 h later. Consequently, the different treatment regimens that were used may explain the observed difference in compound efficacies; while compounds were simultaneously added with L. pneumophila bacteria to host cell monolayers, macrophages infected with Coxiella were not exposed to the compounds until 24 h after the initial bacterial challenge.
We also discovered in our screen two compounds C2 and C6 (Table S2) that met all criteria for being genuine T4SS inhibitor, but that, upon further evaluation, appeared to strongly affect L. pneumophila growth outside the host (Fig. 4A and B), suggesting that they likely had other molecular targets besides the T4SS. Inhibition of L. pneumophila growth in media did not require the presence of Dot/Icm system components as Lp02Δ26 remained sensitive to these two compounds (Fig. 4E). Interestingly, their growth-inhibitory activity was limited to L. pneumophila and did not affect other Gram-negative bacteria such as E. coli or P. aeruginosa, suggesting that these two compounds do not represent general antimicrobials but rather targeted an essential component or mechanism solely present in L. pneumophila.
Notably, none of the five hit compounds discovered here overlapped with the hits found in the study by Charpentier et al. (31). Contributing factors that may lead to this discrepancy in results likely include the different types of host cells used (RAW264.7 macrophages vs J774 macrophages), the aforementioned difference in the duration for which macrophages were exposed to the compounds prior to L. pneumophila challenge, as well as the criteria for selection of positive hits. Despite these differences, the T4SS inhibitors from either study set the stage for the development of a new generation of “smarter” antibiotics that, unlike most conventional antibiotics, could selectively target pathogens while leaving commensals unaffected. By exclusively altering the physiology of pathogenic bacteria that rely on a T4SS for virulence, such compounds have the potential to one day treat infectious diseases while preserving the healthy microbiota, a prerequisite for the prevention of secondary infections. Furthermore, if coupled to chemical handles, the compounds discovered here can be used as laboratory tools to further examine the regulation of bacterial T4SSs during infection. Although recent work has provided first insight into the structural organization of the L. pneumophila T4SS (14–18), much has yet to be learned about the dynamics of these sophisticated protein secretion machines.
MATERIALS AND METHODS
Strains, media, and reagents.
L. pneumophila strains were grown and maintained as described (54). The bacterial strains used in this study are listed in Table S3. L. pneumophila strains Lp02 and Lp03 are thymidine-auxotroph derivatives of L. pneumophila strain Philadelphia-1 (55). Legionella were grown in ACES-buffered yeast extract with thymidine (AYET) supplemented at 100 μg/mL. Whenever indicated, chloramphenicol and kanamycin were used at a final concentration of 5 μg/mL and 20 μg/mL respectively. E. coli and P. aeruginosa were grown with aeration in Luria–Bertani (LB) medium. RAW264.7 macrophages were obtained from American Type Culture Collection and were grown in DMEM supplemented with 10% FBS and incubated in 5% CO2 at 37°C. β-lactamase-specific antibody was obtained from Abcam. Rabbit polyclonal anti-isocitrate dehydrogenase (ICDH) antibody was generously provided by Abraham (Linc) Sonenshein, Tufts University Medical School, Boston, MA.
Strains used in this study. Download Table S3, PDF file, 0.1 MB (94.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Plasmids.
Plasmids and oligonucleotides used in this study are listed in Tables S4 and S5, respectively. Plasmids encoding β-lactamase effector protein fusions were constructed as previously described (35). The pXDC61 plasmid was generously provided by Howard A. Shuman (University of Chicago). Briefly, PCR products of the lidA flanking regions were digested with appropriate restriction enzymes and cloned into the KpnI-SmaI-BamHI-XbaI polylinker of pXDC61. Lp02 or Lp03 were transformed with plasmids by electroporation (56).
Plasmids used in this study. Download Table S4, PDF file, 0.1 MB (63.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Oligonucleotides used in this study. Underlined sequences represent restriction enzyme sites. Download Table S5, PDF file, 0.1 MB (69.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Type IV translocation qHTS assay.
Detection of β-lactamase-LidA fusion protein translocation into infected macrophages was performed using the FRET-based detection method described previously (35), with modifications. Briefly, Lp02 or Lp03 containing the plasmids that encode either β-lactamase or β-lactamase fusion proteins were grown to post-exponential phase in the presence of 0.5 mM IPTG. RAW264.7 macrophages were dispensed by a Multidrop Combi Reagent Dispenser (Thermo Fischer Scientific) into black clear-bottom 1536-well plates (Greiner Bio One) at 2 × 104 cells/well and allowed to attach for 3 h. The compounds assayed were solubilized in DMSO and arrayed as 5- or 6-point inter-plate titrations at final concentrations ranging from 46 μM to 0.18 μM, and 23 nL of compound solution was transferred into a 1,536-well plate using a Kalypsys pintool. After compound addition to macrophages, the cells were challenged with the appropriate strain of L. pneumophila at an multiplicity of infection (MOI) of 20 using a Multidrop Combi Reagent Dispenser. The CCF4/AM substrate (Life Technologies, Inc.) was added to the wells 1 h postinfection and then incubated for an additional 3 h at room temperature. CCF4/AM fluorescence was measured using dual fluorescence intensities (Ex1 = 405 ± 20, Em1 = 460 ± 20, and Ex2 = 405 ± 20, Em2 = 530 ± 20 nm) in an EnVision Multimode Plate Reader (PerkinElmer, Boston, MA, USA). The ratio of fluorescence intensities (Em1/Em2) was calculated as representation for the β-lactamase activity levels within the host cells. When appropriate, individual assay wells were visualized using a Zeiss Observer.Z1 equipped with a βLac, blue/aqua fluorescence filter cube (Chroma Technology Corp).
Compound libraries.
The screening collection of 18,272 members included the following libraries with the number of compounds indicated in parentheses: NCATS Pharmaceutical Collection (NPC, 2,816), NCATS Pharmacologically Active Chemical Toolbox (NIH, 2,108), Mechanism Interrogation PlatE (NIH, 1,912), Library of Pharmacologically Active Compounds (LOPAC, 1,280), Spectrum Collection (MicroSource Discovery Systems, 2,000), Tocris (Tocris Bioscience, 1,408), BioMol (Enzo Life Sciences, 1,408), Cayman natural product (303), Pharmacopeia (3,000), and KINACore Library (ChemBridge, 2,037).
β-lactamase specificity assay.
Purified recombinant β-lactamase (Thermo Fisher) in phosphate-buffered saline (PBS) (0.15 nM) was dispensed into 1,536-well plates using a BioRAPTR FRD Microfluidic Workstation (Beckman Coulter Life Sciences). Compounds or control buffer (DMSO) were transferred using Kalypsys pintool equipped with a 1536-pin array. The plate was incubated for 10 to 15 min at room temperature, and nitrocefin (Calbiochem) was added to a final concentration of 430 μM by the BioRAPTR FRD workstation to initiate the color reaction. After a 20 min incubation at room temperature, hydrolysis of the nitrocefin substrate was measured by absorbance (OD490) using an Envision Multimode Plate Reader.
Cytotoxicity assay.
A Multidrop Combi Reagent Dispenser was used to dispense 5,000 or 50,000 RAW264.7 macrophages per well into either 1,536- or 96-well opaque white plates (Greiner Bio One), respectively. The plates were incubated for 3 h to allow for cell attachment to the substrate to occur. The compounds were transferred into 1,536-well plates using a Kalypsys pintool or manually into 96-well plates. After 24 h, Cell-Titer Glo (Promega) was added following the manufacturer’s instruction using a BioRAPTR FRD. The plates were incubated for 30 min at room temperature, and luminescence was measured using a ViewLux plate reader (Perkin Elmer).
Intracellular growth assay.
RAW264.7 macrophages were dispensed using a BioRAPTR FRD workstation at either 7,000 cells per well in 1,536-well or 14,000 cells/well in 384-well plates with media supplemented with 0.5 mM IPTG. The cells were allowed to attach to the substrate for 3 h before compounds were added using the Kalypsys pintool. The cells were immediately challenged at an MOI of 50 with Lp02 containing pXDC31 (GFP encoding plasmid) grown to post-exponential phase in the presence of 0.5 mM IPTG. The plates were centrifuged at 200 × g for 5 min. After 14 h incubation, the macrophages were chemically fixed for 15 min at room temperature with formaldehyde (4% final concentration) and washed 2× with PBS. The cell nuclei were stained using Hoechst 33342 (Thermo Fisher Scientific). Both 1,536- and 384-well plates were imaged on an automated, widefield high content imager (IN Cell 2200, GE Healthcare) using a 10×/0.45 NA lens and standard DAPI (nuclear stain) and FITC (Legionella infection) excitation and emission filters. The TIFF files were quantitated using the canned Multi Target Analysis protocol (GE Investigator Workstation software v3.7.2). Briefly, nuclei from the DAPI channel were identified using top hat segmentation and a sensitivity setting of 96 and minimum size area of 35 μM2. Bacteria were detected in the FITC channel (cells) using a 2-μm collar dilation from the nuclear bitmap. FITC objects with an average nuclear RFU intensity above 450 (3Stdev above mean negative control wells) were considered as “Legionella positive,” and results were represented as relative infection rates (% of control).
In vitro growth assay.
Lp02, Lp03, JV4044 (a kind gift from Joseph Vogel, Washington University in St. Louis), E. coli K12 MG1655, and P. aeruginosa were diluted in the appropriate growth media to an OD600 between 0.01 to 0.1. The bacteria were treated with 28 μM compound and loaded into 96-well plates at a total volume of 300 μL per well. Growth was measured at an OD600 in 1 h intervals for 18 h or more in a Clariostar Monochromator Microplate Reader (BMG Labtech).
Enumeration of Lamp1 positive LCVs when treated with compounds.
RAW264.7 macrophages were grown in CellVis 8 chambered coverglass slides (#C8-1.5H-N) and challenged at an MOI of 25 with Lp02 producing mCherry from the plasmid pXDC50. DMSO or the indicated compounds (23 M) were added concurrently with Lp02, slides were centrifuged at 200 × g for 5 min. After 2 h of incubation, cells were washed 3× with PBS, fixed with formaldehyde (4% final concentration) for 10 min at room temperature and washed 2× with PBS. Staining for uninfected (outside) Lp02 was performed with rabbit-anti-Legionella primary antibody (1:3,000, 1 h, 37°C), cells were washed 3× with PBS, and stained with goat-anti-rabbit-404 secondary antibody (1:2,000, 1 h, 37°C, Life Technologies #C2764). Cells were then permeabilized with 0.2% TritonX 100 in PBS for 10 min at room temperature and washed with PBS. Cells were stained with rabbit-anti-Lamp1 primary antibody (1:1,000, 1 h, 37°C, Abcam #ab24170), washed 3× with PBS, and stained with goat-anti-rabbit-488 (1:2,000, 1 h, 37°C, Thermo Fisher #656111). Slides were washed with PBS and internalized Lp02 with or without Lamp1 staining were quantified by microscopy using a Zeiss LSM 800 with Airyscan confocal microscope. Experiments were performed 3 to 4 times per compound, counting 50 to 100 internalized Lp02 each. Data were analyzed by dividing Lamp1 positive bacteria by the total number of internalized bacteria and represented as a percentage, averaging the percentages across replicates, and displaying the data as the mean with standard deviation and individual replicate points. A two-way unpaired T-test was performed comparing average counts of each compound to the DMSO control, (*) indicates P < 0.05.
Coxiella effector translocation assay and growth assays.
C. burnetii CyaA translocation assays were performed using THP-1 macrophage-like cells (5 × 105 per well) in 24-well plates as previously described (57). THP-1 cells treated overnight with 200 nM phorbol 12-myristate 13-acetate were washed once with growth medium (RPMI plus 10% FBS), infected with C. burnetii transformants expressing CyaA at an MOI ∼ 50 for 24 h, and then incubated an additional 24h with fresh medium containing 50 μM the indicated compounds. For CyaA translocation assays, the concentration of cAMP in lysates from infected THP-1 cells was determined using the cAMP enzyme immunoassay (GE Healthcare) as previously described (57). Positive secretion of CyaA fusion proteins by C. burnetii was scored as ≥2.5-fold more cytosolic cAMP than the negative control (wild-type C. burnetii producing CyaA alone) (57, 58).
In vitro replication of C. burnetii in the presence of compounds was determined as described (59). Briefly, 1 × 105 per mL of C. burnetii were incubated at 37°C in a 2.5% O2 and 5% CO2 with either 5 μM the indicated compounds or DMSO. The number of C. burnetii genomes measured by qRT-PCR in ACCM-D broth cultures was measured at the start of the experiment (day 0) and after incubation (day 6).
Bacterial uptake assay.
RAW264.7 macrophages (400,000/well) were seeded on coverslips in 24-well plates and grown overnight. The macrophages were treated with compound (57 μM final), Cytochalasin D (10 μM final), or DMSO for 5 min before the addition of Lp02 resuspended in tissue culture media containing compound (57 μM final), Cytochalasin D (10 μM final), or DMSO at an MOI of 1. The plate was centrifuged for 5 min at 200 × g to enhance bacteria-cell contact and incubated for 1h at 37°C in 5% CO2. Extracellular bacteria were removed by washing 3× with PBS. The cells were fixed in PBS containing 3.7% formaldehyde.
The cells were blocked in PBS containing 5% goat serum for 1 h at 37°C. Staining for uninfected (outside) Lp02 was performed with rabbit-anti-Legionella primary antibody, cells were washed 3× with PBS, and stained with goat-anti-rabbit FITC conjugated secondary antibody. Cells were then permeabilized with 100% methanol, blocked with 5% goat serum. Staining for total (inside and outside) Lp02 was performed with rabbit-anti-Legionella primary antibody, cells were washed 3× with PBS, and stained with goat-anti-rabbit Texas Red conjugated secondary antibody. After washing 3× with PBS, the cell nuclei were stained using Hoechst 33342, and the coverslips were mounted on slides before analysis.
ACKNOWLEDGMENTS
We thank all members of the Machner lab and Simeonov lab for their support and feedback. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases (to R.A.H.), National Center for Advancing Translational Sciences (to A.S.), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development as well as the NIH Director’s Challenge Innovation Award (to M.P.M.).
Footnotes
This article is a direct contribution from Robert A. Heinzen, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Hayley Newton, University of Melbourne, and Timothy Foley, Pfizer, DNA-encoded library selection & pharmacology.
Contributor Information
Anton Simeonov, Email: as699p@nih.gov.
Matthias P. Machner, Email: machnerm@nih.gov.
Carmen Buchrieser, Institut Pasteur.
REFERENCES
- 1.Cascales E, Christie PJ. 2003. The versatile bacterial type IV secretion systems. Nat Rev Microbiol 1:137–149. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christie PJ, Vogel JP. 2000. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol 8:354–360. doi: 10.1016/S0966-842X(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandran Darbari V, Waksman G. 2015. Structural biology of bacterial type IV secretion systems. Annu Rev Biochem 84:603–629. doi: 10.1146/annurev-biochem-062911-102821. [DOI] [PubMed] [Google Scholar]
- 4.Segal G, Feldman M, Zusman T. 2005. The Icm/Dot type-IV secretion systems of Legionella pneumophila and Coxiella burnetii. FEMS Microbiol Rev 29:65–81. doi: 10.1016/j.femsre.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Sexton JA, Vogel JP. 2002. Type IVB secretion by intracellular pathogens. Traffic 3:178–185. doi: 10.1034/j.1600-0854.2002.030303.x. [DOI] [PubMed] [Google Scholar]
- 6.Voth DE, Broederdorf LJ, Graham JG. 2012. Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol 7:241–257. doi: 10.2217/fmb.11.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon JE, Costa TRD, Patel RS, Gonzalez-Rivera C, Sarkar MK, Orlova EV, Waksman G, Christie PJ. 2017. Use of chimeric type IV secretion systems to define contributions of outer membrane subassemblies for contact-dependent translocation. Mol Microbiol 105:273–293. doi: 10.1111/mmi.13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD, Cover TL. 2016. Molecular and structural analysis of the Helicobacter pylori cag type IV secretion system core complex. mBio 7:e02001-15–e02015. doi: 10.1128/mBio.02001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung JM, Sheedlo MJ, Campbell AM, Sawhney N, Frick-Cheng AE, Lacy DB, Cover TL, Ohi MD. 2019. Structure of the Helicobacter pylori Cag type IV secretion system. Elife 8 doi: 10.7554/eLife.47644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low HH, Gubellini F, Rivera-Calzada A, Braun N, Connery S, Dujeancourt A, Lu F, Redzej A, Fronzes R, Orlova EV, Waksman G. 2014. Structure of a type IV secretion system. Nature 508:550–553. doi: 10.1038/nature13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462:1011–1015. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fronzes R, Schäfer E, Wang L, Saibil HR, Orlova EV, Waksman G. 2009. Structure of a type IV secretion system core complex. Science 323:266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivera-Calzada A, Fronzes R, Savva CG, Chandran V, Lian PW, Laeremans T, Pardon E, Steyaert J, Remaut H, Waksman G, Orlova EV. 2013. Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J 32:1195–1204. doi: 10.1038/emboj.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosal D, Jeong KC, Chang Y-W, Gyore J, Teng L, Gardner A, Vogel JP, Jensen GJ. 2019. Molecular architecture, polar targeting and biogenesis of the Legionella Dot/Icm T4SS. Nat Microbiol 4:1173–1182. doi: 10.1038/s41564-019-0427-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosal D, Chang YW, Jeong KC, Vogel JP, Jensen GJ. 2017. In situ structure of the Legionella Dot/Icm type IV secretion system by electron cryotomography. EMBO Rep 18:726–732. doi: 10.15252/embr.201643598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubori T, Koike M, Bui XT, Higaki S, Aizawa S-I, Nagai H. 2014. Native structure of a type IV secretion system core complex essential for Legionella pathogenesis. Proc Natl Acad Sci USA 111:11804–11809. doi: 10.1073/pnas.1404506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durie CL, Sheedlo MJ, Chung JM, Byrne BG, Su M, Knight T, Swanson M, Lacy DB, Ohi MD. 2020. Structural analysis of the Legionella pneumophila Dot/Icm type IV secretion system core complex. Elife 9. doi: 10.7554/eLife.59530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheedlo MJ, Durie CL, Chung JM, Chang L, Roberts J, Swanson M, Lacy DB, Ohi MD. 2021. Cryo-EM reveals new species-specific proteins and symmetry elements in the Legionella pneumophila Dot/Icm T4SS. Elife 10. doi: 10.7554/eLife.70427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel JP, Isberg RR. 1999. Cell biology of Legionella pneumophila. Curr Opin Microbiol 2:30–34. doi: 10.1016/S1369-5274(99)80005-8. [DOI] [PubMed] [Google Scholar]
- 20.Horwitz MA, Silverstein SC. 1980. Legionnaires’ disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J Clin Invest 66:441–450. doi: 10.1172/JCI109874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horwitz MA. 1983. The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med 158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horwitz MA. 1983. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med 158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogel JP, Andrews HL, Wong SK, Isberg RR. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- 24.Sadosky AB, Wiater LA, Shuman HA. 1993. Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect Immun 61:5361–5373. doi: 10.1128/iai.61.12.5361-5373.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brand BC, Sadosky AB, Shuman HA. 1994. The Legionella pneumophila icm locus: a set of genes required for intracellular multiplication in human macrophages. Mol Microbiol 14:797–808. doi: 10.1111/j.1365-2958.1994.tb01316.x. [DOI] [PubMed] [Google Scholar]
- 26.Duncan MC, Linington RG, Auerbuch V. 2012. Chemical inhibitors of the type three secretion system: disarming bacterial pathogens. Antimicrob Agents Chemother 56:5433–5441. doi: 10.1128/AAC.00975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christie PJ. 2016. The mosaic type IV secretion systems. EcoSal Plus 7. doi: 10.1128/ecosalplus.ESP-0020-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM, Baron C. 2011. An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect Immun 79:1033–1043. doi: 10.1128/IAI.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith MA, Coinçon M, Paschos A, Jolicoeur B, Lavallée P, Sygusch J, Baron C. 2012. Identification of the binding site of Brucella VirB8 interaction inhibitors. Chem Biol 19:1041–1048. doi: 10.1016/j.chembiol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Ripoll-Rozada J, García-Cazorla Y, Getino M, Machón C, Sanabria-Ríos D, de la Cruz F, Cabezón E, Arechaga I. 2016. Type IV traffic ATPase TrwD as molecular target to inhibit bacterial conjugation. Mol Microbiol 100:912–921. doi: 10.1111/mmi.13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charpentier X, Gabay JE, Reyes M, Zhu JW, Weiss A, Shuman HA. 2009. Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog 5:e1000501. doi: 10.1371/journal.ppat.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo ZQ, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci USA 101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc Natl Acad Sci USA 102:826–831. doi: 10.1073/pnas.0406239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, Shuman HA. 2004. Legionella effectors that promote nonlytic release from protozoa. Science 303:1358–1361. doi: 10.1126/science.1094226. [DOI] [PubMed] [Google Scholar]
- 35.de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. 2008. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog 4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu W, Luo ZQ. 2013. Methods for determining protein translocation by the Legionella pneumophila Dot/Icm type IV secretion system. Methods Mol Biol 954:323–332. doi: 10.1007/978-1-62703-161-5_19. [DOI] [PubMed] [Google Scholar]
- 37.Schroeder GN. 2017. The toolbox for uncovering the functions of Legionella Dot/Icm type IVb secretion system effectors: current state and future directions. Front Cell Infect Microbiol 7:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Derre I, Isberg RR. 2005. LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect Immun 73:4370–4380. doi: 10.1128/IAI.73.7.4370-4380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berger KH, Merriam JJ, Isberg RR. 1994. Altered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol Microbiol 14:809–822. doi: 10.1111/j.1365-2958.1994.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 40.Roy CR, Berger KH, Isberg RR. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol 28:663–674. doi: 10.1046/j.1365-2958.1998.00841.x. [DOI] [PubMed] [Google Scholar]
- 41.Mayr LM, Fuerst P. 2008. The future of high-throughput screening. J Biomol Screen 13:443–448. doi: 10.1177/1087057108319644. [DOI] [PubMed] [Google Scholar]
- 42.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 43.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin CP, Inglese J, Simeonov A. 2008. A robotic platform for quantitative high-throughput screening. Assay Drug Dev Technol 6:637–657. doi: 10.1089/adt.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasgar A, Shinn P, Jadhav A, Auld D, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A. 2008. Compound management for quantitative high-throughput screening. JALA Charlottesv Va 13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. 2006. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA 103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iversen PW, et al. 2004. HTS assay validation. In Sittampalam G.S., et al. (ed), Assay guidance manual, Bethesda, MD. [Google Scholar]
- 47.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. 2002. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem 45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 48.Vogel JP, Roy C, Isberg RR. 1996. Use of salt to isolate Legionella pneumophila mutants unable to replicate in macrophages. Ann N Y Acad Sci 797:271–272. doi: 10.1111/j.1749-6632.1996.tb52975.x. [DOI] [PubMed] [Google Scholar]
- 49.Carey KL, Newton HJ, Luhrmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-11–e00111. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sexton JA, Miller JL, Yoneda A, Kehl-Fie TE, Vogel JP. 2004. Legionella pneumophila DotU and IcmF are required for stability of the Dot/Icm complex. Infect Immun 72:5983–5992. doi: 10.1128/IAI.72.10.5983-5992.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwak M-J, Kim JD, Kim H, Kim C, Bowman JW, Kim S, Joo K, Lee J, Jin KS, Kim Y-G, Lee NK, Jung JU, Oh B-H. 2017. Architecture of the type IV coupling protein complex of Legionella pneumophila. Nat Microbiol 2:17114. doi: 10.1038/nmicrobiol.2017.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. 2006. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol 62:1278–1291. doi: 10.1111/j.1365-2958.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- 54.Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J Clin Microbiol 10:437–441. doi: 10.1128/jcm.10.4.437-441.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol 7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 56.Chen DQ, Huang SS, Lu YJ. 2006. Efficient transformation of Legionella pneumophila by high-voltage electroporation. Microbiol Res 161:246–251. doi: 10.1016/j.micres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, Bastidas RJ, Valdivia RH, Heinzen RA. 2015. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun 83:661–670. doi: 10.1128/IAI.02763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beare PA, Sandoz KM, Larson CL, Howe D, Kronmiller B, Heinzen RA. 2014. Essential role for the response regulator PmrA in Coxiella burnetii type 4B secretion and colonization of mammalian host cells. J Bacteriol 196:1925–1940. doi: 10.1128/JB.01532-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sandoz KM, Sturdevant DE, Hansen B, Heinzen RA. 2014. Developmental transitions of Coxiella burnetii grown in axenic media. J Microbiol Methods 96:104–110. doi: 10.1016/j.mimet.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The production of βLac or βLac_LidA was induced overnight by incubation with IPTG in both strains Lp02 and Lp03. Proteins within the bacterial lysate were separated by SDS-PAGE. After transfer to nitrocellulose membranes, the βLac-tagged proteins were immunologically detected using a β-lactamase specific antibody (top). Isocitrate dehydrogenase (ICDH) served as a loading control (bottom). Download FIG S1, PDF file, 0.6 MB (668.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data of 113 compounds selected from the qHTS reporter translocation assay Table S1, PDF file, 0.5 MB (475.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) β-lactamase specificity assay principle based on hydrolysis of the chromogenic substrate nitrocefin, an indicator of β-Lac enzymatic activity. (B) Cell cytotoxicity assay principle based on ATP abundance, an indicator of metabolically active cells. Download FIG S2, PDF file, 0.4 MB (404.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Legionella growth inhibition in a 1,536-well plate format. RAW264.7 macrophages were challenged with a GFP-producing L. pneumophila for 14 h in the presence of the indicated compounds, and the GFP signal (as a measure for bacterial growth) was determined using an IN Cell Analyzer 2200 Imaging system. Download FIG S3, PDF file, 0.5 MB (473.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data of 7 compounds selected following validation assays. Download Table S2, PDF file, 0.4 MB (387KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Bacterial uptake by macrophages in the presence of compounds. RAW264.7 macrophages were treated with the indicated compounds, Cytochalasin D, or DMSO vehicle prior to infection with Lp02, and intracellular bacteria were enumerated visually. Data shown are the mean with standard deviation from six experimental replicates, with the DMSO control considered as 100%. P-values (t-test) are <0.05 (*), <0.001 (**), or <0.005 (***) relative to the control. Download FIG S4, PDF file, 0.2 MB (198KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
In vitro replication of C. burnetii in the presence of compounds. ACCM-D broth cultures containing 5 μM of the indicated compounds or DMSO and inoculated with ∼ 1 × 105 per mL of C. burnetii were incubated at 37° C in a 2.5% O2 and 5% CO2. The plot depicts the number of C. burnetii genomes measured in ACCM-D broth cultures at the start of the experiment (day 0) and after incubation (day 6) detected by qRT-PCR (K. M. Sandoz, D. E. Sturdevant, B. Hansen, R. A. Heinzen. (2014). Developmental transitions of Coxiella burnetii grown in axenic media. J Microbiol Methods 96:104–110.). Bars indicate the mean ± SEM of combined data from three independent experiments each performed with triplicate samples (N = 9). One-way ANOVA analysis indicated differences between groups at D6 were found not significant (ns) using a 95% confidence interval. Download FIG S5, PDF file, 0.7 MB (754.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Strains used in this study. Download Table S3, PDF file, 0.1 MB (94.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Plasmids used in this study. Download Table S4, PDF file, 0.1 MB (63.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Oligonucleotides used in this study. Underlined sequences represent restriction enzyme sites. Download Table S5, PDF file, 0.1 MB (69.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.