
Fig. 1. (A) Computational scheme was applied to characterize and design potential inhibitors for SARS-CoV-2 Mpro using atomistic simulations and machine learning calculations. (B) A ligand was docked to SARS-CoV-2 Mpro using AutoDock Vina. (C) The protonation states of the catalytic dyad His41 and Cys145. (D) A ligand was dissociated from the bound state using external-harmonic force 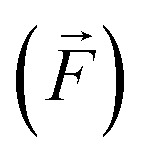 during FPL simulations.
during FPL simulations. 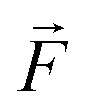 was put on the ligand center of mass in order to force the ligand to mobilize out of the protease binding cavity.
was put on the ligand center of mass in order to force the ligand to mobilize out of the protease binding cavity.
