

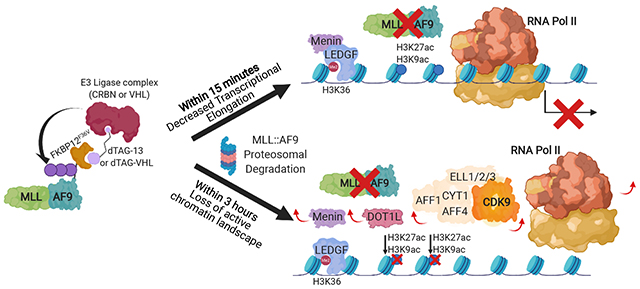
SUMMARY
MLL-rearrangements produce fusion oncoproteins that drive leukemia development, but the direct effects of MLL-fusion inactivation remain poorly defined. We designed models with degradable MLL::AF9 where treatment with small molecules induces rapid degradation. We leveraged the kinetics of this system to identify a core subset of MLL::AF9 target genes where MLL::AF9 degradation induces changes in transcriptional elongation within fifteen minutes. MLL::AF9 degradation subsequently causes loss of a transcriptionally active chromatin landscape. We used this insight to assess the effectiveness of small molecules that target members of the MLL::AF9 multi-protein complex, specifically DOT1L and MENIN. Combined DOT1L/MENIN inhibition resembles MLL::AF9 degradation, whereas single agent treatment has more modest effects on MLL::AF9 occupancy and gene expression. Our data show that MLL::AF9 degradation leads to decreases in transcriptional elongation prior to changes in chromatin landscape at select loci and combined inhibition of chromatin complexes releases the MLL::AF9 oncoprotein from chromatin globally.
Graphical Abstract
eTOC Blurb
Olsen et al. couple MLL::AF9 degradation with genome-wide transcriptional and chromatin analyses to characterize acute consequences of MLL::AF9 loss. MLL::AF9 degradation suppresses transcriptional elongation within minutes, whereas chromatin changes are induced subsequently. Dual targeting of the MLL::AF9-complex via combined DOT1L/MENIN inhibition mimics MLL::AF9 degradation by inducing global MLL::AF9 chromatin loss.
INTRODUCTION
Acute leukemia is the most common pediatric malignancy and remains difficult to treat with most adult patients dying from their disease. Chromosomal translocations involving the mixed lineage leukemia (MLL1, KMT2A) gene on Chromosome 11q23 give rise to a genetically distinct subset of leukemia, namely MLL-rearranged acute myeloid and lymphoblastic leukemia (AML and ALL, respectively) that account for approximately 5-10% of leukemia (Meyer et al., 2018; Muntean and Hess, 2012). MLL-translocations produce a chimeric gene that encodes an oncogenic MLL-fusion protein. The oncogenic fusion protein consists of the N-terminus of the MLL1 protein fused to the C-terminus of one of over 80 known fusion partners (Meyer et al., 2018). One of the most common translocation partners is AF9 on chromosome 9p22 (Winters and Bernt, 2017).
MLL-fusion proteins bind to DNA/chromatin and transform hematopoietic stem and progenitor cells by induction of stem cell associated gene programs (Krivtsov et al., 2006). Multiple chromatin regulators and co-factors interact with MLL-fusion proteins in large, multisubunit complexes. These complexes include transcription and chromatin regulators like MENIN (Yokoyama et al., 2005), disruptor of telomeric silencing 1-like (DOT1L) (Okada et al., 2005), and members of the Super Elongation Complex (SEC) (Lin et al., 2010; Mueller et al., 2007; Yokoyama et al., 2010; Zeisig et al., 2005), many of which have been shown to be important for MLL-fusion driven gene expression and leukemia development.
Since MLL-fusion proteins do not lead to direct activation of an enzyme, identification of small molecules that directly target the fusion protein has been difficult. However, recent studies have shown that it is possible to disrupt critical activities of multi-protein complexes, thereby modulating cancer-specific gene expression patterns (reviewed in Bradner et al., 2017). In MLL-rearranged leukemia, for example, small molecule inhibition of the MENIN:MLL1 protein:protein interaction has been shown to be a potential therapeutic strategy (Borkin et al., 2015; Grembecka et al., 2012; Krivtsov et al., 2019). Similarly, catalytic inhibition of the methyltransferase DOT1L with EPZ-5676 (Pinometostat, Epizyme) has shown promise (Daigle et al., 2011, 2013; Stein et al., 2018). Interestingly, MENIN and DOT1L inhibition have also been reported to synergize to inhibit cell proliferation (Dafflon et al., 2017; Kühn et al., 2016; Okuda et al., 2017), but the mechanism behind this synergy is largely unknown.
In order to define the immediate consequences of MLL::AF9 inactivation and to compare these to DOT1L and MENIN inhibition, we characterized the molecular and phenotypic consequences of direct PROteolysis-TArgeting-Chimeras (PROTACs)-mediated MLL::AF9 degradation. The rapid kinetics of this approach allowed us to assess chromatin state and gene expression changes within minutes after MLL::AF9 degradation, thereby enabling us to distinguish direct immediate effects from potential secondary and compensatory changes. Ultimately, we were able to leverage this mechanistic insight to dissect how DOT1L and MENIN inhibition compare to rapid degradation of MLL::AF9 and why the DOT1L/MENIN combination is so effective.
RESULTS
Degradation of MLL::AF9 induces cellular differentiation and apoptosis
Small molecules that inhibit members of the MLL::AF9 multi-protein complex primarily induce differentiation with only modest induction of apoptosis. Thus, we wanted to assess the phenotypic changes upon degradation of the MLL::AF9 fusion protein itself. We utilized the dTAG system where the MLL::AF9 fusion oncoprotein is fused to mutant FKBP12 (FKBP12F36V) and treatment with hetero-bifunctional molecules that bind to both FKBP12F36V and the E3 ubiquitin ligase Cereblon (dTAG-13) (Erb et al., 2017; Nabet et al., 2018) or VHL (dTAG-VHL) (Nabet et al., 2020) induce proteasomal degradation. We generated murine leukemias with degradable MLL::AF9 by transducing murine hematopoietic stem and progenitor cells (Lin−Sca1+c-kit+, LSK) with either C- or N-terminally HA-FKBP12-tagged MLL::AF9 (Krivtsov et al., 2006). Only the C-terminal tag had the same kinetics of leukemogenesis as the unmodified MLL::AF9 construct (Figure S1A), most likely because the N-terminal tag interfered with MLL::AF9’s chromatin localization since various DNA binding motifs and the MENIN binding domain are located at the N-terminus (Allen et al., 2006; Cierpicki et al., 2010; Hughes et al., 2004; Milne et al., 2010; Yokoyama and Cleary, 2008; Yokoyama et al., 2005). We confirmed that the mice were leukemic (Figure S1B) and that the FKBP12-tagged cells have the same growth kinetics and the same expression level of well-established MLL::AF9 target genes (Braekeleer et al., 2014; Faber et al., 2009; Kumar et al., 2009; Wong et al., 2007) as the non-FKBP12-tagged cells (Figure S1C and D). Intracellular FACs using an HA-directed antibody confirmed that treatment with dTAG-13 induced MLL::AF9 protein degradation in a time-dependent manner (Figure S1E). Moreover, treatment with the negative control compound, dNEG-13, (Nabet et al., 2020), had no effect on MLL::AF9 protein level, confirming that the degradation we observe is mediated by Cereblon (Figure S1F). Co-treatment of dTAG-13 with the proteosome inhibitor carfilzomib or Nedd8 activating enzyme inhibitor MLN-4924 rescued MLL::AF9 degradation, consistent with the mechanism of action of dTAG-13 (Figure S1G). We also transduced human hematopoietic stem and progenitor (CD34+) cells, isolated from human cord blood, with either the unmodified MLL::AF9 construct or the C-terminally tagged MLL::AF9-HA-FKBP12 construct as previously described (Horton et al., 2013; Mulloy et al., 2008; Schneidawind et al., 2018). Unlike the murine system, treatment with dTAG-13 only had modest effects on MLL::AF9 protein level, as visualized by Western blot using an HA-directed antibody (Figure 1A). However, treatment with a newer degrader molecule, dTAG-VHL, which utilizes the E3 ligase VHL instead of Cereblon (Nabet et al., 2020), led to robust MLL::AF9 degradation (Figure 1A). The differential sensitivity to dTAG-13 versus dTAG-VHL is most likely due to differing expression levels of the corresponding E3 ligases, Cereblon and VHL. While both ligases are expressed equally in the murine cells, VHL is more highly expressed in the MLL::AF9 transformed human CD34+ cells (data not shown). Treatment with the negative control compound, dNEG-VHL had no effect on MLL::AF9 protein level, even after 24 hours of treatment (Figure 1B). In summary, we generated both human and murine leukemia systems where we could induce potent degradation of the MLL::AF9 fusion protein.
Figure 1. Characterization of the human MLL::AF9-FKBP12 system.
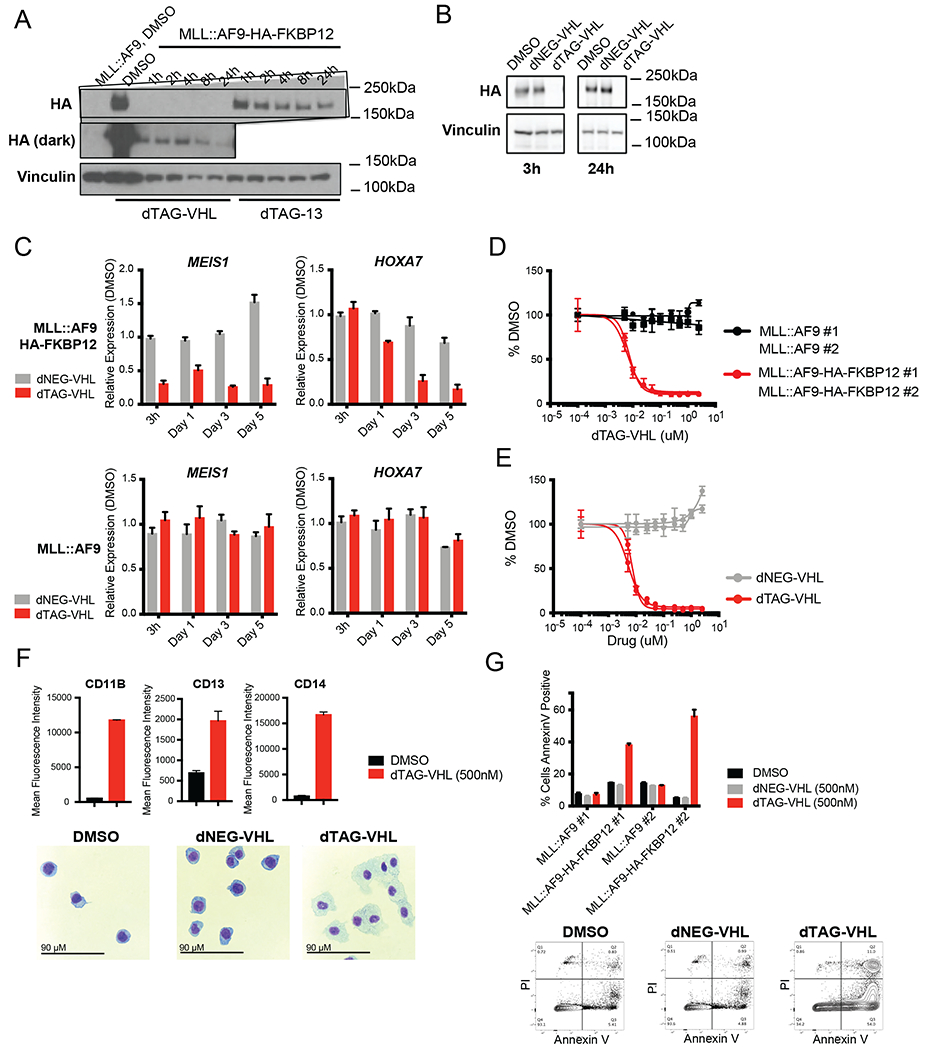
A. Western blot of untagged MLL::AF9 or C-terminal MLL::AF9-HA-FKBP12 cells, treated with dTAG-VHL (500nM) or dTAG-13 (500nM) for the indicated timepoints. HA levels were assessed by immunoblotting.
B. Western blot of cells treated with DMSO, dNEG-VHL (500nM), and dTAG-VHL (500nM) for 3 and 24 hours.
C. Relative expression of MEIS1 and HOXA7 upon dNEG-VHL (500nM) and dTAG-VHL (500nM) treatment for the indicated timepoints. Data are represented as mean + SD of three independent experiments, measured by Taqman.
D. Cell Proliferation assessed as percent DMSO after 6 days with increasing doses of dTAG-VHL. Data are represented as mean ± SD of three independent experiments.
E. Cell Proliferation assessed as percent DMSO after 6 days with increasing doses of dTAG-VHL and dNEG-VHL. Data are represented as mean ± SD of three independent experiments.
F. Mean Fluorescence Intensity of differentiation markers (as indicated) after 5 days of DMSO or dTAG-VHL treatment. Data are represented as mean + SD of triplicates. Cytospins are shown below, scale bar is 90μM.
G. Annexin V positive, PI negative cells 5 days after DMSO, dNEG-VHL, and dTAG-VHL treatment. Data are represented as mean + SD of three independent experiments. Representative FACs plots are shown below.
Next, we assessed how MLL::AF9 degradation affected the expression of previously defined MLL::AF9 targets like MEIS1 and HOXA genes. Treatment with dTAG-VHL, but not dNEG-VHL, led to significant downregulation of MEIS1 as well as HOXA7 (Figure 1C, top). In the untagged MLL::AF9 cells neither treatment with dTAG-VHL nor with dNEG-VHL had any effects (Figure 1C, bottom). Analogous trends were observed in the murine cells, where dTAG-13 treatment induced a dose- and time-dependent reduction in Meisl and HoxA9 expression only in the HA-FKBP12-tagged cells, but not in the untagged MLL::AF9 cells (Figure S1H). Again, no effects on gene expression were observed upon dNEG-13 treatment (Figure S1I). These data indicate that we can degrade MLL::AF9 leading to decreased expression of previously defined MLL::AF9 target genes, with no obvious off-target effects in either the human or murine system.
To assess the cellular consequences of MLL::AF9 degradation, we performed proliferation, differentiation, and apoptosis studies. dTAG-13/dTAG-VHL treatment severely impaired proliferation in the human (Figure 1D and E) as well as the murine model (Figure S1J and K) in a dose-dependent fashion. Moreover, MLL::AF9 degradation led to potent cellular differentiation and apoptosis in both models (Figure 1F and G, Figure S1L and M), while dNEG-13/dNEG-VHL had no effect. These data indicate that the MLL::AF9 fusion protein is crucial for maintaining expression of target genes, cell growth, viability, and survival, as well as for maintaining a blast-like, undifferentiated cell state.
A subset of MLL::AF9 target genes is highly sensitive to MLL::AF9 degradation
To more fully characterize the human and murine MLL::AF9-HA-FKBP12 systems, we first established MLL::AF9 directed gene expression by RNA sequencing (RNA-Seq) at 3 and 24 hours post dTAG-VHL/dTAG-13 treatment. After 3 hours of dTAG-VHL treatment in the human cells there were modest gene expression changes with only 20 genes that had a 25% or greater reduction of expression, many of which are prominent MLL::AF9 target genes such as MEIS1, MEF2C, PBX3, and HOXA9 (Figure 2A, Figure S2A, and Table S1). After 24 hours of dTAG-VHL treatment a much larger set of genes was differentially expressed (Fold Change>2, p<0.05), likely due to both direct and indirect effects of MLL::AF9 degradation (Figure 2A, Figure S2A, and Table S1). Expression of all genes downregulated at the 3-hour timepoint was further decreased after 24 hours of treatment, indicating that there were no compensatory changes within that timeframe. In the murine cells, a similarly small list of differentially expressed genes (Fold Change>1.5, p<0.1) was identified when treating with dTAG-13 for 3 hours (Figure S2B and C, Table S1). Again, many were well-known MLL::AF9 target genes whose expression further decreased after 24 hours of dTAG-13 treatment (Figure S2B and C, Table S1). Although there was only modest overlap when comparing the significantly downregulated genes in human and murine cells after 3 hours of dTAG treatment (Figure 2A and Figure S2C), there was a more notable degree of overlap by gene-set enrichment analysis looking at downregulated genes independent of fold change and p-value cut offs (Figure S2D and Table S1). These findings indicated that while not always significant after 3 hours of treatment, most genes that we identified were differentially expressed upon MLL::AF9 degradation in mouse and human leukemia cells.
Figure 2. Defining MLL::AF9-directed gene expression and chromatin occupancy in human MLL::AF9-FKBP12 cells.
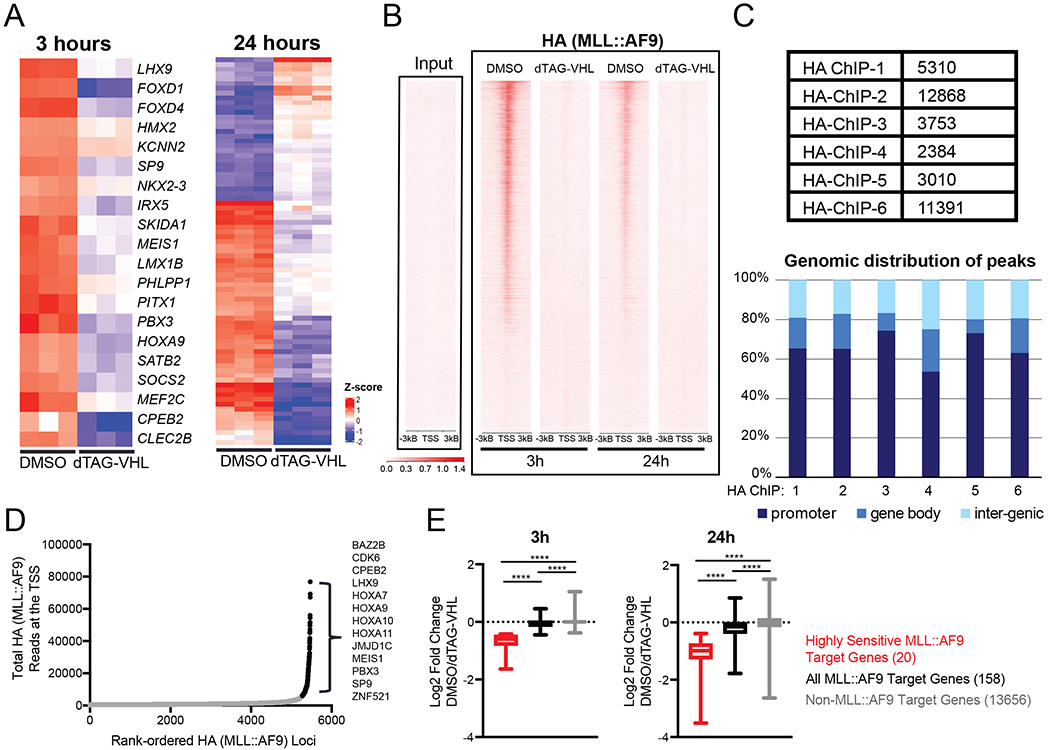
A. (Left) Heatmap of genes with 25% or greater reduction of expression after 3 hours of treatment with either DMSO or dTAG-VHL (500nM, Table S1). (Right) Heatmap of differentially expressed genes (Fold Change > 2, p<0.05, Table S1) after 24 hours of DMSO or dTAG-VHL (500nM) treatment.
B. Genome wide rank-ordered heat map of HA (MLL::AF9) ChIP signal across the promoter (TSS - 3kB/+3kB), 3 and 24 hours following dTAG-VHL (500nM) treatment.
C. Peak number and genomic distribution of HA peaks from six independent HA ChIP-Seq experiments.
D. MLL::AF9 loading at promoters (TSS -1kB/+3kB). HA (MLL::AF9) ChIP-seq reads at each promoter are plotted against their rank among all HA-occupied regions. Black depicts highly bound loci, defined as MLL::AF9 target genes (Table S1).
E. Boxplots of DMSO-normalized Log2 fold change RNA-Seq values after dTAG-VHL (500nM) treatment of ‘highly sensitive genes’ (Table S1), identified in 2A, all other MLL::AF9 target genes (Table S1) identified in 2D, and non MLL::AF9 bound genes. Numbers in parentheses denote the number of genes in each group.
In addition to characterizing the early gene expression changes, we also wanted to assess how long-term gene expression changes induced by dTAG treatment compare to previously published studies. After 5 days of dTAG-13 treatment in the murine cells, known MLL::AF9 (Bernt et al., 2011; Stavropoulou et al., 2016) and MLL-fusion (Guenther et al., 2008) target genes were significantly enriched among our downregulated genes (Figure S2E), confirming that at later timepoints the dTAG approach yielded similar results to previously published work. Moreover, dTAG-13 treatment not only reduced the expression of MLL::AF9-directed stem cell associated gene programs at early timepoints, but it also led to upregulation of genes involved in neutrophil differentiation after 5 days of treatment (Figure S2F and Table S1). This observation was in line with our functional data, where we detected the corresponding cytomorphological changes associated with differentiation (Figure S1L and S2F).
Next, we defined the MLL::AF9 chromatin binding profile by chromatin immunoprecipitation followed by sequencing (ChIP-Seq) using an HA-directed antibody. MLL::AF9 degradation led to complete loss of MLL::AF9 (HA) ChIP signal at 3 hours (Figure 2B). Using a compilation of six independent biological replicates, we on average detected around 6500 MLL::AF9 (HA) peaks, with around 70% of peaks occurring in promoter regions (Figure 2C). Focusing only on promoter regions (Transcriptional Start Site (TSS) −1kB/+3kB) and rank-ordering the MLL::AF9 bound loci, we uncovered an asymmetric distribution of MLL::AF9, in which only around 200 genes had a disproportionally high load of MLL::AF9 around the promoter (Figure 2D and Table S1). These genes were termed MLL::AF9 target genes, as defined by highly bound MLL::AF9 at the promoter. Among them were all the genes identified in Figure 2A (the 20 genes that were downregulated after 3 hours of dTAG-VHL treatment, now referred to as ‘highly sensitive genes’), as well as other well established MLL::AF9 target genes including JMJD1C, CDK6, HOXA7, HOXA10, and HOXA11 (Figure 2D and Table S1). While all highly MLL::AF9 bound genes exhibited decreased gene expression after 3 hours of dTAG-VHL treatment, the changes in the ‘highly sensitive genes’ were significantly more pronounced (Figure 2E). No gene expression changes were detected in the non-MLL::AF9 bound genes upon dTAG-VHL treatment (Figure 2E). After 24 hours of dTAG-VHL treatment, these trends were even more robust, with both the ‘highly sensitive’ as well as all other MLL::AF9 target genes downregulated more prominently (Figure 2E), although at this later timepoint gene expression changes could be due to secondary effects. In summary, we have defined 3 groups of genes in the human MLL::AF9-HA-FKBP12 system based on their level of MLL::AF9 loading/occupancy and the kinetics of gene expression changes after MLL::AF9 degradation. These groups include 1) a subset of 20 genes that are highly bound by MLL::AF9 and show significant changes in gene expression within 3 hours (called ‘highly sensitive genes’) 2) ~180 genes that are highly bound by MLL::AF9 and show only modest changes in gene expression within 3 hours of MLL::AF9 degradation (called MLL::AF9 target genes), and 3) all other genes (non-MLL::AF9 targets).
MLL::AF9 degradation induces changes in transcriptional elongation within minutes
Since we observed changes in expression and MLL::AF9 chromatin binding after 3 hours of dTAG-VHL treatment, we leveraged the rapid kinetics of the dTAG-system to perform shorter time courses to assess the kinetic relationship between MLL::AF9 degradation, changes in transcription, and changes in chromatin architecture. Notably, dTAG-VHL treatment led to significant MLL::AF9 degradation as early as 15 minutes and complete degradation at 30 minutes, as visualized by Western blot using an HA-directed antibody (Figure 3A). Similarly, ChIP-qPCR indicated that MLL::AF9 is displaced from chromatin at these early timepoints, with a ~50% reduction at 15 minutes and complete displacement at 30 minutes post dTAG-VHL treatment (Figure 3B). Importantly, this early loss of binding had immediate effects on gene expression. Standard RT-qPCR using Exon/Exon spanning primers only revealed reductions in expression of MEIS1 and HOXA9 at 60 minutes. However, Intron/Exon primers, which detect pre-spliced, newly transcribed RNA, revealed decreases in MEIS1/HOXA9 expression as early as 15 minutes following dTAG-VHL treatment (Figure 3C).
Figure 3. MLL::AF9 degradation induces transcriptional changes within minutes via increased RNA Polymerase II pausing in human MLL::AF9-FKBP12 cells.
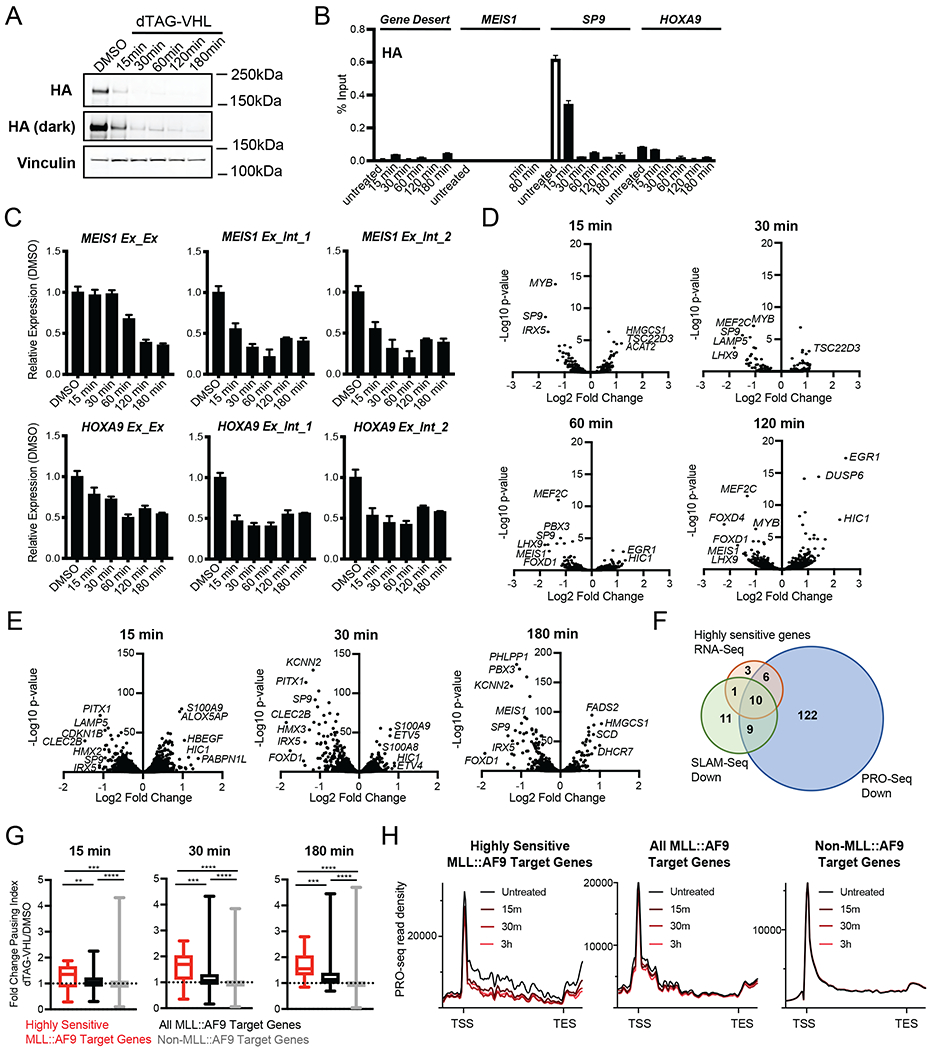
A. Western blot upon dTAG-VHL (500nM) treatment at the indicated timepoints. HA levels were assessed by immunoblotting.
B. ChIP-qPCR of HA (MLL::AF9) at the indicated genes plotted as percent Input following dTAG-VHL (500nM) treatment for the indicated timepoints. Data are represented as mean + SD of experimental triplicates.
C. Relative expression of MEIS1 and HOXA9 upon dTAG-VHL (500nM) treatment for the indicated timepoints, normalized to DMSO. Exon/Exon or Exon/Intron primers were used as indicated. Data are represented as mean + SD of three independent experiments, measured by SYBR green.
D. Volcano plots of differentially transcribed genes following 15, 30, 60, and 120 minutes of dTAG-VHL (500nM) treatment as measured by SLAM-Seq (Table S2).
E. Volcano plots of differentially transcribed genes following 15, 30, and 180 minutes of dTAG-VHL (500nM) treatment as measured by PRO-Seq (Table S2).
F. Venn diagrams depicting the overlap of genes exhibiting decreased transcription upon dTAG-VHL (500nM) treatment by RNA-seq (red, shown in Figure 2A, Table S1), SLAM-Seq (green, Log2 FC>1, p<0.05 at any of the four timepoints shown in Figure 3D, Table S2), and PRO-Seq (blue, FC>1.5, p<0.0001 at any of the three timepoints shown in Figure 3E, Table S2).
G. Boxplots of DMSO-normalized fold change in pausing index (see Figure S3E) after dTAG-VHL (500nM) treatment at the indicated timepoints.
H. Metagene plots of PRO-seq read density after dTAG-VHL (500nM) treatment.
ns p > 0.5; * p ≤ 0.5; * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.001 (Mann Whitney test)
Based on these findings, and since conventional RNA sequencing fails to identify rapid transcriptional changes that are masked by stable RNA species, we performed nascent RNA sequencing using SLAM-Seq (Muhar et al., 2018) following 15, 30, 60 and 120 minutes of dTAG-VHL treatment (Figure 3D) to define the nascent RNA expression changes following MLL::AF9 degradation. While the list of significantly differentially expressed genes (Log2 FC>1, p<0.05) at 15, 30 and 60 minutes was quite small (Table S2), many of the differentially expressed genes at these early timepoints were among the ‘highly sensitive genes’ identified in Figure 2A and over 90% are MLL::AF9 targes, as defined by highly bound MLL::AF9 at the promoter (Figure 2D). A heatmap analysis of all differentially expressed genes (Log2 FC>1, p<0.05) demonstrated that the majority of downregulated genes exhibited decreases in gene expression as early as 15 minutes (Figure S3A). In contrast, there were few upregulated genes at the early timepoints, and most gene induction occurred only after 120 minutes of treatment (Figure S3A). Moreover, while the overlap of significantly downregulated genes at the various timepoints was quite notable and included many of the genes that we identified in Figure 2A (e.g. SP9, PITX1, MEF2C, LHX1, MEIS1), there was almost no overlap of the upregulated genes, with only one common gene, EGR1, upregulated at two of the later timepoints, 60 and 120 minutes (Figure S3B).
Next, we performed PRO-Seq, a technique that reliably detects acute changes in RNA polymerase II dynamics following perturbations of transcription (Aoi et al., 2020; Kwak et al., 2013; Mahat et al., 2016; Stengel et al., 2021; Zhao et al., 2016). PRO-Seq studies at 15, 30 and 180 minutes after dTAG-VHL treatment identified a larger set of differentially transcribed genes (FC>1.5, p<0.0001, Table S2), with more genes downregulated than upregulated (Figure 3E). Evaluation of transcriptional changes at the MLL::AF9 target genes (Table S1) demonstrated that all MLL::AF9 targets exhibited decreased transcription at one of the timepoints (Figure S3C). Again, we observed significant overlap of downregulated genes at the three timepoints, with only modest overlap of upregulated genes (Figure S3D). Assessment of the overlap of downregulated genes identified by SLAM-Seq (Table S2), PRO-Seq (Table S2) and traditional RNA-Seq (Figure 2A, Table S1) not only showed that PRO-Seq is the most sensitive of the three methods, but also revealed that there is significant overlap (Figure 3F). Collectively, these findings demonstrated that MLL::AF9 is a transcriptional activator and upon MLL::AF9 degradation we observed decreased transcription of a biologically important subset of MLL::AF9 target genes as early as 15 minutes.
While previous studies have shown that MLL-fusion proteins can recruit members of the SEC (Lin et al., 2010; Mueller et al., 2007; Yokoyama et al., 2010; Zeisig et al., 2005), a direct functional role of MLL::AF9 in mediating transcriptional elongation has not yet been established. We therefore assessed changes in the RNA Pol II pausing index (Figure S3E) in response to dTAG-VHL treatment. MLL::AF9 degradation induced RNA Pol II pausing at all MLL::AF9 target genes, with significantly more pronounced pausing at the ‘highly sensitive genes,’ as early as 15 minutes upon dTAG-VHL treatment (Figure 3G). By 30 and 180 minutes of dTAG-VHL treatment these changes were even more prominent (Figure 3G). At non-MLL::AF9-bound genes we observed no changes in pausing index, indicating that pausing is specific to MLL::AF9 regulated genes (Figure 3G). A metaplot analysis of PRO-Seq read density along the full gene length demonstrated that RNA Pol II loss occurred predominantly in the gene body, with only modest changes in signal at the TSS. This is consistent with the notion that less RNA Pol II enters the gene body upon MLL::AF9 degradation (Figure 3H). In line with our previous findings, these effects are considerably more noticeable at the ‘highly sensitive genes’ (Figure 3H) as exemplified by MEIS1 (Figure S3F). In summary, we were able to leverage the rapid kinetics of the dTAG system to establish a set of MLL::AF9 target genes that were particularly sensitive to perturbations in MLL::AF9 and whose expression changed within minutes. Mechanistically, our findings demonstrated that MLL::AF9 promotes transcription by maintaining productive RNA Pol II elongation.
MLL::AF9 degradation induces loss of an active chromatin landscape at later timepoints
Next, we assessed whether MLL::AF9 degradation has immediate effects on MLL::AF9 complex members and the chromatin landscape more broadly. ChIP-qPCR at various MLL::AF9-bound loci revealed that DOT1L occupancy changes mirrored MLL::AF9 changes with ~50% loss at 15 minutes and almost complete loss at 30 minutes after MLL::AF9 degradation. MENIN loss was more sluggish, with robust changes only occurring after 3 hours of dTAG-VHL treatment (Figure 4A). Similarly, H3K27ac levels were not affected immediately, and showed a ~50% reduction after 3 hours of dTAG-VHL treatment (Figure 4A). Since transcriptional changes were observed as early as 15 minutes following MLL::AF9 degradation, these findings suggest that occupancy changes of certain MLL::AF9 complex members like MENIN and changes in chromatin state as measured by H3K27ac did not precede transcriptional changes.
Figure 4. Chromatin occupancy changes upon MLL::AF9 degradation in human MLL::AF9-FKBP12 cells.
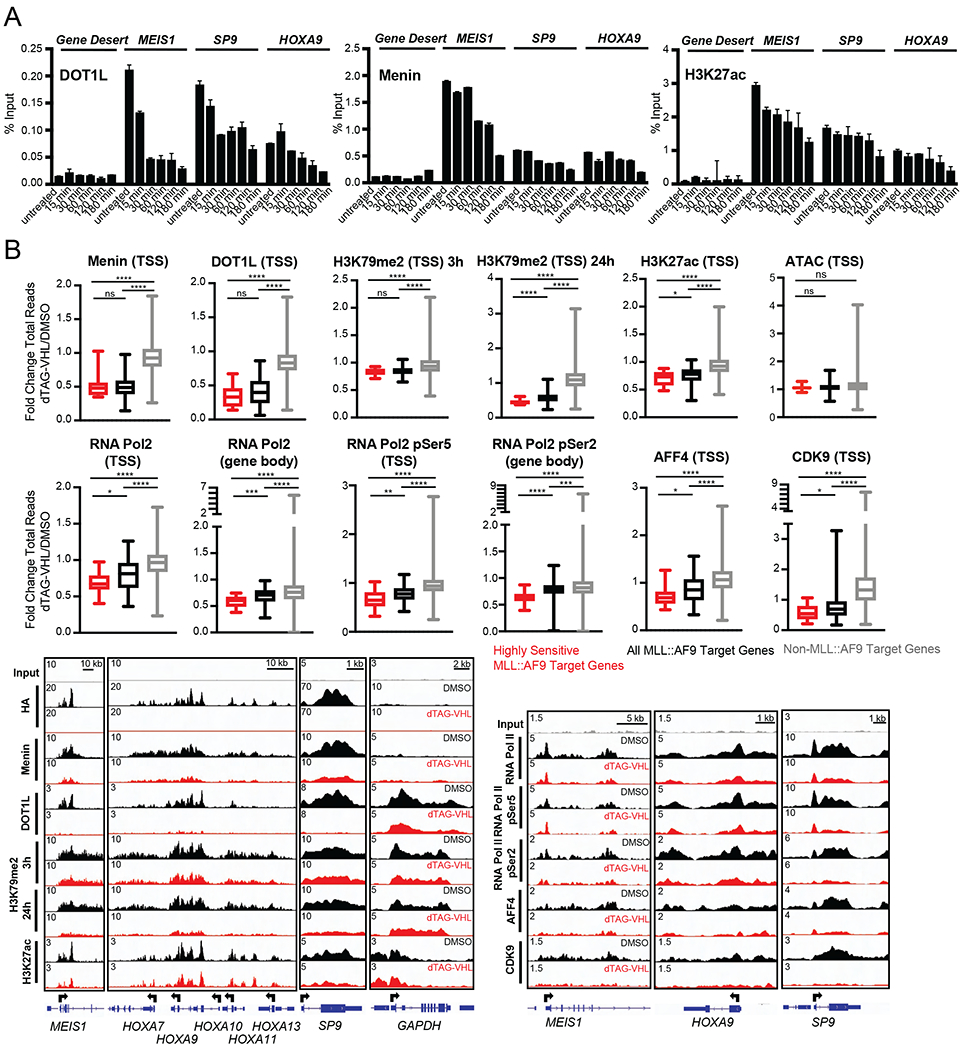
A. ChIP-qPCR of (left) DOT1L, (middle) MENIN, (right) H3K27ac at the indicated genes plotted as percent Input following dTAG-VHL (500nM) treatment for the indicated timepoints. Data are represented as mean + SD of experimental triplicates.
B. (Top) Boxplots of fold change oftotal reads for MENIN, DOT1L, H3K79me2, H3K27ac, ATAC-Seq, RNA Pol II, RNA Pol II S2P, AFF4, CDK9, and RNA Pol II S5P at promoters (TSS -1kB/+3kB, marked as TSS) or gene bodies as indicated, 3 hours upon dTAG-VHL (500nM) treatment, unless otherwise indicated. (Bottom) Gene tracks of ChIP-seq signal at the indicated genes, 3 hours upon dTAG-VHL (500nM) treatment, unless otherwise indicated. The gene tracks were normalized to coverage data (total read counts) using IGV.
ns p > 0.5; * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.001 (Mann Whitney test)
With the kinetics more clearly defined, we assessed chromatin changes after MLL::AF9 degradation more broadly. We performed ChIP-Seq of selected members of the MLL::AF9 protein complex and selected histone modifications after 3 hours of MLL::AF9 degradation, the earliest timepoint at which we saw robust loss of MENIN and H3K27ac by ChIP-qPCR (Figure 4A). After 3 hours of dTAG-VHL treatment, more than 50% of MENIN and almost 70% of DOT1L was lost at MLL::AF9 target genes, while there were no changes in occupancy at non-MLL::AF9-bound genes (Figure 4B). The loss was more prominent at the ‘highly sensitive genes’, compared to all MLL::AF9-bound genes. While we saw DOT1L loss at MLL::AF9 target genes after only 3 hours of dTAG-VHL treatment, we did not see major changes in H3K79me2 at this early timepoint. However, after 24 hours of dTAG-VHL treatment there was a marked reduction of H3K79me2 at the MLL::AF9 target genes, again with more prominent loss at the ‘highly sensitive genes’ (Figure 4B). Of note, total levels of H3K79me2 were significantly higher at MLL::AF9 target genes compared to non-MLL::AF9 bound genes, with generally even higher levels at the ‘highly sensitive genes’ (Figure S4A). Next, we probed histone modifications associated with active transcription, including H3K27ac and H3K9ac, as well as chromatin accessibility. While we did not observe changes in histone acetylation at non MLL::AF9-bound genes, MLL::AF9 degradation led to significant reductions of H3K27ac and H3K9ac at promoters of MLL::AF9 target genes after 3 hours of dTAG-VHL treatment (Figure 4B and Figure S4B) and even more robust reductions after 24 hours of treatment (Figure S4B). Like our observations with MENIN, DOT1L, and H3K79me2, the ‘highly sensitive genes’ exhibited more pronounced reductions compared to all other MLL::AF9 target genes for both H3K27ac as well as H3K9ac. Unlike H3K79me2, the total levels of H3K27acand H3K9ac were similar at the ‘highly sensitive genes’ and all other MLL::AF9 bound genes, yet both exhibited higher levels of acetylation than non-MLL::AF9 bound genes (Figure S4C). Since H3K27ac and H3K9ac are tightly associated with an active chromatin state, we assessed whether MLL::AF9 degradation also induced changes in chromatin accessibility via assay for transposase-accessible chromatin using sequencing (ATAC-Seq). Interestingly, despite reductions of H3K27ac and H3K9ac, we did not observe changes in chromatin accessibility (Figure 4B and Figure S4D) after 3 hours of dTAG-VHL treatment. This lack of change was not due to compensatory effects of wild-type MLL1: 3 hours of dTAG-VHL treatment had no effect on H3K4me3 levels – the mark deposited by MLL1 - at either MLL::AF9 target genes or at non-MLL::AF9 bound genes (Figure S4E). Similarly, ChIP-seq using an antibody recognizing N-terminal MLL1 (which recognizes MLL1 and the MLL::AF9 fusion protein) and C-terminal MLL1 (which only recognizes MLL1) revealed that there were only reductions in MLL::AF9 but not in wild-type MLL1 occupancy at MLL::AF9 target genes (Figure S4F). Collectively, these data showed that MLL::AF9 degradation led to loss of MLL::AF9 complex proteins from chromatin as well as reductions of histone modifications associated with active transcription after three hours but had no corresponding effects on chromatin accessibility. Moreover, there was unlikely to be a compensatory role for wild-type MLL1 at MLL::AF9 target genes following MLL::AF9 degradation.
Since previous work established that MLL::AF9 can directly recruit the SEC (Mueller et al., 2007; Zeisig et al., 2005) and our PRO-Seq data demonstrated that MLL::AF9 directly mediates productive RNA Pol II elongation (Figure 3), we assessed whether there are occupancy changes in the RNA Pol II machinery upon MLL::AF9 degradation. Indeed, after 3 hours of dTAG-VHL treatment, we observed loss of RNA Pol II and pSer2 RNA Pol II throughout the gene body of MLL::AF9 target genes (Figure 4B). RPL3 and TRIM28 tracks confirmed some specificity of the pSer2 RNA Pol II signal, but also revealed some cross-reactivity (Figure S4G). AFF4, a scaffolding protein of the SEC, as well as CDK9, the catalytic subunit of the SEC complex (Lin et al., 2010; Yokoyama et al., 2010), were also specifically lost at promoters of MLL::AF9 target genes (Figure 4B), with more pronounced loss at the ‘highly sensitive genes.’ In addition to effects on SEC occupancy and transcriptional elongation, we also observed modest reductions of RNA Pol II and pSer5 RNA Pol II (marker of transcriptional initiation) at the TSS of MLL::AF9 target genes after 3 hours of dTAG-VHL treatment (Figure 4B). In summary, MLL::AF9 degradation led to loss of a transcriptionally active chromatin state characterized by loss of histone acetylation, MLL::AF9 associated proteins, and the transcriptional machinery itself. At specific loci, this occurred after an immediate decrease in transcriptional elongation.
Combination of DOT1L/MENIN inhibition induces rapid loss of MLL::AF9 from chromatin
The MLL::AF9 degradation system allowed us to compare direct degradation of MLL::AF9 versus small molecule inhibition of DOT1L and MENIN (Figure 5A) (Daigle et al., 2013; Krivtsov et al., 2019). It is well established that DOT1L and MENIN inhibition synergizes to inhibit cell proliferation (Dafflon et al., 2017; Kühn et al., 2016; Okuda et al., 2017), but the mechanism behind this synergy remains largely unknown (Figure 5A). We hypothesized that the EPZ-5676/VTP-50469 combination phenocopies MLL::AF9 degradation by cooperating to displace MLL::AF9 from chromatin faster and more robustly, thus inducing more rapid gene expression and cellular changes than single agent treatment.
Figure 5. Combination of DOT1L and MENIN inhibition induces more rapid MLL::AF9 chromatin loss and gene expression changes compared to single agent treatment.
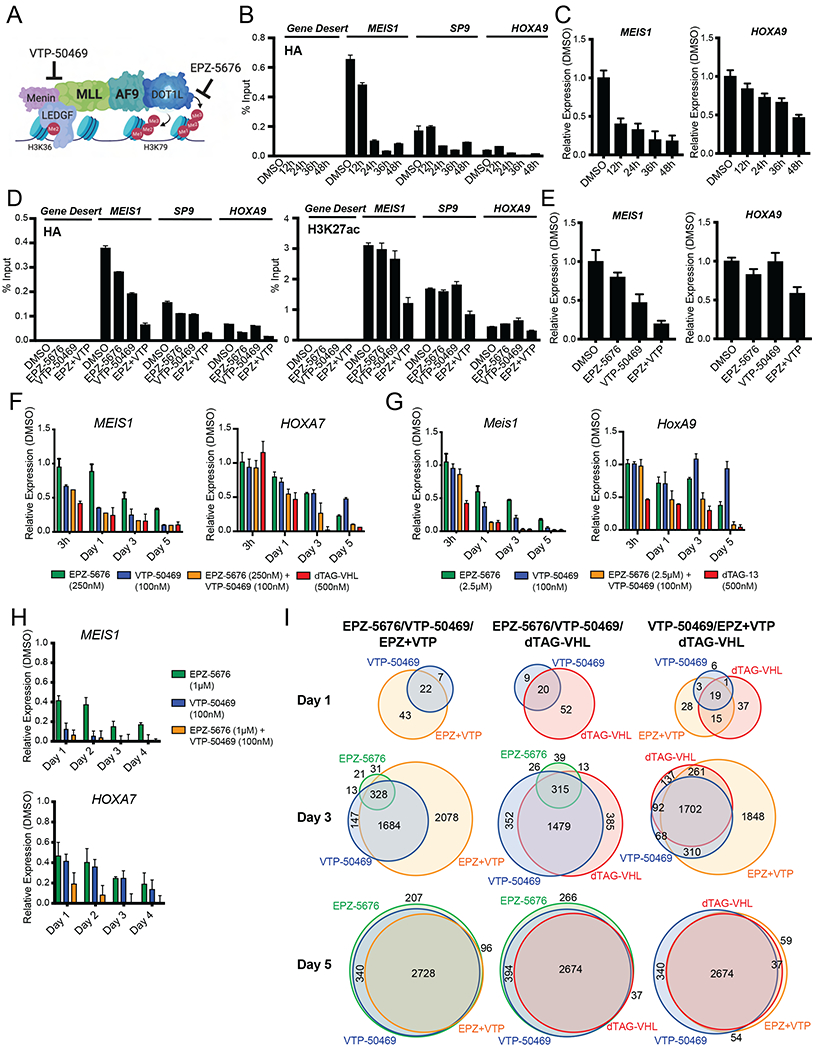
A. Model of the MLL::AF9 multi-protein complex and where EPZ-5676 and VTP-50469 target complex members.
B. ChIP-qPCR of HA (MLL::AF9) at the indicated genes plotted as percent Input following EPZ-5676 (250nM) + VTP-50469 (100nM) treatment for the indicated timepoints. Data are represented as mean + SD of experimental triplicates in the human MLL::AF9-FKBP12 system, measured by SYBR green.
C. Relative expression of MEIS1 and HOXA9 upon EPZ-5676 (250nM) + VTP-50469 (100nM) treatment for the indicated timepoints, normalized to DMSO. Data are represented as mean + SD of experimental triplicates in the human MLL::AF9-FKBP12 system, measured by SYBR green.
D. ChIP-qPCR of (left) HA (MLL::AF9) and (right) H3K27ac at the indicated genes plotted as percent Input following DMSO, EPZ-5676 (250nM), VTP-50469 (100nM), or the combination of EPZ-5676 (250nM) + VTP-50469 (100nM) treatment for 24 hours. Data are represented as mean + SD of experimental triplicates in the human MLL::AF9-FKBP12 system, measured by SYBR green.
E. Relative expression of MEIS1 and HOXA9 upon DMSO, EPZ-5676 (250nM), VTP-50469 (100nM), or the combination of EPZ-5676 (250nM) + VTP-50469 (100nM) treatment for 24 hours, normalized to DMSO. Data are represented as mean + SD of experimental triplicates in the human MLL::AF9-FKBP12 system, measured by SYBR green.
F. Relative expression of MEIS1 and HOXA7 in MLL::AF9-HA-FKBP12 human cells, normalized to DMSO. Data are represented as mean + SD of three independent experiments, measured by Taqman.
G. Relative expression of Meis1 and HoxA9 in MLL::AF9-HA-FKBP12 murine cells, normalized to DMSO treatment. Data are represented as mean + SD of three independent experiments, measured by Taqman.
H. Relative expression of MEIS1 and HOXA7 in MOLM13 cells, normalized to DMSO treatment. Data are represented as mean + SD of three independent experiments, measured by Taqman.
I. Venn diagrams of RNA-Seq from human MLL::AF9-FKBP12 cells depicting the overlap of genes with increased or decreased transcription (FC>2, p<0.05, Table S3) following treatment with EPZ-5676 (250nM, green), VTP-50469 (100nM, blue), EPZ-5676 (250nM) + VTP-50469 (100nM, orange) or dTAG-VHL (500nM, red) at the indicated timepoints.
First, we confirmed that combination treatment with EPZ-5676 and VTP-50469 showed enhanced effects on cell proliferation. We observed strong cooperativity on cell growth in the human MLL::AF9 transformed CD34+ cells (Figure S5A), as well as human MLL::AF9 rearranged MOLM13 cells (Figure S5B), with no toxicity to human hematopoietic stem and progenitor (CD34+) cells (Figure S5C) or non-/MLL-rearranged HL60 cells (Figure S5D). Notably, proliferation (Figure S5E), differentiation (Figure S5F, top) and apoptosis (Figure S5F, bottom) time course experiments showed that the EPZ-5676/VTP-50469 combination also had more rapid effects than the single agents. Specifically, the combination induced cellular effects with comparable kinetics to dTAG-VHL treatment (Figure S5E and F). Moreover, co-treatment with the pan-caspase inhibitor Z-VAD-FMK rescued dTAG-VHL and EPZ-5676/VTP-50469-induced cell death, supporting the notion that dTAG-VHL and EPZ-5676/VTP-50469 work in similar ways by inducing early caspase-mediated apoptosis (Figure S5G and H).
Since the EPZ-5676/VTP-50469 combination had faster cellular effects than single agent treatment, we next assessed the kinetics of MLL::AF9 displacement with the combination of EPZ-5676/VTP-50469. Combination treatment led to modest reductions in MLL::AF9 occupancy at MEIS1 after 12 hours and complete loss from chromatin at MEIS1, SP9 and HOXA9 after 24 hours as measured by ChIP-qPCR using an HA-directed antibody (Figure 5B). Displacement from chromatin had implications for gene expression: MEIS1 RNA was reduced at 12 hours post EPZ-5676/VTP-50469 treatment in line with early MLL::AF9 loss from MEIS1 (Figure 5B and C). Because we observed consistent MLL::AF9 displacement from chromatin with combination treatment after 24 hours, we assessed the effects of single agent EPZ-5676 and VTP-50469 treatment on MLL::AF9 occupancy by ChIP-qPCR after 24 hours. While EPZ-5676 and VTP-50469 had some effect on MLL::AF9 occupancy and gene expression at 24 hours, the effects were modest (Figure 5D and 5E). Only the combination was able to completely displace MLL::AF9 from chromatin and suppress gene expression robustly (Figure 5D and 5E). Similarly, only the combination led to reduced H3K27ac levels at 24 hours (Figure 5D).
To dissect the kinetics of gene expression changes induced by either single agent or combination EPZ-5676/VTP-50469 treatment, we performed RT-qPCR time course experiments in the human and murine FKBP12-MLL::AF9 systems (Figure 5F and G, respectively) as well as in MOLM13 cells (Figure 5H). In all three systems did the EPZ-5676/VTP-50469 combination have faster and more robust effects than single agents. To assess the kinetics of gene expression changes more globally and to assess potential off-target effects of DOT1L/MENIN inhibition in the human MLL::AF9 FKBP12 system, we performed RNA-Seq after 1, 3, and 5 days of single agent or combination treatment and analyzed the full overlap of differentially expressed genes (FC>2, p<0.05, Table S3). Consistent with the RT-qPCR results, this analysis demonstrated that the EPZ-5676/VTP-50469 combination had more rapid effects on gene expression than single agent treatment (Figure 5I). Moreover, these weren’t “off target” effects since after 5 days of EPZ-5676/VTP-50469 or single agent treatment the differentially expressed genes were almost identical to dTAG-VHL treatment (Figure 5I, Table S3). These findings indicated that while the EPZ-5676/VTP-50469 combination did not necessarily affect a larger set of genes, it affected gene expression changes more rapidly than single agent treatment.
Combination of DOT1L/MENIN inhibition induces global MLL::AF9 chromatin dissociation
In addition to affecting MLL::AF9 chromatin occupancy and gene expression changes more rapidly, we hypothesized that the EPZ-5676/VTP-50469 combination might also affect MLL::AF9 occupancy more globally. Since EPZ-5676 and VTP-50469 treatment had more delayed effects than the combination treatment (Figure 5), we assessed MLL::AF9 occupancy after 4 days of either single agent or combination treatment to measure the maximum effect of single agent treatment. We confirmed that the overall chromatin architecture and gene expression remained intact in the various treatment conditions at this timepoint by assessing H3K27ac ChIP-Seq and RNA-Seq at housekeeping genes, as exemplified by ACTB (Figure S6A). As hypothesized, we observed strong cooperativity of EPZ-5676 and VTP-50469: the EPZ-5676/VTP-50469 combination led to global loss of MLL::AF9 binding to chromatin, similar to treatment with dTAG-VHL, whereas EPZ-5676 and VTP-50469 treatment alone had more modest effects (Figure 6A). At certain loci where single agent treatment had no effect on MLL::AF9 occupancy, like SP9, APOLD1, and CDKN2C, the combination treatment led to complete loss of MLL::AF9 from chromatin (Figure 6B). Importantly, only the combination was able to suppress expression of those genes (Figure 6C). Additionally, looking at all MLL::AF9 target genes as defined by highly bound MLL::AF9 at the promoter, we found that MLL::AF9 loss from chromatin was more pronounced upon combination treatment compared to single agent treatment and the extent of MLL::AF9 loss from chromatin in the combination treatment closely resembled the extent observed upon MLL::AF9 degradation (Figure 6D), a phenomenon nicely illustrated at the HOXA locus (Figure 6B and Figure S6A).
Figure 6. Combination of DOT1L and MENIN inhibition displaces MLL::AF9 from chromatin globally in human MLL::AF9-FKBP12 cells.
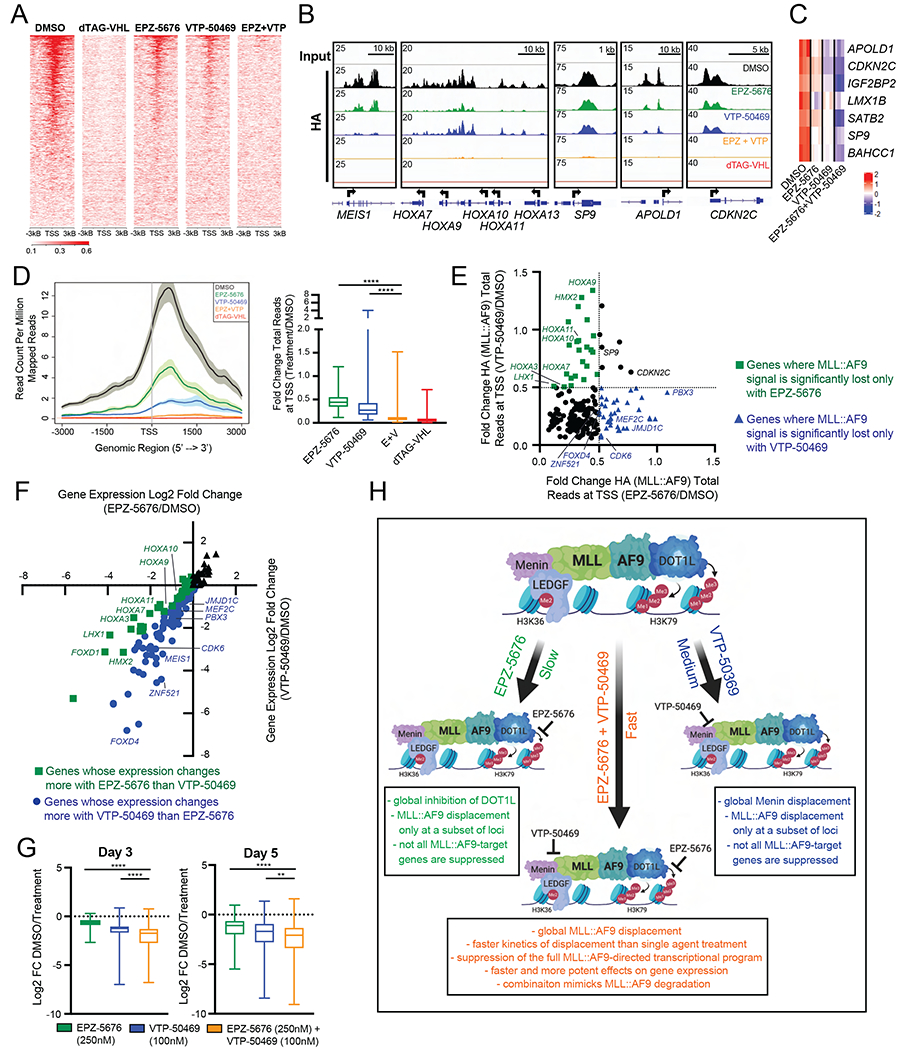
A. Genome wide rank-ordered heat map of HA (MLL::AF9) ChIP signal across the promoter (TSS -3kB/+3kB), 4 days following DMSO, dTAG-VHL (500nM), EPZ-5676 (250nM), VTP-50469 (100nM) and EPZ-5676 (250nM) + VTP-50469 (100nM) treatment.
B. Gene tracks of HA (MLL::AF9) ChIP-seq signal at representative genes following 4 days of DMSO (black), EPZ-5676 (250nM), VTP-50469 (100nM), EPZ-5676 (250nM) + VTP-50469 (100nM) or dTAG-VHL (500nM) treatment. Gene tracks were normalized to coverage data (total read counts) using IGV.
C. Heatmap of genes 5 days upon treatment with DMSO, EPZ-5676 (250nM), VTP-50469 (100nM), or the combination EPZ-5676 (250nM) + VTP-50469 (100nM).
D. 4 days of treatment with EPZ-5676 (250nM), VTP-50469 (100nM), EPZ-5676 (250nM) + VTP-50469 (100nM), or dTAG-VHL (500nM). (Left) Average/meta plot of HA (MLL::AF9) ChIP reads across the promoter (TSS -3kB/+3kB) of MLL::AF9 target genes (Table S1). (Right) Boxplot of fold change in ChIP-seq reads for HA (MLL::AF9) at MLL::AF9 target genes (Table S1) around the TSS (TSS -1kB/+3kB).
E. Fold change of HA (MLL::AF9) total reads at promoters (TSS -1kB/+3kB) of MLL::AF9 target genes (Table S1) 4 days upon EPZ-5676 (250nM) and VTP-50469 (100nM) treatment, relative to DMSO (Table S4).
F. Log2 fold change of gene expression of MLL::AF9 target genes (Table S1) induced by EPZ-5676 (250nM) or VTP-50469 (100nM), relative to DMSO 5 days upon treatment (Table S4).
G. Boxplots of DMSO-normalized Log2 fold change RNA-Seq values of MLL::AF9 target genes.
H. Model of how DOT1L and MENIN inhibition cooperate to induce cellular changes. Created with BioRender.com
ns p > 0.5; * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.001 (Mann Whitney test)
To determine whether DOT1L and MENIN inhibition also cooperate in destabilizing MLL::AF9 on chromatin in non-engineered cell systems, we assessed the human MLL::AF9 rearranged MOLM13 cell line. While single agent EPZ-5676 and VTP-50469 treatment had some effect on MLL occupancy at MLL::AF9 target genes (Table S4, definition of MLL::AF9 target genes in MOLM13 cells can be found in the Methods section), the combination treatment almost completely evicted MLL from chromatin (Figure S6B and C). Moreover, not only was the loss of MLL more pronounced in the combination, but the combination treatment also induced MLL dissociation from chromatin at loci that were relatively resistant to single agent treatment, again a phenomenon nicely illustrated at the HOXA locus (Figure S6B). Collectively, these studies revealed that single agent DOT1L and MENIN inhibition influence MLL::AF9 occupancy much less than the EPZ-5676/VTP-50469 combination treatment.
To further assess single agent EPZ-5676 and VTP-50469 treatment, we closely examined MLL::AF9 occupancy changes at MLL::AF9 target genes in response to single agent treatment in the human MLL::AF9 FKBP12 system. While EPZ-5676 and VTP-50469 treatment led to a reduction of MLL::AF9 occupancy at a shared subset of genes, at many genes EPZ-5676 and VTP-50469 had distinct and separable effects on MLL::AF9 occupancy (Figure 6E and Table S4) and gene expression (Figure 6F and Table S4). Neither DOT1L nor MENIN inhibition alone completely disrupted MLL::AF9 occupancy at all loci or suppressed expression of all MLL::AF9 target genes. In contrast, the combination induced global MLL::AF9 chromatin loss (Figure 6A) and led to more robust gene suppression (Figure 6G). While the single agent DOT 1L and MENIN inhibitor studies do not directly address the cause-and-effect relationship of MLL::AF9 occupancy and gene expression changes, our data nonetheless demonstrate that the combination more closely phenocopied MLL::AF9 degradation (Figure 6H).
DISCUSSION
PROTAC technologies are providing new insight across all areas of biology. A particularly powerful use of PROTACs is to engineer proteins for small molecule mediated degradation to assess the effects of rapid target degradation (Jaeger and Winter, 2021). The fast kinetics of degradation, compared to gene editing tools, allows for the identification of direct and primary effects while avoiding the confounding consequences of secondary and compensatory changes. This is particularly relevant in the context of a transcriptional regulator like the MLL::AF9 fusion oncoprotein. Since MLL::AF9 drives the expression of genes like MEIS1, PBX3 and the HOXA genes, which themselves are transcription factors, it has been difficult to conclusively define the MLL::AF9-directed transcriptional program and the direct effects of MLL::AF9 inactivation. By coupling rapid degradation of MLL::AF9 with genome-wide transcriptional and chromatin occupancy assays, we were able to define the direct targets of MLL::AF9 and assess the immediate gene expression and chromatin state consequences of MLL::AF9 degradation. Notably, we identified a small subset of MLL::AF9 bound genes that are particularly sensitive to MLL::AF9 degradation, many of which are key targets in MLL-rearranged leukemia, like MEIS1, MEF2C, PBX3 and HOXA9. Interestingly, these ‘highly sensitive genes’ have on average twice as much promoter-bound MLL::AF9, compared to all other MLL::AF9 target genes. They also exhibit higher levels of activating histone modifications like H3K79me2 at their promoters and higher levels of RNA Pol II throughout their gene body. Moreover, on average, the ‘highly sensitive genes’ have higher PRO-Seq read densities compared to all other MLL::AF9 target genes. These observations, higher promoter load of MLL::AF9, higher levels of H3K79me2, higher load of RNA Pol II, and higher PRO-Seq read density indicate that these genes are transcribed at very high rates, providing a possible explanation for why they are particularly dependent on continued MLL::AF9 occupancy. Moreover, we speculate that many of the other ~180 highly bound MLL::AF9 loci that exhibit more modest immediate changes in gene expression rely on other regulatory mechanisms to maintain gene expression in the short term (15-30 minutes) but are dependent on MLL::AF9 for continued expression as shown by robust changes in gene expression after 24 hours.
Leveraging the fast kinetics of the dTAG system we were able to gain new insight into how MLL::AF9 mediates transcription and leukemogenic chromatin architecture. We found that MLL::AF9 directly maintains transcription and MLL::AF9 degradation induces rapid decreases in RNA Pol II elongation at the ‘highly sensitive genes’ as early as 15 minutes upon dTAG treatment. At later timepoints, namely 3 hours after MLL::AF9 degradation, loss of the fusion protein also induced loss of an active chromatin landscape at MLL::AF9 target genes. Interestingly, these studies revealed that DOT1L dissociates from chromatin faster than MENIN upon MLL::AF9 degradation. This may be because the AF9 fusion partner directly binds and recruits DOT1L to chromatin (Biswas et al.,2011; Kuntimaddi et al., 2015). In contrast to DOT1L, MENIN is thought to be tethered to chromatin via LEDGF (Yokoyama and Cleary, 2008) independently of MLL::AF9. Additional and/or compensatory anchorage by LEDGF could explain why MENIN dissociates from chromatin more slowly upon MLL::AF9 degradation. Nonetheless, our studies indicate that MLL::AF9 contributes to retaining MENIN on chromatin at certain loci, suggesting that there are multiple interactions that maintain MENIN occupancy, a point that is relevant as MENIN-directed drugs continue to be developed.
In addition to yielding basic insight into the MLL::AF9 driven transcriptional and epigenetic program, the dTAG approach also allowed us to characterize the similarities and differences between direct degradation of MLL::AF9 versus indirect targeting of the fusion protein complex via catalytic inhibition of DOT1L (EPZ-5676) and inhibition of the MENIN:MLL protein:protein interaction (VTP-50469). Notably, only the combination of EPZ-5676 and VTP-50469 induced MLL::AF9 chromatin loss with similar kinetics as MLL::AF9 degradation and only the combination evicted MLL::AF9 from chromatin globally. This had relevant implications for MLL::AF9-directed gene expression as well as important cellular consequences: to date, DOT1L and MENIN inhibitors have primarily induced cell differentiation with only modest induction of apoptosis at late timepoints of treatment (Borkin et al., 2015; Daigle et al., 2013). In contrast, in this study we observe potent induction of apoptosis after only 2 to 3 days of EPZ-5676/VTP-50469 combination treatment, mimicking apoptosis-induction upon MLL::AF9 degradation. These findings indicate that the kinetics and the extent of destabilization of the MLL::AF9 protein complex may be critical to drive cellular consequences. Importantly, this could have relevant clinical implications - despite exciting evidence that DOT1L and MENIN inhibitors have anti-leukemic effects, the responses may not be long-lasting thus leading to resistance. The fact that single agent treatments fail to phenocopy MLL::AF9 degradation may present a mechanistic rational for assessing the combination of DOT1L and MENIN inhibitors in MLL-rearranged leukemias in patients with this devastating disease.
Limitations of the Study
A potential limitation of this work is that it uses retroviral overexpression to define the MLL::AF9 directed transcriptional program. In both systems two wild-type copies of MLL1 remain, which could play compensatory roles upon MLL::AF9 degradation. We tried to address this by looking at occupancy changes of wild-type MLL1 and H3K4me3, the modification deposited by wild-type MLL1. While we did not observe any compensation from the wild-type MLL1 complex, future studies should include endogenously degron-tagged MLL::AF9. Another potential caveat is the difficulty comparing dTAG treatment to DOT1L and MENIN inhibition due to their different kinetics. Despite being small molecule inhibitors, EPZ-5676 and VTP-50469 have relatively slow effects on gene expression. EPZ-5676 and VTP-50469-induced chromatin and gene expression changes are typically measured after a few days and significant proliferative changes are usually found after 4 to 9 days (Krivtsov et al., 2019; Perner et al., 2020). Thus, by the time these drugs have robust effects on gene expression, dTAG-treated cells have undergone state changes and differentiation, making direct comparisons difficult. We attempted to circumvent this problem by performing detailed phenotypic and gene expression time course experiments.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Scott A. Armstrong (scott_armstrong@dfci.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
RNA-seq, ChIP-seq, ATAC-seq, SLAM-seq, and PRO-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Publicly available data used for GSEA analyses are listed in the key resource table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-HA, rabbit | Cell Signaling Technology | Cat# 3724 (C29F4) |
| anti-cleaved PARP (Asp214), rabbit | Cell Signaling Technology | Cat# 5625 (D64E10) |
| anti-PARP, rabbit | Cell Signaling Technology | Cat# 9542 |
| anti-GAPDH, mouse | Cell Signaling Technology | Cat# 97166 (D4C6R) |
| anti-Vinculin, rabbit | Cell Signaling Technology | Cat# 13901 (E1E9V) |
| anti-KMT2A/MLL1, rabbit | Bethyl | Cat# A300-086A |
| anti-MLL/HRX, rabbit | Active Motif | Cat# 61296 |
| anti-MENIN, rabbit | Bethyl | Cat# A300-105A |
| anti-DOT1L, rabbit | Cell Signaling Technology | Cat# 77087 (D1W4Z) |
| anti-H3K79me2, rabbit | Cell Signaling Technology | Cat# 5427 (D15E8) |
| anti-H3K27ac, rabbit | Diagenode | Cat# C15410196 |
| anti-H3K9ac, rabbit | Abcam | Cat# ab4441 |
| anti-H3K4me3, rabbit | Abcam | Cat# ab8580 |
| anti-RNA Pol II (Rpb1 CTD), mouse | Cell Signaling Technology | Cat# 2629 (4H8) |
| anti-RNA Pol II pSer2 (Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2)), rabbit | Abcam | Cat# ab5095 |
| anti-RNA Pol II pSer5 (Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5)), rabbit | Abcam | Cat# ab5131 |
| anti-AFF4/MCEF | Bethyl | Cat# A302-538A |
| anti-CDK9, mouse | Santa Cruz Biotechnology | Cat# SC-13130 |
| anti-human CD11b, APC-conjugated | BioLegend | Cat# 301309 (ICRF44) |
| anti-human CD13, PerCP/Cy5.5-conjugated | BioLegend | Cat# 301714 (WM15) |
| anti-human CD14, PE/Cy7-conjugated | BioLegend | Cat# 301814 (M5E2) |
| anti-mouse sca-1, PE/Cy7 | Invitrogen | Cat# 25-5981-82 (D7) |
| anti-mouse CD117 (c-Kit), Alexa Fluor® 647 | BioLegend | Cat# 105818 (2B8) |
| Rabbit IgG, HRP-linked | Cytiva | Cat# NA934V |
| anti-rabbit, Alexa Fluor®647-Conjugate | Cell Signaling Technology | Cat# 4414 |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine Serum Albumin | Sigma Aldrich | Cat# A7906 |
| UltraPure BSA (50 mg/mL) | Invitrogen | Cat# AM2616 |
| EPZ-5676 | Selleckchem | Cat# S7062 |
| VTP-50469 | Syndax | provided by Syndax Pharmaceuticals |
| dTAG-13 | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dNEG-13 | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dTAG-VHL | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dNEG-VHL | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| Z-VAD-FMK | Selleckchem | Cat# 7023 |
| DSG (disuccinimidyl glutarate) | Thermo Fisher Scientific | Cat# 20593 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28906 |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | Cat# D2650 |
| Biotin-11-ATP | PerkinElmer | NEL544001EA |
| Biotin-11-CTP | PerkinElmer | NEL542001EA |
| Biotin-11-GTP | PerkinElmer | NEL545001EA |
| Biotin-11-UTP | PerkinElmer | NEL543001EA |
| Critical commercial assays | ||
| Amicon Ultra-15 | Millipore | Cat# UFC910024 |
| X-tremeGene 9 DNA Transfection Reagent | Millipore | Cat #06365809001 |
| Polybrene Transfection Reagent | Millipore | Cat# TR-1003-G |
| RetroNectin GMP grade | Takara | Cat# T202 |
| Cell Dissociation Buffer Enzyme-Free | Gibco | Cat# 13151-014 |
| EasySep Mouse Neutrophil Enrichment Kit | StemCell Technologies | Cat# 19762 |
| EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit | StemCell Technologies | Cat# 19856 |
| StemSpan SFEM | StemCell Technologies | Cat# 09600 |
| Mouse Recombinant SCF | StemCell Technologies | Cat# 78064 |
| Mouse Recombinant Flt3/Flk-2 Ligand | StemCell Technologies | Cat# 78011 |
| Mouse Recombinant TPO | StemCell Technologies | Cat# 78072 |
| Mouse Recombinant IL-3 | StemCell Technologies | Cat# 78042 |
| Mouse Recombinant IL-6 | StemCell Technologies | Cat# 78052 |
| Human Recombinant SCF | StemCell Technologies | Cat# 78062 |
| Human Recombinant Flt3/Flk-2 Ligand | StemCell Technologies | Cat# 78009 |
| Human Recombinant TPO | StemCell Technologies | Cat# 78210 |
| Human Recombinant IL-3, ACF | StemCell Technologies | Cat# 78146 |
| Human Recombinant IL-6 | StemCell Technologies | Cat# 78050 |
| BD Pharm Lyse Buffer | BD Biosciences | Cat# 555899 |
| EasySep Human Cord Blood CD34 Positive Selection Kit II | StemCell Technologies | Cat# 17896 |
| Lymphoprep | StemCell Technologies | Cat# 07801 |
| SepMate-50 (IVD) | StemCell Technologies | Cat# 85450 |
| DAPI | Santa Cruz Biotechnology | Cat# 28718-90-3 |
| JorVet DipQuick Stain | Jorgensen Laboratories, Inc. | Cat# J-322 |
| Annexin V Apoptosis Detection Kit APC | eBioscience | Cat# 88-8007-74 |
| BD Cytofix/Cytoperm | BD Biosciences | Cat# 554722 |
| BD Perm/Wash | BD Biosciences | Cat# 554723 |
| BD Permeabilization Buffer Plus | BD Biosciences | Cat# 561651 |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen | Cat# NP0007 |
| Precision Plus Dual Color Standards | BioRad | Cat #12620S |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34095 |
| RNeasy Mini RNA Extraction Kit | Qiagen | Cat# 74106 |
| RNase-Free DNase | Qiagen | Cat# 79254 |
| SuperScript™ First-Strand Synthesis System | Invitrogen | Cat# 11904018 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat# 4369542 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Glycogen, ultrapure | Thermo Fisher Scientific | Cat# 10814010 |
| mRNA decapping enzyme (MDE) | New England Biolabs | Cat# M0608S |
| NEBNext Ultra II Q5 Master mix | New England Biolabs | Cat# M0544L |
| Q5 DNA polymerase (using included high GC enhancer) | New England Biolabs | Cat# M0491L |
| SUPERase-In RNase inhibitor | Thermo Fisher Scientific | Cat# AM2696 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18090200 |
| SYBR Gold nucleic acid stain | Thermo Fisher Scientific | Cat# S11494 |
| T4 Polynucleotide kinase | New England Biolabs | Cat# M0201L |
| T4 RNA Ligase 1 (High Conc.) | New England Biolabs | Cat# M0204L |
| T4 RNA Ligase 2, truncated KQ | New England Biolabs | Cat# M0373L |
| Trizol Reagent | Thermo Fisher Scientific | Cat# 15596026 |
| Pronex Size-Selective Purification System | Promega | Cat# NG2001 |
| Total RNA purification kit | Norgen Biotek Corp | Cat# 17250 |
| Dynabeads MyOne Streptavidin C1 beads | Thermo Fisher Scientific | Cat# 65002 |
| NEB 5′ DNA adenylation kit | New England Biolabs | Cat# E2610S |
| NEBNext Library Quant Kit for Illumina | New England Biolabs | Cat# E7630L |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7530 |
| SLAMseq Kinetics Kit - Anabolic Kinetics Module | Lexogen | Cat# 061.24 |
| RNeasy Plus Mini RNA Extraction Kit | Qiagen | Cat# 74134 |
| QIAshredder | Qiagen | Cat# 79654 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | Cat # 20034197 M0541L |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat # M0541L |
| QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina | Lexogen | Cat# 015.96 |
| Ampure XP | Beckman Coulter | Cat# A63880 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| ThruPlex DNA-seq Kit | Takara | Cat# R400675 |
| Dynabeads Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat #10001D |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat #10003D |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat #Q32851 |
| D5000 Reagents | Agilent | Cat# 5067-5589 |
| D5000 Screen Tape | Agilent | Cat# 5067-5588 |
| High Sensitivity D5000 Reagents | Agilent | Cat# 5067-5593 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067-5592 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| NextSeq 500 High Output v2 | Illumina | Cat #FC-404-2005 |
| Deposited data | ||
| Mendeley Dataset DOI URL | This paper | doi: 10.17632/n55f7xfx79.1 |
| GEO Superseries | This paper | GSE173600 |
| Raw and analyzed RNA-Seq Data | This paper | GSE173574 |
| Raw and analyzed ChIP-Seq Data | This paper | GSE173599 |
| Raw and analyzed ATAC-Seq Data | This paper | GSE189098 |
| Raw and analyzed SLAM-Seq Data | This paper | GSE173571 |
| Raw and analyzed PRO-Seq Data | This paper | GSE189105 |
| Bernt MLL::AF9 Targets | Bernt et al., 2011 | GSE29130 |
| Guenther MLL-AF4 targets | Guenther et al., 2008 | GSE13313 |
| Stavropoulou MLL::AF9 Removal | Stavropoulou et al., 2016 | GSE65384 |
| LSK Gene Set | Uckelmann et al., 2020 | GSE129638 |
| Experimental models: Cell lines | ||
| HL-60 | ATCC | CCL-240 |
| MOLM13 | DSMZ | ACC 554 |
| 293T | ATCC | CRL-3216 |
| murine MLL::AF9 leukemia | This paper | MLL::AF9 #1, MLL::AF9 #2 |
| murine MLL::AF9-HA-FKBP12 leukemia | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| hCD34+ MLL::AF9 leukemia | This paper | MLL::AF9 #1, MLL::AF9 #2 |
| hCD34+ MLL::AF9-HA-FKBP12 leukemia | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| Drosophila melanogaster S2 cells | Nascent Transcriptomics Core at Harvard Medical School | Drosophila melanogaster S2 cells for spike-in controls |
| Experimental models: Organisms/strains | ||
| C57BL/6NCrl | Charles River Laboratories | C57BL/6 |
| Oligonucleotides | ||
| 3′ adapter | IDT | /5Phos/rGrArUrCrGrUrCrGrGrArCrUrGrUrArGrArArCrUrCrUrGrArArC/3InvdT/ |
| 5′ adapter | IDT | rCrCrUrUrGrGrCrArCrCrCrGrArGrArArUrUrCrCrA |
| Illumina TruSeq small RNA indexing PCR primers | IDT | CAAGCAGAAGACGGCATACGAGATxxxxxxGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA (where Xs indicate index sequences) |
| RT and PCR primer | IDT | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA |
| Recombinant DNA | ||
| pMSCV-MLL::AF9-IRES-GFP | Armstrong Lab | MLL::AF9 #1, MLL::AF9 #2 |
| pMSCV-HA-FKBP12-MLL::AF9-IRES-GFP | This paper | HA-FKBP12-MLL::AF9 |
| pMSCV-MLL::AF9-HA-FKBP12-IRES-GFP | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| pUMVC | Addgene | Plasmid #8449 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| pCL-Eco | Addgene | Plasmid #12371 |
| Software and algorithms | ||
| bcl2fastq Conversion Software, v2.20.0.422 | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastqconversion-software.html |
| Bowtie2, v2.3.4.3 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR, v2.7.5a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| CEAS sitepro | Shin et al., 2009 | https://liulab-dfci.github.io/software/ |
| bedtools v2.28.0 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| ngs.plot | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| Slamdunk SLAMseq pipeline | Neumann et al., 2019 | https://t-neumann.github.io/slamdunk/ |
| Homo sapiens GRCh38/hg38 reference source | Broad Institute CTAT GRCh38_gencode_v33_CTAT_lib_Apr062020 | https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/ |
| Mus musculus M24/mm10 reference source | Broad Institute CTAT Mouse_gencode_M24_CTAT_lib_Apr062020 | https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/ |
| IGVtools, 2.3.75 | Robinson et al., 2011, 2017; Thorvaldsdóttir et al., 2013 | https://software.broadinstitute.org/software/igv/igvtools |
| Samtools, v1.95 | Li et al., 2009 | http://www.htslib.org/ |
| HTSeq, htseq-count, v0.11.2 | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| Picard tools, v2.9.4 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GSEA, v.3.160 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| R Bioconductor DESeq2 package v1.24.0 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MACS2 v2.1.4 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| bedgraphs2stdBedGraph | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/bedgraphs2stdBedGraph |
| Bowtie 1.2.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| bowtie2stdbedgraph | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/bowtie2stdbedgraph |
| Cutadapt 1.14 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| GetGeneAnnotation_GGA | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/GetGeneAnnotation_GGA |
| make_heatmap | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/make_heatmap |
| samtools 1.3.1 | Danecek et al., 2021 | https://www.htslib.org/ |
| trim_and_filter_PE | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/trim_and_filter_PE |
| TSScall | Lavender et al., 2017 | https://github.com/lavenderca/TSScall |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All animal experiments were performed with the approval of Dana-Farber Cancer Institute’s Institutional Animal Care and Use Committee (IACUC). Hematopoietic stem cell-enriched Lineage-Sca1+c-KIT/CD117+(LSK) cells were isolated from the bone marrow of 8-10 week old female C57BL/6 mice (Charles River Laboratories). Upon MLL::AF9 transduction, 2.5x105 GFP positive cells, alongside 2.5x105 helper cells (crude cells isolated from bone marrow of female C57BL/6 mice), were injected into lethally irradiated (900Gy) 12-week old C57BL/6 female recipient mice (Charles River Laboratories). Engraftment in the peripheral blood was monitored every 3-4 weeks. When animals developed signs of leukemia, the bone marrow and spleen were harvested.
Cell lines
Human cell lines were acquired from American Type Culture Collection (ATCC) or Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and were cultured in RPMI 1640 (HL60, MOLM13) or DMEM (293T) supplemented with 10% fetal bovine serum, 1xpenicillin/streptomycin (Gibco) at 37°C and 5% CO2. Identification of all cell lines was independently confirmed by cytogenetics profiling. The human CD34+ MLL::AF9 transformed cells were maintained in IMDM with 20% FBS, 1xpenicillin/streptomycin (Gibco),1xβ-mercaptoethanol (Gibco), 6 μg/mL hIL3, 10 μg/mL hIL6, 10 μg/mL hSCF, 10 μg/mL TPO, and 10 μg/mL FLT3 (Stemcell Technologies) at 37°C and 5% CO2. The murine MLL::AF9 transformed leukemic cells were maintained in IMDM with 20% FBS, 1xpenicillin/streptomycin (Gibco), 1xβ-mercaptoethanol (Gibco), 6 μg/mL mIL3, 10 μg/mL mIL6, and 20 μg/mL mSCF (Stemcell Technologies) at 37°C and 5% CO2.
METHOD DETAILS
Virus production and transduction
Viral supernatants were generated by co-transfection of HEK293-T cells with retroviral (MSCV-MLL::AF9-IRES-GFP or MSCV-MLL::AF9-HA-FKBP12F36V-IRES-GFP) expression vectors with packaging and envelope vectors (Human Retro: pUMVC and VSV-G; Mouse Retro: pCL-Eco) and X-tremeGene transfection reagent (Millipore). The viral supernatant was filtered through 0.45μm and was concentrated using Amicon Ultra centrifugal filters (Millipore). The murine LSK cells were spin infected at a 1:1 dilution of virus:media and 0.8μg/ml polybrene (Millipore) at 2000rpm at 37°C for 1.5h and the supernatant was replaced with fresh medium immediately after the spin infection. The human CD45+ cells were plated on retronectin-coated plates (Takara) and were spin infected at a 1:1 dilution of virus:media at 2000rpm at 37°C for 1.5h. The cells were dissociated from the plates using enzyme-free dissociation buffer (Gibco) and were plated in fresh media.
Neutrophil isolation, LSK cell sort, and MLL::AF9 leukemia generation
Neutrophils and hematopoietic stem cell-enriched Lineage-Sca1+c-KIT/CD117+(LSK) cells were isolated from the bone marrow of 8-10 week old C57BL/6mice (Charles River Laboratories). Mice were sacrificed, and tibiae, femurs, coxae and vertebral columns were crushed in PBS supplemented with 2% fetal bovine serum. Neutrophils were isolated using the EasySep™ Mouse Neutrophil Enrichment Kit according to the manufacturer’s instructions. For the LSK cells, lineage depletion was performed using EasySep™ Mouse Hematopoietic Progenitor Cell Isolation Kit (Stemcell Technologies) and cells were subsequently stained with Sca-1 (Invitrogen) and c-kit (Biolegend) in 2% PBS for 30 minutes on ice. FACSAria cell sorters were used (BD Bioscience) to sort Sca-1 and c-kit double positive cells. LSK cells were cultured in StemSpan SFEM (Stemcell Technologies) containing 100 ng/ml mSCF/mFLT3/mTPO (Stemcell Technologies) and were transduced with concentrated MLL::AF9 virus the following day. Two days after transduction, GFP positive cells were sorted using FACSAria cell sorters (BD Bioscience). GFP positive cells were expanded in IMDM media supplemented with FBS and cytokines (described above) for 2-4 days. 2.5x105 GFP positive cells, alongside 2.5x105 helper cells (crude cells isolated from bone marrow of C57BL/6 mice), were injected into lethally irradiated (900Gy) 12-week old C57BL/6 recipient mice (Charles River Laboratories). Engraftment in the peripheral blood was monitored every 3-4 weeks. For analysis of peripheral blood, four to five drops of blood were collected from the vena facialis into an EDTA-coated tube. Erythrocytes were lysed with 1 ml of 1xPharm Lyse buffer (BD Bioscience), washed with PBS and stained in PBS with 2%FBS. Cells were analyzed for GFP positivity using an LSR Fortessa flow cytometer (BD Bioscience). Data were analyzed with FlowJo software (Tree Star). Once the mice reached their endpoint according to our protocol, the mice were sacrificed and leukemic cells were extracted from the bone marrow (as described above) and were analyzed for GFP positivity using an LSR Fortessa flow cytometer (BD Bioscience). Data were analyzed with FlowJo software (Tree Star). The cells were maintained in IMDM media (as described above).
Isolation and transformation of human CD34+ cells
Human CD34+ cells were isolated from human cord blood (New York Blood Center) using the EasySep™ Human Cord Blood CD34 Positive Selection Kit II (Stemcell Technologies). For the pre-enrichment, Lymphoprep (Stemcell Technologies) and SepMate™ columns (Stemcell Technologies) were used, according to the manufacturer’s instructions. Human CD34+ cells were cultured and transduced as described above. Two days after transduction, GFP positive cells were sorted using FACSAria cell sorters (BD Bioscience) and cells were maintained in culture.
Cell viability, differentiation, apoptosis, and Z-VAD-FMK-mediated rescue
For proliferation studies, cells were plated at 1-5x105 cells/mL and treated with limiting dilutions of drug as indicated or 0.1% DMSO. The cells were re-plated every 3-4 days and new drug/DMSO was added. Viable (DAPI-, Santa Cruz) cells were counted in 30μL of media plus 120uL PBS/DAPI using an LSR Fortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star). Ratios of cell numbers in drug to DMSO were then plotted to calculate IC50 values.
For differentiation studies, human cells were stained with APC-conjugated CD11b (BioLegend), PerCPCy5.5-conjugated CD13 (BioLegend), and PE-Cy7-conjugated CD14 (BioLegend). Samples were then analyzed using an LSR Fortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star). For assessment of morphology, 5-10x104 cells were collected after drug treatment and centrifuged onto slides using the CytoCentrifuge (Thermo Scientific) and stained for microscopy using the JorVet DipQuick Stain (Jorgensen Laboratories, Inc.).
For apoptosis studies, cells were harvested following drug treatment and were stained with APC-conjugated Annexin V according to the manufacturer’s instructions (eBioscience). Cells were counter-stained with Propidium Iodide. Samples were then analyzed using an LSR Fortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star).
For the Z-VAD-FMK rescue experiments, cells were pre-treated with 50μM Z-VAD-FMK for 6 hours and where then treated with DMSO, EPZ-5676, VTP-50469, EPZ-5676+VTP-50469, or dTAG-VHL at the indicated concentrations and for the indicated timepoints.
Intracellular FACs
1x106 cells were harvested following drug treatment and cells were fixed using BD Cytofix/Cytoperm, BD Perm/Wash Buffer, and BD Cytoperm Permealization Buffer Plus according to the manufacturer’s instructions (BD Bioscience). After fixation/permealization, cells were incubated with 1x Wash Buffer supplemented with 5% BSA (Sigma Aldrich) for 30 minutes on ice. Subsequently, cells were incubated with 1:100 anti-HA antibody (CST) for 30 minutes on ice, washed 3 times with Wash Buffer and then incubated with 1:200 Anti-rabbit IgG Alexa Fluor 647 (CST) at room temperature for 30 minutes. Subsequently, cells were washed 3 times with Wash Buffer and resuspended in PBS for FACs analysis. Samples were analyzed using an LSR Fortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star).
Western blot analysis and immunoblotting
1x106 cells were washed twice in cold PBS and were lysed in 100μL of 1X LDS Sample Buffer (Invitrogen) supplemented with 10 mM DTT. The samples were sonicated on ice for 40 seconds (with 5 seconds rest after every 10 second sonication pulse at 25% Amplitude) with a 125-watt probe sonicator (Qsonica, Q125) and were subsequently denatured for 10 min at 95°C. Samples were loaded (40μL per lane) onto 3-8% Tris-Acetate gels (Thermo Fisher) with a protein ladder (BioRad), proteins were separated by electrophoresis for 2 hours and then transferred onto nitrocellulose membranes using semi-dry transfer (Thermo Fisher). The membranes were blocked in 5% dry milk for 1 hour and incubated overnight with anti-HA (CST), anti-cleaved PARP (CST), anti-total PARP (CST), anti-GAPDH (CST), or anti-Vinculin (CST). The next day membranes were washed in TBST and developed using a secondary rabbit anti-HRP (Cytiva) and chemiluminescence kit (Thermo Scientific).
RNA isolation and quantitative real-time PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions and DNase treatment (Qiagen) was performed on the column. RNA was reverse transcribed with the Superscript™ First-Strand Synthesis System (Invitrogen). For qRT-PCR, the ViiA 7 Real-Time PCR system (Applied Biosystems) was used with 384-well plates. Taqman gene expression assays (Applied Biosystems) were used for real-time quantitative PCR where indicated in the Figure legends. GAPDH was used as the housekeeping gene for normalization. Relative gene expression was calculated by the comparative ΔΔ cycle threshold method. Mouse probes used: Gapdh (Mm99999915_g1), Hoxa7 (Mm00657963_m1), Hoxa9 (Mm00439364_m1), HoxA10 (Mm00433966_m1), Meis1 (Mm00487664_m1); Human probes used: GAPDH (Hs02786624_g1), HOXA7 (Hs00600844_m1), MEIS1 (Hs01017441_m1). SYBR green gene expression assays (Applied Biosystems) were used for real-time quantitative PCR where indicated in the Figure legends. For Intron/Exon qRT-qPCRs cDNA synthesis reactions were set up with and without Reverse Transcriptase to assess contaminating genomic DNA. GAPDH was used as the housekeeping gene for normalization. Relative gene expression was calculated by the comparative ΔΔ cycle threshold method. Probes used: GAPDH (F: TGCACCACCAACTGCTTAGC, R: GGCATGGACTGTGGTCATGAG), MEIS1 Exon/Exon (F: CACGCTTTTTGTGACGCTT, R: GGACAACAGCAGTGAGCAAG), MEIS1 Intron/Exon (F: AGACGATAGAGAAGGAGGATCAA R: CACGCTTTTTGTGACGCTT), HOXA9 Exon/Exon (F: AATGCTGAGAATGAGAGCGG, R: GTATAGGGGCACCGCTTTTT), HOXA9 Intron/Exon (F: AACAACCCAGCGAAGGC, R: TTGAAGGGAGGAGACACTTACT).
For ChIP-qPCRs, the ViiA 7 Real-Time PCR system (Applied Biosystems) was used with 384-well plates using SYBR green (Applied Biosystems). The human ChIP-qPCR primers used were: GAPDH (F: CAGTCAGCCGCATCTTCTTT, R: CCTTCAGGCCGTCCCTA), MEIS1 (F: GAGGCGCGACGAATGAA, R: TCCAGACCGGAGAACTGG), HOXA9 (F: GGGTGAGAGAAGGGAGAAGG, R: AAAGACCGAGCAAAAGACGA), SP9 (F: GCTATGGCCACGTCTATACTC, R: TGCAGTCTAGCAGCAACAG), gene desert (F: AACCTCACTTTCATTGTTACTAGCCATA, R: CGCTCAAGGATGTCAGTAGCAT)
RNA sequencing
For RNA-seq experiments, the cells were plated at 1x105 cells/mL, treated with drug or 0.1% DMSO for the indicated time. Cells were sub-cultured to the original density after 3 days in fresh media with the same concentration of drug or DMSO. RNA was isolated as described above. RNA quality for RNA sequencing was checked on the Agilent TapeStation (Agilent) and quantified by Qubit (ThermoFisher). RNA (1μg) was used to make Illumina compatible libraries by doing Poly-A tail selection and library preparation using the NEBNext Ultra™ RNA Library Prep Kit for Illumina (New England Biolabs). Sequencing was done using the Illumina Next Gen Sequencing NextSeq platform (Illumina) with 20-30 million 37bp, paired-end reads.
SLAM sequencing
The procedure was performed using the Lexogen SLAM-Seq Metabolic Labeling kit. Cells were plated at 5x105 cells/mL in the appropriate culture medium and were pre-treated either with 0.1% DMSO or with dTAG-VHL (500nM) for 15, 30, 60, or 120 minutes. After the appropriate pretreatment, cells were incubated with 4SU (Lexogen) at a final concentration of 100μM and were incubated with continuous dTAG-VHL/DMSO treatment for 60 min at 37°C / 5% CO2. Upon addition of 4SU, cells were kept away from light. Cells were harvested at 300 g for 5 min at 4°C, washed with PBS and the RNA was extracted from the cell pellet using QIAshredders (Qiagen) and RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. 5ug of the extracted RNA was lodoacetamide treated according to the manufacturer’s instructions (Lexogen). Libraries were prepared using 500ng RNA and the QuantSeq 3’ mRNA-Seq Library Preparation kit (Lexogen). Sequencing was done using the Illumina Next Gen Sequencing NextSeq platform (Illumina) with 20-30 million 75bp, single-end reads.
ATAC sequencing
60,000 cells were harvested and washed in PBS. Cell pellets were then resuspended in 50μL cold lysis buffer (10 mM Tris-HCl, pH 7.4; 10 mM NaCl; 3 mM MgCl2; 0.1% IGEPAL CA-630). Cells were spun down immediately at 500 χ g for 10 min at 4 °C. Following this, the pellet was then resuspended in a transposase reaction mixture (2X TD buffer, 20X transposase (100nM final)) (Illumina) at 37 °C for 30 minutes. DNA was purified using a MinElute kit (Qiagen) as per the manufacturer’s instructions. The DNA fragments were amplified in a PCR reaction with 2X NEBNext High-Fidelity PCR Master Mix (New England Biolabs) for 11 cycles and purified using AMPure XP beads (Beckman Coulter). DNA fragments were quantified by TapeStation 4200 (Agilent) using HSD5000 Tape and Reagent (Agilent) and Qubit (ThermoFisher). This was followed by sequencing using the NextSeq550 (Illumina) to obtain 20-30 million 37bp, paired-end reads.
PRO sequencing
10 million cells were harvested and washed once with ice cold PBS in 50mL conical tubes. Cell pellets were resuspended in 250μL wash buffer (10 mM Tris-Cl, pH 8.0, 10 mM KCl, 250 mM Sucrose, 5 mM MgCl2, 1 mM EGTA, 10 % (v/v) Glycerol, supplemented freshly with 0.5mM DTT, 0.2μL/mL RNase inhibitor and protease inhibitor) to get single-cell suspensions. 10mL permealization buffer (10 mM Tris-Cl, pH 8.0, 10 mM KCl, 250 mM Sucrose, 5 mM MgCl2, 1 mM EGTA, 0.1 % (v/v), Igepal CA-630, 0.05 % (v/v) Tween-20, 10 % (v/v) Glycerol, supplemented freshly with 0.5mM DTT, 0.2μL/mL RNase inhibitor and protease inhibitor) was added and samples were incubated on ice for 5 min. Cells were spun down and pellets were washed in 10mL wash buffer. Cells were spun again and resuspended in freezing buffer (50 mM Tris-Cl, pH 8.0, 40 % (v/v) glycerol, 5 mM MgCl2, 1.1 mM EDTA, supplemented freshly with 0.5 mM DTT and 1μL/mL RNase inhibitor). Cells were counted and permealization was visualized using tryphan blue. 5 million permealized cells were snap-frozen in 500ul freezing buffer in liquid nitrogen and stored at −80°C. All subsequent steps were performed by the Nascent Transcriptomics Core at Harvard Medical School, Boston, MA.
Aliquots of frozen (−80°C) permeabilized cells were thawed on ice and pipetted gently to fully resuspend. Aliquots were removed and permeabilized cells were counted using a Luna II, Logos Biosystems instrument. For each sample, 1 million permeabilized cells were used for nuclear run-on, with 50,000 permeabilized Drosophila S2 cells added to each sample for normalization. Nuclear run on assays and library preparation were performed essentially as described in Reimer et al. (Reimer et al., 2021)with modifications noted: 2X nuclear run-on buffer consisted of (10 mM Tris (pH 8), 10 mM MgCl2, 1 mM DTT, 300mM KCI, 40uM/ea biotin-11-NTPs (Perkin Elmer), 0.8U/uL SuperaseIN (Thermo), 1% sarkosyl). Run-on reactions were performed at 37°C. Adenylated 3′ adapter was prepared using the 5′ DNA adenylation kit (NEB) and ligated using T4 RNA ligase 2, truncated KQ (NEB, per manufacturers instructions with 15% PEG-8000 final) and incubated at 16°C overnight. 180uL of betaine blocking buffer (1,42g of betaine brought to 10mL with binding buffer supplemented to 0.6 uM blocking oligo (TCCGACGATCCCACGTTCCCGTGG/3InvdT/)) was mixed with ligations and incubated 5 min at 65°C and 2 min on ice prior to addition of streptavidin beads. After T4 polynucleotide kinase (NEB) treatment, beads were washed once each with high salt, low salt, and blocking oligo wash (0.25XT4 RNA ligase buffer (NEB), 0.3uM blocking oligo) solutions and resuspended in 5′ adapter mix (10 pmol 5′ adapter, 30 pmol blocking oligo, water). 5′ adapter ligation was per Reimer but with 15% PEG-8000 final. Eluted cDNA was amplified 5-cycles (NEBNext Ultra II Q5 master mix (NEB) with Illumina TruSeq PCR primers RP-1 and RPI-X) following the manufacturer’s suggested cycling protocol for library construction. A portion of preCR was serially diluted and for test amplification to determine optimal amplification of final libraries. Pooled libraries were sequenced using the Illumina NovaSeq platform.
For the analysis, all custom scripts described herein are available on the AdelmanLab Github (https://github.com/AdelmanLab/NIH_scripts). Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 41 bp per mate, and read pairs with a minimum average base quality score of 20 retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). R1 reads, corresponding to RNA 3’ ends, were then aligned to the spiked in Drosophila genome index (dm3) using Bowtie 1.2.2 (-v 2 -p 6 --best --un), with those reads not mapping to the spike genome serving as input to the primary genome alignment step (using Bowtie 1.2.2 options -v 2 --best). Reads mapping to the hg38 reference genome were then sorted, via samtools 1.3.1 (-n), and subsequently converted to bedGraph format using a custom script (bowtie2stdBedGraph.pl). Because R1 in PRO-seq reveals the position of the RNA 3’ end, the “+” and “−” strands were swapped to generate bedGraphs representing 3’ end position at single nucleotide resolution.
A table of statistics, including raw read counts, mappable read counts to the spike in and reference genomes, and Spearman’s correlation coefficient between promoter reads in replicate samples is below:
| Samples | Number Raw Reads | Number Spike Reads Mapped | % Spike Reads Mapped | Number Ref Reads Mapped | % Ref Reads Mapped |
|---|---|---|---|---|---|
| untreated_1 | 96340680 | 4,102,096 | 4.72 | 78,324,815 | 90.05 |
| untreated_2 | 89419830 | 3,227,398 | 4.05 | 73,022,054 | 91.57 |
| 15m_1 | 101587715 | 4,254,875 | 4.63 | 83,390,781 | 90.65 |
| 15m_2 | 79114293 | 2,938,173 | 4.13 | 65,096,557 | 91.58 |
| 30m_1 | 100887984 | 4,406,459 | 4.88 | 81,511,750 | 90.36 |
| 30m_2 | 104201353 | 3,840,702 | 4.09 | 86,094,121 | 91.71 |
| 3h_1 | 83906090 | 3,607,935 | 4.81 | 68,036,834 | 90.7 |
| 3h_2 | 81542428 | 2,908,145 | 3.96 | 67,611,970 | 92.08 |
| untreated_1 | 15m_1 | 30m_1 | 3h_1 | untreated_2 | 15m_2 | 30m_2 | 3h_2 | |
|---|---|---|---|---|---|---|---|---|
| untreated_1 | NA | NA | NA | NA | NA | NA | NA | NA |
| 15m_1 | 0.98 | NA | NA | NA | NA | NA | NA | NA |
| 30m_1 | 0.98 | 0.98 | NA | NA | NA | NA | NA | NA |
| 3h_1 | 0.98 | 0.98 | 0.98 | NA | NA | NA | NA | NA |
| untreated_2 | 0.98 | 0.98 | 0.98 | 0.98 | NA | NA | NA | NA |
| 15m_2 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | NA | NA | NA |
| 30m_2 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | NA | NA |
| 3h_2 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | NA |
For promoter reads, annotated transcription start sites were obtained from human (GRCh38.99) GTFs from Ensembl. After removing transcripts with {immunoglobulin, Mt, Mt_tRNA, rRNA} biotypes, PRO-seq signal in each sample was calculated in the window from the annotated TSS to +150 nt downstream, using a custom script, make_heatmap.pl. This script counts each read one time, at the exact 3’ end location of the nascent RNA.
Given good agreement between replicates and similar return of spike-in reads, bedGraphs were merged within conditions, and depth-normalized, to generate bigWig files binned at 10bp.
For the refinement of gene annotation (GGA), paired-end SLAM-seq reads were mapped to the hg38 reference genome via HISAT2 v2.2.1 (--known-splicesite-infile). To select gene-level features for differential expression analysis, and for pairing with PRO-seq data, we assigned a single, dominant TSS and transcription end site (TES) to each active gene. This was accomplished using a custom script, get_gene_annotations.sh (available at https://github.com/AdelmanLab/GeneAnnotationScripts), which uses SLAM-seq read abundance and PRO-seq R2 reads (RNA 5’ ends) to identify dominant TSSs. SLAM-seq and PRO-seq data from all conditions were used for this analysis, to comprehensively capture gene activity in these samples.
For the differential expression analysis, reads were summed within the TSS to TES window for each active gene using the using the make_heatmap script (https://github.com/AdelmanLab/NIH_scripts), which counts each read one time, at the exact 3’ end location of the nascent RNA. DEseq2, using the Wald test, was used to determine statistically significant differentially expressed genes. Unless otherwise noted, the default size factors determined by DEseq2 were used.
For the average metagene plot from TSS to TES, with adjacent upstream and downstream regions, average metagene plots of PRO-seq read density from TSS to TES of indicated gene groups were generated using make_heatmap as described above. Each gene was divided from TSS to TES positions into 100 bins of equal length, and read density calculated in each length scaled bin, as reads per kilobase. Read density flanking each gene were also calculated (2kb upstream of TSS and 2kb downstream of TES), in 20bins of 50pb each. Average values for each gene group are shown. Values were smoothed by averaging over 3 nearest neighbors.
Pausing indices were generated as follows: read counts were summed in the window from TSS to +150 for each active gene, and from +250 downstream of the TSS to the TES, using the refined annotations described above. The ratio of promoter proximal signal (TSS to +150) over gene body signal (+250 to TES) is the Pausing Index.
Chromatin immunoprecipitation and sequencing
Following the appropriate drug treatment, cells were washed twice in cold PBS. For CDK9 ChIPs, cells were double crosslinked, first with 2mM disuccinimidyl glutarate (ThermoFisher) for 30 min at room temperature, then spun down at 600g for 5 min and subsequently crosslinked in 1% methanol-free formaldehyde (ThermoFisher) for 8 min at room temperature. All other ChIPs were only crosslinked with 1% methanol-free formaldehyde (ThermoFisher) for 8 min at room temperature. Following crosslinking, cells were quenched using 100 mM Tris pH 8.0 and 250 mM Glycine and washed twice with room temperature PBS. 20 million cells per mL were then lysed using 50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM EDTA, 1% SDS and chromatin was collected by centrifugation at 15,000g for 10 min. Chromatin from 20 million cells was then resuspended in 1mL 66mM Tris-HCl pH 8.0, 100mM NaCl, 5mM EDTA, 1.7% Triton X-100, 0.5% SDS and sheared using an E100S sonicator (Covaris) to chromatin fragments of 200-400 base-pair DNA size. 5μL of sonicated chromatin was de-crosslinked with 100 mM NaHCO3, 100 mM NaCl, 1% SDS in a total volume of 50uL and was incubated at 65°C for 4-6 hours. Following de-crosslinking, DNA was purified with AMPure XP beads (Beckman Coulter). Input DNA fragments were run on a TapeStation 4200 (Agilent), using D5000 Tape and Reagents (Agilent) to ensure proper shearing, and quantified using Qubit (ThermoFisher). Sheared chromatin from 20 million cells was used in each immunoprecipitation using the following antibodies: anti-HA (CST), MENIN (Bethyl), CDK9 (Santa Cruz), DOT1L (CST), RNA Pol II (CST), RNA Pol2 pSer2 (Abcam), RNA Pol2 pSer5 (Abcam), AFF4 (Bethyl), and KMT2A/MLL1 (N-terminal, Bethyl). Sheared chromatin from 60 million cells was used in the MLL1-C-terminal immunoprecipitation using the following antibody: MLL/HRX (Active Motif). Sheared chromatin from 10 million cells was used in each immunoprecipitation using the following antibodies: anti-H3K79me2 (CST), H3K27ac (Diagenode), H3K9ac (Abcam), and H3Kme3 (Abcam). Antibodies were conjugated to protein-A or protein-G magnetic beads (Dynabeads) for 4-6 hours on a rotator at 4°C with 0.5μg/μL BSA (Invitrogen). Subsequently, the sheared chromatin was added to the beads+antibody and was incubated on a rotator at 4°C over night. Immunoprecipitated DNA fragments were eluted and de-crosslinked in 100 mM NaHCO3, 100 mM NaCl, 1% SDS, and was incubated at 65°C over night. Following de-crosslinking, DNA was purified with AMPure XP beads (Beckman Coulter). DNA fragments were quantified by TapeStation 4200 (Agilent) using HSD1000 Tape and Reagent (Agilent) and Qubit (ThermoFisher). 1-10 ng of DNA was used in preparation of Illumina compatible libraries using SMARTer ThruPLEX DNA-Seq Kit (Takara) followed by sequencing using NextSeq550 (Illumina) to obtain 20-30 million 37bp, paired-end reads.
Bioinformatics analysis
For all ChIP-Seq analyses and ATAC-Seq, raw Illumina sequencer output was converted to FASTQ format using bcl2fastq (v2.20.0.422). Reads (paired-end 37-mers) were aligned to the human (Gencode GRCh38/hg38) genome using STAR (v2.7.5a; params --alignlntronMax 1 --alignEndsType EndToEnd --alignMatesGapMax 2000 for ChIPseq analysis), sorted and duplicates marked/removed with picard pipeline tools (v2.9.4). Final “deduped” .BAM files were indexed using SAMtools (v1.95). ChIP-seq data visualizations were produced using IGVtools (TDF signal pileups; v2.3.75) and ngs.plot (pileup heatmaps).
For the initial HA ChIP-Seq analysis MACS2 (v2.1.1) was used to call peaks, with q-value cut-off of 0.01 and broad peak mode. Peaks that are shorter than 100 bp or are overlapped with blacklisted regions were filtered out. To compare the genomic distribution of the peaks of the six replicate HA ChIP samples, we then unified the genome coordinates of the peaks called in each sample. First, we pooled peaks from the six samples and merged the overlapped peaks using “bedtools merge -d 0,” which resulted in a unified set of peaks without overlapping. Next, we compared the peaks from each sample with the unified peak set, and any peaks showing overlap (≥1bp) with the peaks in the unified set were added to the peak set using the coordinates of the unified peak.
For all other ChIP-Seq analyses and ATAC-Seq, peaks were also called using MACS2. In the case of ChIP-seq, appropriate input samples were used as controls for peak calling. Gene body and TSS-associated signals were assessed using sitepro from the CEAS suite, using annotated gene body genomic intervals and TSS region intervals (−1kb to +3kb for each proteincoding transcript TSS, using gene orientation). A single representative TSS region was chosen for each gene, based on the site with the highest average signal in DMSO control samples. Genes with relevant signal at the TSS (2-fold enrichment over input and > 200 reads within TSS −1kb to +3kb) were kept for further analysis.
For 3h and 24h samples (Figure 3 and Figure S3), treated sample interval counts were normalized using a ratio of TSS signal values among a set of selected housekeeping genes (for list of housekeeping genes and calculation of the normalization factor see GEO: GSE173599). For later timepoints (Figure 6 and Figure S6), reads were normalized using ratios of total read counts between treated and control sample pairs. For read counts and calculation of the normalization factor see GEO: GSE173599).
To define MLL::AF9 targets in MOLM13 cells an N-terminal MLL1 antibody was used, which detects both wild-type MLL1 as well as MLL::AF9. MLL::AF9 target genes were defined by overlapping the MLL::AF9 bound genes identified in Figure 2 (listed in Table S1) with the MLL1 ChIP signal in MOLM13 cells (Table S4) – genes that had MLL1 bound and were defined as an MLL::AF9 target gene in Figure 2 were considered MLL::AF9 target genes in the MOLM13 cells (Table S4).
For RNAseq, raw Illumina sequencer output was converted to FASTQ format using bcl2fastq (v2.20.0.422). Reads (paired-end 37-mers) were aligned to the human (Gencode GRCh38/hg38) or mouse (Gencode M24/mm10) genome using STAR (v2.7.5a), sorted and duplicates marked/removed with picard pipeline tools (v2.9.4). Final “deduped” .BAM files were indexed using SAMtools (v1.95). RNA-seq data visualizations were produced using IGVtools (TDF signal pileups; v2.3.75). Raw per-gene counts were calculated with HTSeq (htseq-count, v0.6.1pl). Differential RNA-seq expression was calculated using the BioConductor DESeq2 package (v1.24,0), using raw unnormalized per-gene counts from deduplicated BAMs.
For SLAMseq, SLAMseq FASTQ files were analyzed using the Slamdunk pipeline, described in (Neumann et al., 2019). Differential SLAMseq expression was calculated using the BioConductor DESeq2 package (v1.24,0), using Slamdunk converted counts.
QUANTIFICATION AND STATISTICAL ANALYSIS
For proliferation assays, dose-response curves and IC50 values were calculated in a variable slope model as a four-parameter dose-response curve (GraphPad Software). Gene set enrichment analysis (GSEA v3.0 software/Broad Institute) was used to identify functional associations from the described RNA-seq profiles. Significance cut-offs were assessed on the basis of the GSEA standard recommendations: absolute NES ≥ 1, P ≤ 0.05, Benjamini-Hochberg FDR ≤ 0.25. Mann Whitney tests (nonparametric t-test, GraphPad Software) were performed as indicated in the Figure Legends.
Supplementary Material
Table S3. Gene expression changes upon DOT1L/MENIN inhibition and MLL::AF9 degradation, related to Figure 5
Table S4. MLL::AF9 occupancy changes upon DOT1L and MENIN inhibition, related to Figure 6
Highlights.
Rapid degradation of MLL::AF9 identifies direct MLL::AF9 target genes
MLL::AF9 maintains transcription by supporting productive RNA Pol II elongation
MLL::AF9 degradation induces chromatin state changes after decreases in elongation
Combined DOT1L/MENIN inhibition cooperatively destabilizes MLL::AF9 on chromatin
ACKNOWLEDGEMENTS
S.A.A. is supported by NIH grants CA176745, CA206963, CA204639 and CA066996 and by the Leukemia and Lymphoma Society (LLS). S.N.O. is a Damon Runyon Cancer Research Foundation Sohn Fellow (DRSG: 26-18). B. N. is supported by NIH grant K22 CA258805. We thank all members of the Armstrong lab for invaluable discussions. We thank Johannes Zuber for insight regarding SLAM-Seq. We thank Nathanael Gray for graciously supplying the dNEG/dTAG compounds for these studies. We would also like to thank Karen Adelman, Seth Goldman, and the Nascent Transcriptomics Core at Harvard Medical School, Boston, MA for assistance with PRO-Seq library construction and data analysis. We are also grateful for BioRender - the graphical abstract and Figure 6H were created with BioRender.com.
DECLARATION OF INTERESTS
S.A.A. has been a consultant and/or shareholder for Neomorph Inc, Imago Biosciences, Vitae/Allergan Pharma, Cyteir Therapeutics, C4 Therapeutics, Accent Therapeutics, and Mana Therapeutics. S.A.A. has received research support from Janssen, Novartis, and Syndax. SAA is an inventor on patent applications related to MENIN inhibition WO/2017/132398A1. B.N. is an inventor on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148440, WO/2018/148443 and WO/2020/146250).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allen MD, Grummitt CG, Hilcenko C, Min SY, Tonkin LM, Johnson CM, Freund SM, Bycroft M, and Warren AJ (2006). Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. Embo J 25, 4503–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi Y, Smith ER, Shah AP, Rendleman EJ, Marshall SA, Woodfin AR, Chen FX, Shiekhattar R, and Shilatifard A (2020). NELF Regulates a Promoter-Proximal Step Distinct from RNA Pol II Pause-Release. Mol Cell 78, 261–274.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. (2011). MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KSJ, Allis CD, and Roeder RG (2011). Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc National Acad Sci 108, 15751–15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, et al. (2015). Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer Cell 27, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braekeleer ED, Douet-Guilbert N, Basinko A, Bris M-JL, Morel F, and Braekeleer MD (2014). Hox gene dysregulation in acute myeloid leukemia. Future Oncol 10, 475–495. [DOI] [PubMed] [Google Scholar]
- Cierpicki T, Risner LE, Grembecka J, Lukasik SM, Popovic R, Omonkowska M, Shultis DD, Zeleznik-Le NJ, and Bushweller JH (2010). Structure of the MLL CXXC domain–DNA complex and its functional role in MLL::AF9 leukemia. Nat Struct Mol Biol 17, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafflon C, Craig VJ, Méreau H, Gräsel J, Engstler BS, Hoffman G, Nigsch F, Gaulis S, Barys L, Ito M, et al. (2017). Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 31, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. (2011). Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. (2013). Potent inhibition of DOTIL as treatment of MLL-fusion leukemia. Blood 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo H-S, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, Heuvel-Eibrink M. van den, Zwaan CM, Kung AL, and Armstrong SA (2009). HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 113, 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. (2012). Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, Croce CM, Nakamura T, Canaani E, and Young RA (2008). Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Gene Dev 22, 3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton SJ, Jaques J, Woolthuis C, Dijk J. van, Mesuraca M, Huls G, Morrone G, Vellenga E, and Schuringa JJ (2013). MLL–AF9-mediated immortalization of human hematopoietic cells along different lineages changes during ontogeny. Leukemia 27, 1116–1126. [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. (2004). Menin Associates with a Trithorax Family Histone Methyltransferase Complex and with the Hoxc8 Locus. Mol Cell 13, 587–597. [DOI] [PubMed] [Google Scholar]
- Jaeger MG, and Winter GE (2021). Fast-acting chemical tools to delineate causality in transcriptional control. Mol Cell 81, 1617–1630. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, Ross KN, Perner F, Olsen SN, Pritchard T, et al. (2019). A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 36, 660–673.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn MWM, Song E, Feng Z, Sinha A, Chen C-W, Deshpande AJ, Cusan M, Farnoud N, Mupo A, Grove C, et al. (2016). Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov 6, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AR, Li Q, Hudson WA, Chen W, Sam T, Yao Q, Lund EA, Wu B, Kowal BJ, and Kersey JH (2009). A role for MEIS1 in MLL-fusion gene leukemia. Blood 113, 1756–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, Adli M, Zeleznik-Le NJ, and Bushweller JH (2015). Degree of Recruitment of DOTIL to MLL::AF9 Defines Level of H3K79 Di- and Tri-methylation on Target Genes and Transformation Potential. Cell Reports 11, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise Maps of RNA Polymerase Reveal How Promoters Direct Initiation and Pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender CA, Shapiro AJ, Burkholder AB, Bennett BD, Adelman K, and Fargo DC (2017). ORIO (Online Resource for Integrative Omics): a web-based platform for rapid integration of next generation sequencing data. Nucleic Acids Res 45, 5678–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, and Shilatifard A (2010). AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Mol Cell 37, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo-de-Oliveira MS, et al. (2018). The MLL recombinome of acute leukemias in 2017. Leukemia 32, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KSJ, Roeder RG, et al. (2010). Multiple Interactions Recruit MLL1 and MLL1 Fusion Proteins to the HOXA9 Locus in Leukemogenesis. Mol Cell 38, 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar M-P, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, et al. (2007). A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. (2018). SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, eaao2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulloy JC, Wunderlich M, Zheng Y, and Wei J (2008). Transforming human blood stem and progenitor cells: A new way forward in leukemia modeling. Cell Cycle 7, 3314–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean AG, and Hess JL (2012). The Pathogenesis of Mixed-Lineage Leukemia. Annu Rev Pathology Mech Dis 7, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Ferguson FM, Seong BKA, Kuljanin M, Leggett AL, Mohardt ML, Robichaud A, Conway AS, Buckley DL, Mancias JD, et al. (2020). Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun 11, 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann T, Herzog VA, Muhar M, Haeseler A von Zuber, J., Ameres SL, and Rescheneder P (2019). Guantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets. Bmc Bioinformatics 20, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, and Zhang Y (2005). hDOHL Links Histone Methylation to Leukemogenesis. Cell 121, 167–178. [DOI] [PubMed] [Google Scholar]
- Okuda H, Stanojevic B, Kanai A, Kawamura T, Takahashi S, Matsui H, Takaori-Kondo A, and Yokoyama A (2017). Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J Clin Invest 127, 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner F, Gadrey JY, Xiong Y, Hatton C, Eschle BK, Weiss A, Stauffer F, Gaul C, Tiedt R, Perry JA, et al. (2020). Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 136, 1983–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer KA, Mimoso CA, Adelman K, and Neugebauer KM (2021). Co-transcriptional splicing regulates 3’ end cleavage during mammalian erythropoiesis. Mol Cell 81, 998–1012.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidawind C, Jeong J, Schneidawind D, Kim I-S, Duque-Afonso J, Wong SHK, Iwasaki M, Breese EH, Zehnder JL, Porteus M, et al. (2018). MLL leukemia induction by t(9; 11) chromosomal translocation in human hematopoietic stem cells using genome editing. Blood Adv 2, 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, Tzankov A, Iacovino M, Lau I-J, Milne TA, et al. (2016). MLL::AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 30, 43–58. [DOI] [PubMed] [Google Scholar]
- Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, Jongen-Lavrenic M, Altman JK, Thomson B, Blakemore SJ, et al. (2018). The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131, 2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel KR, Ellis JD, Spielman CL, Bomber ML, and Hiebert SW (2021). Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol Cell 81, 530–545.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, Krivtsov AV, Rücker FG, Döhner K, McGeehan GM, et al. (2020). Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 367, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters AC, and Bernt KM (2017). MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Frontiers Pediatrics 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TCP, So CWE, and Cleary ML (2007). Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Gene Dev 21, 2762–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, and Cleary ML (2008). Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O, Meyerson M, and Cleary ML (2005). The Menin Tumor Suppressor Protein Is an Essential Oncogenic Cofactor for MLL-Associated Leukemogenesis. Cell 123, 207–218. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Lin M, Naresh A, Kitabayashi I, and Cleary ML (2010). A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell 17, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisig DT, Bittner CB, Zeisig BB, García-Cuéllar M-P, Hess JL, and Slany RK (2005). The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 24, 5525–5532. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Liu Q, Acharya P, Stengel KR, Sheng Q, Zhou X, Kwak H, Fischer MA, Bradner JE, Strickland SA, et al. (2016). High-Resolution Mapping of RNA Polymerases Identifies Mechanisms of Sensitivity and Resistance to BET Inhibitors in t(8;21) AML. Cell Reports 16, 2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Gene expression changes upon DOT1L/MENIN inhibition and MLL::AF9 degradation, related to Figure 5
Table S4. MLL::AF9 occupancy changes upon DOT1L and MENIN inhibition, related to Figure 6
Data Availability Statement
RNA-seq, ChIP-seq, ATAC-seq, SLAM-seq, and PRO-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Publicly available data used for GSEA analyses are listed in the key resource table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-HA, rabbit | Cell Signaling Technology | Cat# 3724 (C29F4) |
| anti-cleaved PARP (Asp214), rabbit | Cell Signaling Technology | Cat# 5625 (D64E10) |
| anti-PARP, rabbit | Cell Signaling Technology | Cat# 9542 |
| anti-GAPDH, mouse | Cell Signaling Technology | Cat# 97166 (D4C6R) |
| anti-Vinculin, rabbit | Cell Signaling Technology | Cat# 13901 (E1E9V) |
| anti-KMT2A/MLL1, rabbit | Bethyl | Cat# A300-086A |
| anti-MLL/HRX, rabbit | Active Motif | Cat# 61296 |
| anti-MENIN, rabbit | Bethyl | Cat# A300-105A |
| anti-DOT1L, rabbit | Cell Signaling Technology | Cat# 77087 (D1W4Z) |
| anti-H3K79me2, rabbit | Cell Signaling Technology | Cat# 5427 (D15E8) |
| anti-H3K27ac, rabbit | Diagenode | Cat# C15410196 |
| anti-H3K9ac, rabbit | Abcam | Cat# ab4441 |
| anti-H3K4me3, rabbit | Abcam | Cat# ab8580 |
| anti-RNA Pol II (Rpb1 CTD), mouse | Cell Signaling Technology | Cat# 2629 (4H8) |
| anti-RNA Pol II pSer2 (Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2)), rabbit | Abcam | Cat# ab5095 |
| anti-RNA Pol II pSer5 (Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5)), rabbit | Abcam | Cat# ab5131 |
| anti-AFF4/MCEF | Bethyl | Cat# A302-538A |
| anti-CDK9, mouse | Santa Cruz Biotechnology | Cat# SC-13130 |
| anti-human CD11b, APC-conjugated | BioLegend | Cat# 301309 (ICRF44) |
| anti-human CD13, PerCP/Cy5.5-conjugated | BioLegend | Cat# 301714 (WM15) |
| anti-human CD14, PE/Cy7-conjugated | BioLegend | Cat# 301814 (M5E2) |
| anti-mouse sca-1, PE/Cy7 | Invitrogen | Cat# 25-5981-82 (D7) |
| anti-mouse CD117 (c-Kit), Alexa Fluor® 647 | BioLegend | Cat# 105818 (2B8) |
| Rabbit IgG, HRP-linked | Cytiva | Cat# NA934V |
| anti-rabbit, Alexa Fluor®647-Conjugate | Cell Signaling Technology | Cat# 4414 |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine Serum Albumin | Sigma Aldrich | Cat# A7906 |
| UltraPure BSA (50 mg/mL) | Invitrogen | Cat# AM2616 |
| EPZ-5676 | Selleckchem | Cat# S7062 |
| VTP-50469 | Syndax | provided by Syndax Pharmaceuticals |
| dTAG-13 | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dNEG-13 | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dTAG-VHL | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| dNEG-VHL | provided by Nathanael Gray lab | provided by Nathanael Gray lab |
| Z-VAD-FMK | Selleckchem | Cat# 7023 |
| DSG (disuccinimidyl glutarate) | Thermo Fisher Scientific | Cat# 20593 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28906 |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | Cat# D2650 |
| Biotin-11-ATP | PerkinElmer | NEL544001EA |
| Biotin-11-CTP | PerkinElmer | NEL542001EA |
| Biotin-11-GTP | PerkinElmer | NEL545001EA |
| Biotin-11-UTP | PerkinElmer | NEL543001EA |
| Critical commercial assays | ||
| Amicon Ultra-15 | Millipore | Cat# UFC910024 |
| X-tremeGene 9 DNA Transfection Reagent | Millipore | Cat #06365809001 |
| Polybrene Transfection Reagent | Millipore | Cat# TR-1003-G |
| RetroNectin GMP grade | Takara | Cat# T202 |
| Cell Dissociation Buffer Enzyme-Free | Gibco | Cat# 13151-014 |
| EasySep Mouse Neutrophil Enrichment Kit | StemCell Technologies | Cat# 19762 |
| EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit | StemCell Technologies | Cat# 19856 |
| StemSpan SFEM | StemCell Technologies | Cat# 09600 |
| Mouse Recombinant SCF | StemCell Technologies | Cat# 78064 |
| Mouse Recombinant Flt3/Flk-2 Ligand | StemCell Technologies | Cat# 78011 |
| Mouse Recombinant TPO | StemCell Technologies | Cat# 78072 |
| Mouse Recombinant IL-3 | StemCell Technologies | Cat# 78042 |
| Mouse Recombinant IL-6 | StemCell Technologies | Cat# 78052 |
| Human Recombinant SCF | StemCell Technologies | Cat# 78062 |
| Human Recombinant Flt3/Flk-2 Ligand | StemCell Technologies | Cat# 78009 |
| Human Recombinant TPO | StemCell Technologies | Cat# 78210 |
| Human Recombinant IL-3, ACF | StemCell Technologies | Cat# 78146 |
| Human Recombinant IL-6 | StemCell Technologies | Cat# 78050 |
| BD Pharm Lyse Buffer | BD Biosciences | Cat# 555899 |
| EasySep Human Cord Blood CD34 Positive Selection Kit II | StemCell Technologies | Cat# 17896 |
| Lymphoprep | StemCell Technologies | Cat# 07801 |
| SepMate-50 (IVD) | StemCell Technologies | Cat# 85450 |
| DAPI | Santa Cruz Biotechnology | Cat# 28718-90-3 |
| JorVet DipQuick Stain | Jorgensen Laboratories, Inc. | Cat# J-322 |
| Annexin V Apoptosis Detection Kit APC | eBioscience | Cat# 88-8007-74 |
| BD Cytofix/Cytoperm | BD Biosciences | Cat# 554722 |
| BD Perm/Wash | BD Biosciences | Cat# 554723 |
| BD Permeabilization Buffer Plus | BD Biosciences | Cat# 561651 |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen | Cat# NP0007 |
| Precision Plus Dual Color Standards | BioRad | Cat #12620S |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34095 |
| RNeasy Mini RNA Extraction Kit | Qiagen | Cat# 74106 |
| RNase-Free DNase | Qiagen | Cat# 79254 |
| SuperScript™ First-Strand Synthesis System | Invitrogen | Cat# 11904018 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat# 4369542 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Glycogen, ultrapure | Thermo Fisher Scientific | Cat# 10814010 |
| mRNA decapping enzyme (MDE) | New England Biolabs | Cat# M0608S |
| NEBNext Ultra II Q5 Master mix | New England Biolabs | Cat# M0544L |
| Q5 DNA polymerase (using included high GC enhancer) | New England Biolabs | Cat# M0491L |
| SUPERase-In RNase inhibitor | Thermo Fisher Scientific | Cat# AM2696 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18090200 |
| SYBR Gold nucleic acid stain | Thermo Fisher Scientific | Cat# S11494 |
| T4 Polynucleotide kinase | New England Biolabs | Cat# M0201L |
| T4 RNA Ligase 1 (High Conc.) | New England Biolabs | Cat# M0204L |
| T4 RNA Ligase 2, truncated KQ | New England Biolabs | Cat# M0373L |
| Trizol Reagent | Thermo Fisher Scientific | Cat# 15596026 |
| Pronex Size-Selective Purification System | Promega | Cat# NG2001 |
| Total RNA purification kit | Norgen Biotek Corp | Cat# 17250 |
| Dynabeads MyOne Streptavidin C1 beads | Thermo Fisher Scientific | Cat# 65002 |
| NEB 5′ DNA adenylation kit | New England Biolabs | Cat# E2610S |
| NEBNext Library Quant Kit for Illumina | New England Biolabs | Cat# E7630L |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7530 |
| SLAMseq Kinetics Kit - Anabolic Kinetics Module | Lexogen | Cat# 061.24 |
| RNeasy Plus Mini RNA Extraction Kit | Qiagen | Cat# 74134 |
| QIAshredder | Qiagen | Cat# 79654 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | Cat # 20034197 M0541L |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat # M0541L |
| QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina | Lexogen | Cat# 015.96 |
| Ampure XP | Beckman Coulter | Cat# A63880 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| ThruPlex DNA-seq Kit | Takara | Cat# R400675 |
| Dynabeads Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat #10001D |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat #10003D |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat #Q32851 |
| D5000 Reagents | Agilent | Cat# 5067-5589 |
| D5000 Screen Tape | Agilent | Cat# 5067-5588 |
| High Sensitivity D5000 Reagents | Agilent | Cat# 5067-5593 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067-5592 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| NextSeq 500 High Output v2 | Illumina | Cat #FC-404-2005 |
| Deposited data | ||
| Mendeley Dataset DOI URL | This paper | doi: 10.17632/n55f7xfx79.1 |
| GEO Superseries | This paper | GSE173600 |
| Raw and analyzed RNA-Seq Data | This paper | GSE173574 |
| Raw and analyzed ChIP-Seq Data | This paper | GSE173599 |
| Raw and analyzed ATAC-Seq Data | This paper | GSE189098 |
| Raw and analyzed SLAM-Seq Data | This paper | GSE173571 |
| Raw and analyzed PRO-Seq Data | This paper | GSE189105 |
| Bernt MLL::AF9 Targets | Bernt et al., 2011 | GSE29130 |
| Guenther MLL-AF4 targets | Guenther et al., 2008 | GSE13313 |
| Stavropoulou MLL::AF9 Removal | Stavropoulou et al., 2016 | GSE65384 |
| LSK Gene Set | Uckelmann et al., 2020 | GSE129638 |
| Experimental models: Cell lines | ||
| HL-60 | ATCC | CCL-240 |
| MOLM13 | DSMZ | ACC 554 |
| 293T | ATCC | CRL-3216 |
| murine MLL::AF9 leukemia | This paper | MLL::AF9 #1, MLL::AF9 #2 |
| murine MLL::AF9-HA-FKBP12 leukemia | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| hCD34+ MLL::AF9 leukemia | This paper | MLL::AF9 #1, MLL::AF9 #2 |
| hCD34+ MLL::AF9-HA-FKBP12 leukemia | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| Drosophila melanogaster S2 cells | Nascent Transcriptomics Core at Harvard Medical School | Drosophila melanogaster S2 cells for spike-in controls |
| Experimental models: Organisms/strains | ||
| C57BL/6NCrl | Charles River Laboratories | C57BL/6 |
| Oligonucleotides | ||
| 3′ adapter | IDT | /5Phos/rGrArUrCrGrUrCrGrGrArCrUrGrUrArGrArArCrUrCrUrGrArArC/3InvdT/ |
| 5′ adapter | IDT | rCrCrUrUrGrGrCrArCrCrCrGrArGrArArUrUrCrCrA |
| Illumina TruSeq small RNA indexing PCR primers | IDT | CAAGCAGAAGACGGCATACGAGATxxxxxxGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA (where Xs indicate index sequences) |
| RT and PCR primer | IDT | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA |
| Recombinant DNA | ||
| pMSCV-MLL::AF9-IRES-GFP | Armstrong Lab | MLL::AF9 #1, MLL::AF9 #2 |
| pMSCV-HA-FKBP12-MLL::AF9-IRES-GFP | This paper | HA-FKBP12-MLL::AF9 |
| pMSCV-MLL::AF9-HA-FKBP12-IRES-GFP | This paper | MLL::AF9-HA-FKBP12 #1, MLL::AF9-HA-FKBP12 #2 |
| pUMVC | Addgene | Plasmid #8449 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| pCL-Eco | Addgene | Plasmid #12371 |
| Software and algorithms | ||
| bcl2fastq Conversion Software, v2.20.0.422 | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastqconversion-software.html |
| Bowtie2, v2.3.4.3 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR, v2.7.5a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| CEAS sitepro | Shin et al., 2009 | https://liulab-dfci.github.io/software/ |
| bedtools v2.28.0 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| ngs.plot | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| Slamdunk SLAMseq pipeline | Neumann et al., 2019 | https://t-neumann.github.io/slamdunk/ |
| Homo sapiens GRCh38/hg38 reference source | Broad Institute CTAT GRCh38_gencode_v33_CTAT_lib_Apr062020 | https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/ |
| Mus musculus M24/mm10 reference source | Broad Institute CTAT Mouse_gencode_M24_CTAT_lib_Apr062020 | https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/ |
| IGVtools, 2.3.75 | Robinson et al., 2011, 2017; Thorvaldsdóttir et al., 2013 | https://software.broadinstitute.org/software/igv/igvtools |
| Samtools, v1.95 | Li et al., 2009 | http://www.htslib.org/ |
| HTSeq, htseq-count, v0.11.2 | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| Picard tools, v2.9.4 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GSEA, v.3.160 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| R Bioconductor DESeq2 package v1.24.0 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MACS2 v2.1.4 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| bedgraphs2stdBedGraph | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/bedgraphs2stdBedGraph |
| Bowtie 1.2.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| bowtie2stdbedgraph | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/bowtie2stdbedgraph |
| Cutadapt 1.14 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| GetGeneAnnotation_GGA | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/GetGeneAnnotation_GGA |
| make_heatmap | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/make_heatmap |
| samtools 1.3.1 | Danecek et al., 2021 | https://www.htslib.org/ |
| trim_and_filter_PE | Nascent Transcriptomics Core at Harvard Medical School | https://github.com/AdelmanLab/NIH_scripts/tree/main/trim_and_filter_PE |
| TSScall | Lavender et al., 2017 | https://github.com/lavenderca/TSScall |