

Abstract
The ability to detect pathogens specifically and sensitively is critical to combat infectious diseases outbreaks and pandemics. Colorimetric assays involving loop‐mediated isothermal amplification (LAMP) provide simple readouts yet suffer from the intrinsic non‐template amplification. Herein, a highly specific and sensitive assay relying on plasmonic sensing of LAMP amplicons via DNA hybridization, termed as plasmonic LAMP, is developed for the severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) RNA detection. This work has two important advances. First, gold and silver (Au–Ag) alloy nanoshells are developed as plasmonic sensors that have 4‐times stronger extinction in the visible wavelengths and give a 20‐times lower detection limit for oligonucleotides over Au counterparts. Second, the integrated method allows cutting the complex LAMP amplicons into short repeats that are amendable for hybridization with oligonucleotide‐functionalized Au–Ag nanoshells. In the SARS‐CoV‐2 RNA detection, plasmonic LAMP takes ≈75 min assay time, achieves a detection limit of 10 copies per reaction, and eliminates the contamination from non‐template amplification. It also shows better detection specificity and sensitivity over commercially available LAMP kits due to the additional sequence identification. This work opens a new route for LAMP amplicon detection and provides a method for virus testing at its early representation.
Keywords: hollow nanostructures, homogeneous assays, loop‐mediated isothermal amplification, plasmonics, SARS‐CoV‐2
Plasmonic sensing of isothermally amplified SARS‐CoV‐2 RNA by sequence‐specific hybridization allows differentiating the amplicons generated from template versus non‐template amplification and thus improving the detection sensitivity and specificity.
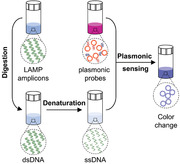
1. Introduction
Developing sensitive and reliable diagnostic methods is critical to accurately screen potential cases of patients and combat infectious disease outbreaks, as evidenced by the current coronavirus disease 2019 (COVID‐19) pandemic.[ 1 , 2 , 3 , 4 ] Polymerase chain reaction (PCR) has been the gold standard for molecular tests.[ 5 ] However, its time‐consuming protocols and the need for laboratory infrastructure largely preclude it from point‐of‐care (POC) testing.[ 6 , 7 ] Isothermal amplification methods, such as loop‐mediated isothermal amplification (LAMP), have emerged as an alternative to PCR and allow POC testing without the need for thermal cycling.[ 8 ] Although simple, LAMP is susceptible to non‐template amplification and its simple readout (e.g., based on pH change) cannot distinguish template versus non‐templated amplification, thus leads to false‐positive results.[ 8 , 9 , 10 , 11 , 12 ] Previous efforts in reducing the non‐template amplification focused on primer design, additives introduction (e.g., betaine), and closed‐tube detection.[ 13 , 14 , 15 ] However, it is still difficult to prevent the non‐template amplification caused by the formation of primer dimers, considering that a number of primers (up to 6) are used in LAMP assays and the potential contamination from isothermal conditions (e.g., water bath).[ 15 ] It is therefore important to develop techniques that can accurately identify the amplified sequences via an accessible detection scheme, in order to provide diagnostic platforms with simple readouts and high detection specificity and sensitivity.
So far, there have been several approaches that can detect isothermal amplicons via sequence recognition. For example, the utilities of molecular beacons and strand displacement probes allow detection of single‐stranded loops that are formed among the primer regions of LAMP amplicons.[ 16 , 17 , 18 ] However, extensive expertise in molecule design (i.e., primers and probes) is required to identify those specific regions. Also, it has been reported that the detection of LAMP amplicons by molecular beacons has a poor sequence specificity.[ 19 ] Recently, clustered regularly interspaced short palindromic repeats (CRISPR)‐based diagnostic methods that combine nucleic acid amplification with CRISPR‐associated protein (Cas) assisted enzymology have been developed for specific recognition of target DNA or RNA.[ 20 , 21 , 22 , 23 ] Taking Cas12 as an example, it couples with single guide RNA (sgRNA) for the recognition of the protospacer‐adjacent motif of double‐stranded DNA (dsDNA) as LAMP amplicons and seamlessly activates its cleavage activity for the signal generation.[ 20 , 24 ] Although the ability to directly detect LAMP amplicons without the need for generating single‐stranded DNA (ssDNA) is a truly advantage in molecular diagnostics, CRISPR‐based diagnosis has an off‐target phenomenon that is caused by the non‐specific binding of sgRNA and can lead to false positive results.[ 25 , 26 ] A precise selection of sgRNAs through specialized bioinformatics tools is thus needed in order to reduce the off‐target effects. Motivated by this challenge, we aim to develop an approach that integrates isothermal amplification and amplicon sensing with simple detection readouts, and thus provide a more accessible method using readily available reagents.
In this study, we report a robust method for nucleic acid detection, based on plasmonic sensing of LAMP amplicons via DNA hybridization, termed as plasmonic LAMP. Using severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) RNA as a model, we demonstrate that the plasmonic LAMP method achieved a detection limit of ten copies per reaction via colorimetric analysis, which is a desirable diagnostic toolkit to reduce the severity of COVID‐19 pandemic.[ 2 , 27 , 28 ] Plasmonic LAMP has two distinctive features. First, we developed gold–silver (Au–Ag) nanoshells as sensitive plasmonic labels. The Au–Ag shells have 4‐times stronger plasmonic extinction in the visible wavelengths and provide 20‐times sensitivity enhancement in plasmonic sensing of oligonucleotides over Au nanoparticles (NPs). Second, we introduced restrict enzyme digestion and heat denaturation in order to cut the concatemer‐like LAMP amplicons into short repeats that are amendable for subsequent hybridization with oligonucleotides‐functionalized Au–Ag shells as plasmonic sensors (Scheme 1 ). This plasmonic LAMP approach has several advantages over other molecular tests: 1) the restriction enzymes can recognize a plethora of specific sequences (dsDNA) and digest them at specific sites;[ 29 ] 2) it has an additional sequence identification enabled by the plasmonic sensing and thus improves the detection specificity and sensitivity over the conventional LAMP and AuNP‐assisted LAMP;[ 30 , 31 , 32 , 33 , 34 ] 3) compared with amplification‐free approaches, it eliminates the contamination from non‐specific interactions (e.g., highly ionic strength) that cause false signals and saturation of probes;[ 35 , 36 ] and 4) despite the multistep operation, it detects the target‐specific sequence, instead of non‐specific molecular labels (e.g., biotin‐labeled primers) that are frequently used in a number of isothermal amplification and lateral flow assays.[ 27 ] Therefore, our study is an important step toward advancing sensitive and reliable molecular tests.
Scheme 1.
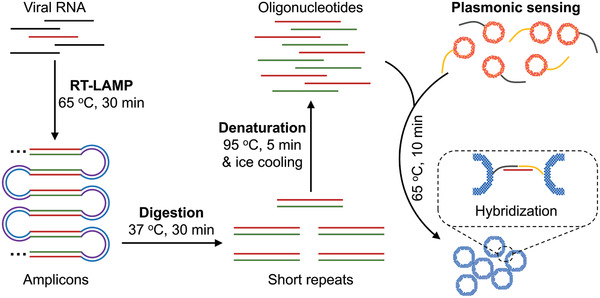
Schematic illustration of the major steps involved in the plasmonic LAMP for viral RNA detection. The RNA is first reverse transcribed and amplified into LAMP amplicons that are subjected to restriction enzymes digestion, forming short repeats that can be denatured into oligonucleotides for subsequent DNA hybridization linked with plasmonic sensors. RT‐LAMP is reverse transcription loop‐mediated isothermal amplification.
2. Results and Discussion
2.1. Controllable Synthesis and Characterization of Au–Ag Nanoshells
We started with the synthesis of Au–Ag nanoshells through titrating AgNPs with gold chloride trihydrate (HAuCl4). In a standard synthesis, 10 mL of HAuCl4 is injected at a speed of 6 mL h−1 into a matrix solution containing AgNPs with an average size of 32 nm as the sacrificial templates (Figure S1, Supporting Information) and 2 mm sodium citrate (Na3CA) as a reductant and a colloidal stabilizer (see Experimental Section for details). In this case, AuCl2 − is first generated from AuCl4 − via Na3CA reduction and directs the galvanic replacement reaction with favored high standard reduction potential.[ 37 ] Figure 1A illustrates the growth of Au–Ag shells, where a pitting process can be observed in the beginning of reaction, followed by the cavity evolution and simultaneous shell formation as more HAuCl4 are injected. Figure 1B–D shows the transmission electron microscopy (TEM) images of products taken from the reaction, confirming the morphological transformation. The Au–Ag shells as final products have an inner diameter of 30.0 ± 4.78 nm and outer diameter of 45.9 ± 4.45 nm by randomly measuring 200 particles, indicating a relative low inhomogeneity (10–15%). Despite this, Na3CA as a capping ligand facilitates the conjugation of oligonucleotides as discussed later. Notably, the absence of Na3CA leads to the formation of thin and porous cages at a HAuCl4 load of 3.3 mL (Figure S2, Supporting Information, in equilibrium to 10 mL AuCl2 −). Interestingly, the colloidal stabilities of hollow nanostructures (i.e., shells and cages) were barely affected by the low amount or absence of Na3CA, as evidenced by the increased zeta potential magnitudes over the Ag seeds (Figure S3A, Supporting Information).[ 38 ] They could also remain colloidally stable against 50 mm salt solution (Figure S3B,C, Supporting Information). Furthermore, the different pathways for the growth of shell and cage can be attributed to the reaction stoichiometry, where one Ag atom is replaced by one Au atom in the case of AuCl2 −, instead of three Ag atoms for AuCl4 −. Also, the dissolution of the Ag template requires a longer time in the case of AuCl2 − since less openings are preserved on the surface.[ 37 ] As a result, the slow pitting process increased shell thickness and thus limits the red shift of the localized surface plasmon resonance (LSPR) peak as compared to that of cages (more in next section). The energy‐dispersive X‐ray (EDX) mapping of an individual Au–Ag shell (Figure 1E) shows that the Au and Ag elements are distributed across the entire particle, confirming the alloyed structure. Figure S4, Supporting Information shows an atomic‐resolution high‐angle annular dark‐field scanning TEM (HAADF‐STEM) image of a single particle, where the lattice spacing (1.43 Å) can be attributed to the interplane distance of the (110) planes of the face‐centered cubic Au and Ag. The alloy structure is expected to improve the corrosion resistance of Au–Ag nanoshells over the AgNPs.[ 39 ]
Figure 1.
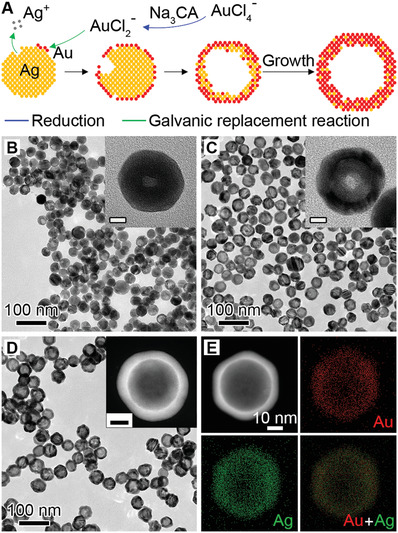
Synthesis and characterization of Au–Ag nanoshells. A) Schematic illustration of the Au–Ag shells growth based on galvanic replacement reaction in the presence of 2 mm sodium citrate. TEM images of aliquots taken from the reaction when B) 1.0, C) 2.5, and D) 10.0 mL HAuCl4 was injected. Insets show the magnified TEM images (B,C) and HAADF‐STEM image (D) of individual particles. Scale bars in insets are 10 nm. E) EDX mapping image of an individual particle shown in (D).
We then evaluated the role of Na3CA in the shell growth by increasing its concentration from 0 to 20 mm while keeping other conditions the same. Due to the reducing ability endowed by Na3CA, we assume that the different reaction species of Au precursor (i.e., AuCl4 − and AuCl2 −) would influence the reaction stoichiometry (Figure S5A, Supporting Information). Specifically, without Na3CA, the reaction proceeds between AuCl4 − and AgNPs and results in porous cages. At low concentrations, Na3CA turns a small portion of AuCl4 − into AuCl2 − and leads to the mixture of cages and shells. In contrast, above a critical concentration (1–2 mm), Na3CA converts the majority of AuCl4 − into AuCl2 − and yields shells as the final product. As expected, the synthesized particles (Figure S5B–G, Supporting Information) have a cage‐like morphology below 1 mm Na3CA and shell‐like structure above 2 mm, supporting the proposed growth mechanism. We also found that the reaction kinetics affect the shell growth (Figure S6A, Supporting Information). Replacing the Na3CA with stronger reducing agent like l ‐ascorbic acid leads to a mixture of small NPs and shells (Figure S6B, Supporting Information). This is because the l ‐ascorbic acid can also reduce AuCl4 − into atoms for the self‐nucleation growth of NPs. On the other hand, boosting the loading rate of HAuCl4 by increasing its concentration and injection speed led to the formation of branched Au–Ag shells (Figure S6C, Supporting Information). When either the HAuCl4 concentration or injection speed increases, Au–Ag shells with a few holes on the surface can be observed (Figure S6D,E, Supporting Information), suggesting a small portion of AuCl4 − participated in the reaction. In all cases, the increased deposition rate of Au atoms is the key to the preferential overgrowth of branched or porous shells, given that the diffusion rate remains constant during the growth.[ 40 ]
2.2. LSPR Properties of Au–Ag Nanoshells
Next, we investigated the LSPR properties of the Au–Ag shells since they are critical for plasmonic sensing. Figure S7, Supporting Information shows the photographs of aliquots taken from the standard synthesis (top panel, nanoshells) and in the absence of Na3CA (bottom panel, nanocages). The suspension of Au–Ag shells remains red‐purple, while the cages change from yellow to blue with lower amount of Au precursor injected. The corresponding extinction spectra clearly reveal a different peak shift during the formation of these two structures. The LSPR extinction peak (LSPR λmax) of Au–Ag shells shifted from 392 to 530 nm (Figure 2A), while that of cages shifted to a longer wavelength in near‐infrared region (Figure 2B). Both nanostructures show split LSPR peaks during the galvanic replacement reaction due to co‐existence of Ag cores (for peak at 400 nm wavelength) and Au–Ag walls (for peak at a longer wavelength). The limited and slow peak shifting of Au–Ag shells (Figure 2C) arises from the formation of thick and solid shells during the reaction. In contrast, the appearance of holes on surface led to the quick decrease of Ag content and thus made the peak shift rapidly and widely.[ 37 , 41 ] Furthermore, the extinction intensity change of the cages and shells differs from each other, with a monotonic decrease and falling‐rising trend, respectively (Figure 2D). This suggests the Au–Ag shells have higher extinction than the cages. Similar trends were observed for the Au–Ag shells and cages synthesized with different Na3CA concentrations (Figures S4,S8, Supporting Information).
Figure 2.
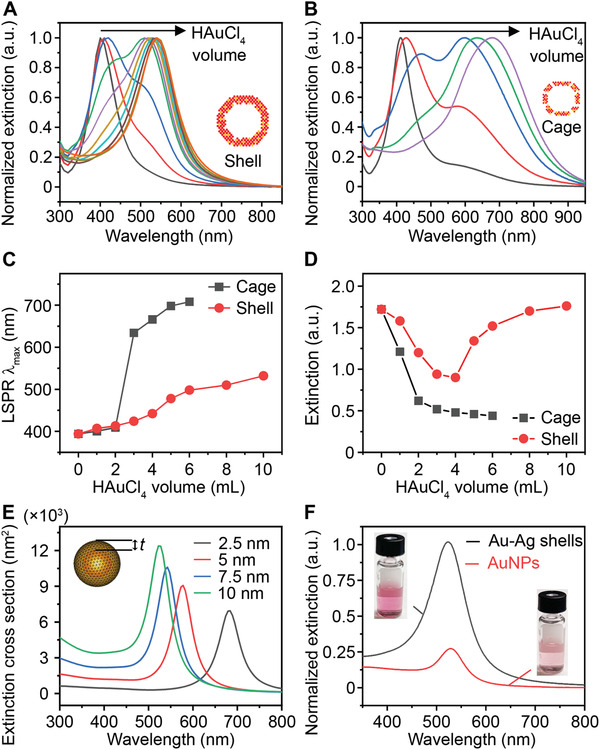
The LSPR properties of Au–Ag nanoshells and nanocages. LSPR extinction spectra of aliquots taken from the A) shell‐growth and B) cage‐growth reaction at an injection increment of 1 mL HAuCl4 (0.004% w/v). Insets show the models of shell and cage. Comparison of C) LSRP extinction peak λmax and D) maximal extinction intensity change during the growth of shells and cages. E) Simulation results of the extinction cross section area of a hollow shell with varied thickness (t). Inset shows the model. F) Normalized LSPR extinction of Au–Ag shells and 50 nm AuNPs at same particle concentration. Insets show the photographs of corresponding suspensions.
To study the impact of wall thickness on the LSPR property of the Au–Ag shells, we performed a numerical simulation using the boundary element method (BEM).[ 42 , 43 ] Based on the TEM image of the Au–Ag shell (Figure 1D) and the Au:Ag molar ratio of 76%:24% obtained from inductively coupled plasma‐mass spectrometry (ICP‐MS), we first set a geometric model with Au:Ag molar ratio of 80%:20% (homogeneously distributed), 30 nm interior diameter, and varied wall thickness (t) ranging from 2.5 to 10 nm (Figure 2E inset). The simulation results suggest that increasing the shell thickness leads to blue shift of LSPR λmax and higher extinction cross section area (Figure 2E). We also set a model to understand the peak splitting in the early stage of growth. As shown in Figure S9, Supporting Information, a sandwich Ag–water–AuAg core–void‐shell structure shows dual peaks located before and after 400 nm. Increasing the Au content in the shell leads to the intensity change for both peaks, where the first peak decreases and second one increases. This is in good agreement with our experimental observation. Additionaly, both the electric field intensity and distribution continuously increase with the shell thickness (Figure S10, Supporting Information). The enhancement in local field effect of Au–Ag shells is mainly ascribed to the collective effect of surface plasmons of unique shell structure, which condenses the interacting light to the structural center.[ 44 ] It should be pointed out that in recent studies, it has been reported that the holes on the surface of NPs have an influence on the LSPR λmax.[ 45 ] This phenomenon is also observed in the shells, as evidenced by the extinction spectra, where the shells with and without holes show peak shift (Figure S6F, Supporting Information). We also compared the LSPR properties of Au–Ag shells and 50 nm AuNPs (Figure S11, Supporting Information) at the same particle concentration (8.77 × 109 mL−1) and observed a 4‐times higher peak extinction while maintaining similar peak position at visible wavelengths (500–600 nm, Figure 2F). Taken together, the Au–Ag nanoshells possess stronger plasmonic extinction at visible range compared with AuNPs and have the potential to be a class of highly sensitive plasmonic sensors.
2.3. Sensitive Colorimetric Detection of Oligonucleotides by Au–Ag Nanoshells
We further examined the Au–Ag nanoshells that were obtained from standard synthesis as labels for oligonucleotide detection and compared the detection performance with 50 nm AuNPs. The rationale is that metallic nanostructures with hollow interior show superior plasmonic activities compared to their solid counterparts, due to the plasmon hybridization that induces field enhancement from the inner and outer space.[ 46 , 47 ] On the other hand, nanoshells with enhanced peak extinction and LSPR peaks in the visible wavelength is more desired over nanocages for colorimetric sensing. We first designed a sequence associated with the SARS‐CoV‐2 N gene (nucleocapsid phosphoprotein gene) as the target and two complementary sequences as probes (Table S1, Supporting Information). A pH‐assisted method was adopted, where the Au–Ag shells were incubated with thiol‐modified oligonucleotides in an acidic buffer for 30 min.[ 48 ] Spectra measurements confirm that the sufficient coating of oligonucleotides on NPs’ surface yields stable NP‐oligonucleotides complexes and leads to the disappearance of secondary LSPR peak ≈700 nm (Figure S12A, Supporting Information). The purified products show a red shift of the main LSPR peak to ≈530 nm (Figure S12B, Supporting Information), increased hydrodynamic size (Figure S12C, Supporting Information), and high stability in an aqueous solution including up to 4 m sodium chloride solution (Figure S12D, Supporting Information). To perform the plasmonic sensing, we mixed serial dilutions of the target oligonucleotides with a pair of shells‐based sensors in a hybridization buffer and subjected them to naked‐eye observation or UV–vis measurements (Figure 3A). Figure 3B shows the digital photo taken from the completed assay solutions, where a color change can be observed at a target concentration of 250 pm against the blank sample (0 pm) as a reference. For quantitative analysis, the corresponding LSPR extinction spectra were recorded and normalized at 537 nm (Figure 3C). A calibration curve was obtained by plotting the extinction intensity (I ext) ratio between λmax = 675 and 537 nm against target concentration (Figure 3D). A good linear relationship (R 2 = 0.983) was observed in 1–100 pm (Figure 3E). The limit of detection (LOD) was determined to be 3.6 pm, by calculating the concentration corresponding to a signal that is 3 times standard deviation above the zero calibrator.[ 49 ] Notably, this LOD is ≈20 times lower than that of using 50 nm AuNPs as probes under same conditions (Figure S13, Supporting Information). Because the Au–Ag shells and AuNPs have similar size and shape, the improved LSPR property including the four‐fold higher extinction intensity of the shells is likely the key to the sensitivity enhancement. It is worth noting that a ratiometric quantification method is adopted in the present work, which also contributes to the improved analytical performance.
Figure 3.
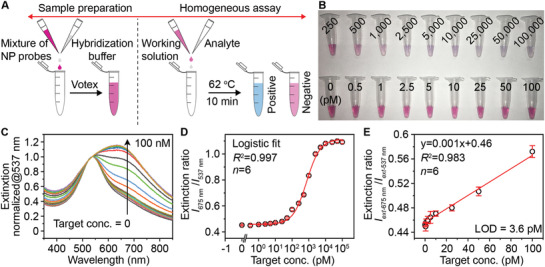
Oligonucleotide detection by the Au–Ag‐shells‐based plasmonic coupling assay. A) Schematic illustration of the major steps involved in the plasmonic coupling assay. The operation can be simplified in one step (mixing probes and analyte in the buffer). B) Representative photographs of the completed assay solutions for the detection of target standards with varied concentrations. C) Normalized LSPR extinction spectra of detection results taken from (B). D) Corresponding calibration curve generated by plotting the extinction intensity (I ext) ratio against target concentration. E) A linear range of the calibration curve shown in (D). Error bars indicate the standard deviations (n = 6).
2.4. Ultrasensitive and Reliable RNA Detection by Plasmonic LAMP
Lastly, we evaluated the diagnostic performance of plasmonic LAMP for the detection of SARS‐CoV‐2. A set of primers was first designed targeting six sites flanking a conserved region in the N gene of SARS‐CoV‐2 (Figure 4A, sequence information in Table S2, Supporting Information). With reverse transcription LAMP (RT‐LAMP, 65 °C, 30 min), the viral RNA was reverse transcribed into complementary DNA and subsequently amplified via self‐priming loop structures and a high strand‐displacing DNA polymerase that generates large cauliflower‐like concatemers.[ 8 ] The RT‐LAMP products were verified by agarose gel electrophoresis that reveals multiple bands in a ladder‐like pattern indicative of successful LAMP amplification, whereas a sample without RNA input lacked any such bands (red box of Figure 4B). The concatemers were then subjected to restriction endonuclease digestion by HincII and EaeI (37 °C, 30 min), which recognize and cleave GTY/RAC and Y/GGCCR sites, respectively. These sites are conserved throughout the long concatemers such that their cleavage by digestion collapses the concatemer bands and produces significantly shorter sequences more suitable for detection. The gel image (blue box of Figure 4B) reveals clean bands located below 200 base‐pair (BP) length, confirming the effective and uniform digestion. Next, the reaction product was denatured into ssDNA by heating to 95 °C for 5 min and cooling on ice for 2 min, followed by mixing with a set of shells‐based probes and incubated at 65 °C for 10 min or room temperature for 30 min. Significantly, a color change can be observed at RNA input ≥10 copies µL−1 or 80 copies per reaction (8 µL RNA sample per reaction, Figure 4C). Figure 4D shows corresponding LSPR extinction spectra normalized at 537 nm. The spectral signal obtained from 10 RNA copies µL−1 input can be faithfully differentiated from the background, indicating the empirically range of measured LOD in 1–10 copies µL−1. A linear relationship (R 2 = 0.99) was observed covering 3 logs from 1 to 1000 copies µL−1 (Figure 4E). The LOD was determined to be 1.3 copy/µL, equivalent of 10 copies per reaction. This result confirmed the potential capability of plasmonic LAMP approach for the single‐molecule RNA detection upon protocol optimization. When used respiratory syncytial virus (RSV) RNA as initial target, it led to insignificant spectral change as compared to the reference sample, indicating the high specificity of this assay.
Figure 4.
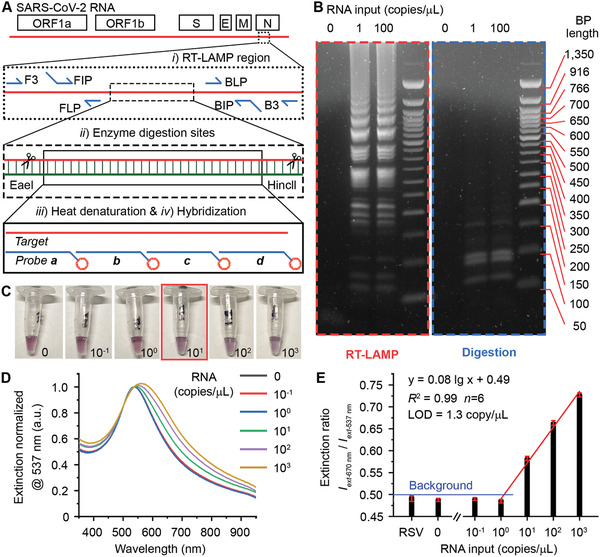
Plasmonic LAMP for sensitive detection of SARS‐CoV‐2 RNA. A) Schematic illustration of the SARS‐CoV‐2 genome architecture and reaction proceeding. Target region, primers, cutting sites, and probe sequences are highlighted. B) Gel images showing the representative products after RT‐LAMP and enzyme digestion at different RNA input. C) Photographs taken from the completed assay at varied RNA input (copies µL−1). The red box highlights the visual detection limit. D) Corresponding LSPR extinction spectra (normalized at 537 nm) of the detection results shown in (C). E) Linear region of the calibration curve shown in (D). Error bars indicate the standard deviations of six parallel measurements. RSV is respiratory syncytial virus, and its RNA was extracted and cleaned before use.
It should be emphasized that the enzyme digestion and heat denaturation (steps ii and iii, Figure 4A) involved in the plasmonic LAMP are necessary. Without heat denaturation (step iii), directly detecting the RT‐LAMP products via oligonucleotide sensors results in no color change since RT‐LAMP amplicons are double‐strand DNA (dsDNA) with no available binding sites (samples c, d, Figure S14, Supporting Information). Without enzyme digestion (step ii), the produced ssDNA from denatured RT‐LAMP amplicons could easily re‐anneal back as dsDNA in the hybridization buffer due to the extensive stem‐loop and cauliflower‐like structure.[ 50 ] Likewise, this would decrease the binding efficiency between the NP probes and amplicons, hence does not lead to color change (sample e, Figure S14, Supporting Information). On the contrary, with step ii and iii, the amplicons can be cut into uniform and short repeats, which are amendable for subsequent hybridization detection with short oligonucleotides as probes, leading to highly specific and sensitive detection. Different from our method, previous efforts in implementing AuNPs‐based plasmonic sensing of LAMP products rely on indirect methods, where the ionic strength change in a salt solution or molecule‐labeled on primers essentially induce the aggregation of AuNPs‐based sensors.[ 30 , 31 , 32 , 33 , 34 , 51 , 52 , 53 ] Those methods cannot differentiate the template versus non‐template amplification and lead to false‐positive results.
To demonstrate the advances of plasmonic LAMP over commercially available colorimetric LAMP kits, we compared their detection performance for SARS‐CoV‐2 RNA using the same RT‐LAMP protocol. Figure 5A shows the photographs of colorimetric RT‐LAMP incubating with different SARS‐CoV‐2 RNA inputs and RSV RNA as a negative control. The reaction was conducted for up to 45 min at 65 °C. It can be seen that it only shows color change for SARS‐CoV‐2 RNA molecules of 50 copies µL−1 after 30 min incubation, while no color change can be observed for a short reaction time (e.g., 15 min). However, at time point of 45 min, all samples turned into yellow color, resulting in undistinguished positive and negative controls. We then performed plasmonic LAMP for the samples processed with 15 and 45 min RT‐LAMP reaction. Spectral analysis of the completed assay solutions suggests that plasmonic LAMP pushes down the detection limit to 50 copies µL−1 at 15 min and discriminates signals for 5 copies µL−1 against control sample at 45 min (Figure 5B). It should be pointed out that for both RT‐LAMP reactions with 45 min incubation, the positive and negative controls (50 and 0 copies µL−1 RNA input) show ladder patterns in the gel image (Figure 5C), confirming the products formed through the presence of the template and non‐template amplification. In this case, due to the incapability to differentiate those amplicons by a pH indicator, colorimetric RT‐LAMP has compromised detection specificity and sensitivity at prolonged incubation. In contrast, integrating the hybridization‐based plasmonic sensing with RT‐LAMP effectively discriminates the template versus non‐template amplification and thus enhances the detection sensitivity and specificity of LAMP. Taken together, we believe that the plasmonic LAMP is a highly specific and sensitive approach for nucleic acid detection, although it adds additional steps for enzymatic cutting and adds additional temperature steps. Further optimization of the workflow and protocol may allow improving the detection sensitivity and reducing the operation steps. For example, integrating a portable cartridge with pre‐stored reagents and a heating dock (similar to the Accula SARS‐CoV‐2 Test)[ 27 ] is a potential way to enable the plasmonic LAMP with a sample‐to‐answer workflow and a minimal hands‐on time.
Figure 5.
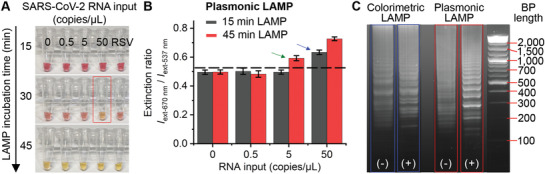
Comparison of plasmonic LAMP and a commercially available colorimetric LAMP kit for SARS‐CoV‐2 RNA detection. A) Photograph of colorimetric LAMP reaction with different RNA inputs and incubation time at 65 °C. Red box highlights the visually detectable sample. RSV RNA (50 copies µL−1) was used as a negative control. B) Detection results of plasmonic LAMP with different LAMP incubation times. At indicated times, LAMP products were processed for plasmonic sensing using Au–Ag‐shells‐based probes. Dashed line marks the background (3 times standard deviation above mean of zero indicator). Blue and green arrows indicate the detection limits for 15 and 45 min, respectively. C) Gel image of positive and negative controls obtained by colorimetric LAMP (blue) and plasmonic LAMP (red). The symbols (−) and (+) indicate 0 and 50 SARS‐CoV‐2 RNA copies µL−1 inputs, respectively. Both reactions were conducted at 65 °C for 45 min.
To demonstrate the potential clinical use, we applied the integrated plasmonic LAMP approach to detect nasal swab samples that were spiked with SARS‐COV‐2 RNA. RNA of six different concentrations in the range of 0–100 copies µL−1 were spiked in healthy nasal swab samples and tested. The analytical performance shows sensitive detection (Table 1 ) and suggests that the Au–Ag‐shells‐based sensors work well in a complex sample matrix and have potential uses in clinical setting. Taken together, the established diagnostic approach should be readily adopted as a nucleic‐acid detection platform simply by changing the set of primers and probing sequences.
Table 1.
Analytical performance of plasmonic LAMP detection of SARS‐CoV‐2 RNA‐spiked nasopharyngeal swab samples
| Viral copies in sample [copies µL−1] | Plasmonic LAMP |
|---|---|
| 50 | 6/6 a) |
| 25 | 6/6 |
| 5 | 6/6 |
| 2.5 | 4/6 |
| 0.5 | 1/6 |
| 0 | 0/6 |
Total positive samples versus total testing sample.
3. Conclusions
In summary, we have developed the plasmonic LAMP approach for the sensitive detection of SARS‐CoV‐2 RNA with improved specificity. With enzyme digestion and heat denaturation, plasmonic LAMP allows identifying amplicons via DNA hybridization utilizing Au–Ag nanoshells as sensors, allowing differentiation of template versus non‐template amplification and yielding a detection limit of 10 RNA copies per reaction. The Au–Ag shells can be prepared from AgNP templates through titration with Au+ ions and have thick and hole‐free surface. They show LSPR peak around 530 nm and high peak extinction, offering substantially enhanced detection sensitivity compared to that of conventional AuNPs‐based assay. Plasmonic LAMP has improved detection performance and only requires easily accessible heat blocks. Our work provides a diagnostic toolkit with simple readouts and highly specific and sensitive detection that may has potential for clinical applications.
4. Experimental Section
Chemicals and Materials
Gold (III) chloride trihydrate (HAuCl4·3H2O), silver nitrate (AgNO3), sodium citrate dihydrate (Na3CA⋅2H2O), sodium borohydride (NaBH4). L‐ascorbic acid (AA), gold and silver standards, tuning solution for ICP‐MS tests, and nuclease‐free water were ordered from FisherScientific Inc. HPLC purified probe sequences for oligo and RNA detection and oligo target sequences including the non‐complementary sequence and single BP mismatch sequences were ordered from BioBasic Inc. (Markham, ON, Canada). Primers for loop‐mediate isothermal amplification were ordered from Sigma Aldrich (St. Louis, MO, USA). Sherlock CRISPR SARS‐CoV‐2 were ordered from Integrated DNA Technologies, Inc. Synthetic RNA positive controls were Twist Bioscience SARS‐CoV‐2 RNA Control 2 ordered from Fisher Scientific Inc. Restriction enzymes (EaeI and HincII) and WarmStart Colorimetric LAMP 2X Master Mix (DNA & RNA) were ordered from New England Biolabs Inc. Hydroquinone, formamide, dextran sulfate, and sodium chloride (NaCl) were purchased from Sigma Aldrich (St. Louis, MO, USA). The procured chemicals were used as received. All aqueous solutions were prepared using deionized (DI) water with a resistivity of 18.0 MΩ·cm. All glassware for the synthesis of nanoparticle was cleaned using aqua regia (3:1 ratio of hydrochloric acid and nitric acid).
Preparation of 4 nm Ag Nanoparticles as Seeds
In a typical synthesis, ≈4 nm AgNPs could be prepared by reducing aqueous AgNO3 solution with NaBH4 according to a previous report.[ 54 ] Briefly, 2 mL of 1% w/v Na3CA solution and 6 mL of DI water were added to a 20 mL vial and preheated at 70 °C in an oil bath under magnetic stirring for 15 min, followed by sequentially adding 0.17 mL of 1% w/v AgNO3 solution and 0.2 mL of 0.1% w/v NaBH4 solution. The reaction was kept at 70 °C under vigorous stirring for 1 h. After cooling down to room temperature, the ≈4 nm AgNPs were diluted to 10 mL using DI water and stored in dark for further use.
Preparation of 32 nm Ag Nanoparticles as Templates
32 nm AgNPs were prepared based on a seeded‐growth method.[ 55 ] In brief, 5 mL of the prepared ≈4 nm AgNPs as seeds, 1 mL of 1% w/v sodium citrate aqueous solution, 1 mL of 1% w/v AA aqueous solution, and 35 mL of DI water were mixed in a 100 mL flask and preheated at 80 °C in an oil bath under magnetic stirring for 15 min. Then, 0.85 mL of 1% w/v AgNO3 solution was added into the mixture using a pipette. The reaction was kept at 80 °C under vigorous stirring for 1 h. The product was centrifuged and washed with DI water three times, redispersed in 50 mL of DI water, and stored in the dark for further use (0.224 nm in particle concentration).
Preparation of ≈50 nm Au–Ag Nanoshells
Approximately 50 nm Au–Ag nanoshells were prepared via galvanic replacement reaction. In brief, 3 mL of the prepared 32 nm AgNPs as templates, 1 mL of sodium citrate aqueous solution with varied concentration (0–10 mm), and 6 mL of DI water were mixed in a 50 mL flask and preheated at 95 °C in an oil bath under magnetic stirring for 15 min. Then, 0.004% w/v HAuCl4 solution was injected into the mixture using a syringe pump at a speed of 6 mL h−1 with total amount of 10 mL. After injection, the reaction was kept at 95 °C under vigorous stirring for 10 min. For the growth of nanocages, 0 mL of sodium citrate was added and only 5 mL of 0.004% w/v HAuCl4 solution was injected. The final product was centrifuged and washed with DI water three times, redispersed in 3 mL of DI water, and stored in the dark for further use.
Preparation of ≈50 nm Au Nanoparticles
15 nm AuNP was first synthesized according to the previous publication and subsequently used as seeds for the larger AuNP synthesis.[ 48 ] Briefly, water was heated to boiling under magnetic stirring. Immediately after, 1% w/v Na3CA⋅2H2O and 0.01% w/v HAuCl4·3H2O were sequentially added. After a prominent color change from purple to red, the solution was heated for additional 30 min before cooling down to room temperature. 50 nm AuNPs were synthesized using hydroquinone as a reducing agent. Specifically, hydroquinone was added to a solution of HAuCl4·3H2O, Na3CA⋅2H2O, and the 15 nm AuNPs (used as seed particles) at room temperature. This solution was stirred continually overnight to allow for AuNPs growth.
Preparation of Nanoparticle‐Oligonucleotide as Probes
Nanoparticle‐oligonucleotides conjugation was performed according to the previous work.[ 48 ] The poly A‐tail probe oligonucleotides were first resuspended in DI water per vendor suggestions before use. The SH‐capped oligonucleotides were conjugated separately to NPs in acidic buffer. Briefly, nanoparticle suspension was mixed with oligonucleotide solution before adding a 50 mm citrate‐HCl buffer with pH of 3.0 ± 0.1 in a 1:1 volumetric ratio. After 30 min of incubation at room temperature, the nanoparticle‐oligonucleotides conjugates were centrifuged and washed with DI water for three times. The purified products were redispersed in DI water and stored in the 4 °C refrigerator for further use.
Plasmonic Coupling Assay of Oligonucleotide
The assay was performed according to the previous work.[ 48 ] In a standard approach, a hybridization buffer (20% formamide, 16% dextran sulfate, and 0.6 m NaCl solution) was mixed with probe A and B solution in a volume ratio of 4:3:3. The freshly prepared working solution was then added to the target samples at different concentrations (volume ratio = 2:1). The solution was then incubated at 62 °C for 10 min or room temperature (22 °C) for 30 min prior to the UV–vis measurement.
Detection of SARS‐CoV‐2 RNA by Plasmonic LAMP
The protocol was performed per the manufacturer's recommendation. In a standard approach, 8 µL of RNA sample with varied copy number was first mixed with RT‐LAMP mixture containing 10 µL of 2× Warm Start RT‐LAMP mix and 2 µL of 10× customized primers, followed by incubation at 65 °C for 30 min. 2 µL of the product was loaded into a solution containing 1.25 µL of EaeI, 1.25 µL of HincII, 5 µL of rCutSmart buffer, and 40.5 µL of water. The mixed sample was incubated at 37 °C for 30 min and then heated to 95 °C for denaturation. Afterward, the sample was quickly cooled down on ice and ssDNA was formed. To process the plasmonic coupling assay, 50 µL of working solution containing probe suspensions a–d, and hybridization buffer with volume ratio of 1.5:1.5:1.5:1.5:4 was first prepared. Then 25 µL of the ssDNA sample was mixed with the working solution and incubated at 65 °C for 10 min or room temperature (22 °C) for 30 min before the UV–vis measurement or naked eye discrimination.
Detection of SARS‐CoV‐2 RNA by a Commercially Available LAMP Kit
The reaction was performed according to the manufacturer's instruction. Briefly, 1 µL of RNA sample, 9 µL of water, and 2.5 µL of 10× customized primers were added into the 12.5 µL colorimetric LAMP buffer and the mixture was incubated at 65 °C for 15, 30, and 45 min. For comparison, the same protocol was used for plasmonic LAMP.
RNA Extraction and Purification of Respiratory Syncytial virus (RSV)
RSV RNA as a negative control was extracted using a commercially available kit (Viral RNA Extraction Buffer, VRE100, Millipore‐Sigma). Briefly, 10 µL viral stock (e.g., 103 PFU mL−1 as pre‐quantified by plaques assay) was mixed with 5 µL extraction buffer at room temperature for 10 min and subjected to purification. A commercially available kit (Monarch Kits for RNA Cleanup, New England Biolabs Inc.) was used as per the manufacturer's protocol. The concentration of RNA was then quantified to be 5.9 ng µL−1 using NanoDrop 2000 (Thermo Scientific).
Gel Image Analysis
For gel electrophoresis, a 2% agarose gel was made with Low‐EEO Agarose powder (FISHER BP160‐500) dissolved in 1× Tris‐Borate‐EDTA (TBE) solution (FISHER BP1333‐1) and stained with 0.5 µg mL−1 of ethidium bromide (FISHER BP1302‐10). The gel ran at 150 V in a 1× TBE buffer solution for 30 min powered by a Bio‐Rad PowerPac Basic (300 V 400 mA 75 W) power supply.
Clinical‐Mock Sample Tests
Nasal swab samples were collected from a healthy individual with SARS‐CoV‐2 negative results. BD universal viral transport medium were gifts from UT Southwestern Medical Center and used as collecting buffer. The medium was first pipetted out carefully and loaded with varied amount of RNA (volume ratio = 1:1). The samples were freshly prepared before use. To proceed the sample test, plasmonic LAMP protocol was used. The use of human nasal swab sample was approved by Institutional Review Board (IRB) at University of Texas at Dallas (ID: 20MR0093).
Boundary Element Method Simulation
The plasmonic properties of Au–Ag nanoshells were simulated by means of the BEM, which solved Maxwell's equations for the geometry of a metallic nanoparticle only in terms of the boundaries that separate different media described by homogeneous and isotropic dielectric functions. All simulations were conducted via the MNPBEM Matlab toolbox.[ 42 ] The dielectric functions of silver and gold alloy were obtained from a modified Drude–Lorentz model developed by Rioux et al.[ 43 ] In the extinction spectra and electromagnetic field simulation, the Au–Ag shell model was built to be immersed in water as dielectric background. The electric field maps were computed from the surface charges where the particles were excited by plane wave at wavelength of 532 nm.
Characterizations
The extinction spectra in microtiter plates were read using microplate reader (Synergy 2, BioTek). The TEM images were taken using a JEOL JEM‐2010 microscope operated at 120 kV. The pH values of buffer solutions were measured using a pH Meter (Accumet AP71). Agarose gel imaging was performed using a Bio‐Rad Molecular Imager ChemiDoc XRS+. A FEI 200 kV Titan Themis scanning TEM was used to acquire the HADDF‐STEM images and EDX mapping images. A Perkin‐Elmer Sciex Elan 6100 DRC ICP‐MS was used to determine the amounts of Ag and Au elements in various nanostructures. Gel image analysis was performed using Bio‐Rad Image Lab v5.2.1 software. Digital photographs were taken by iPhone 12 ProMax. Dynamic light scattering and Zeta potential tests were performed using Malvern Zetasizer Nano ZS.
Conflict of Interest
Z.Q., L.B., and H.Y. are the inventors on a provisional patent related to this work filed by University of Texas at Dallas. The remaining authors declare no competing interests.
Supporting information
Supporting Information
Acknowledgements
This work was supported in part by National Institutes of Health (NIH) grant R01AI151374, and U.S. Department of Defense (DOD) grant PR192581, NSF grant 1361355, and Cecil H. and Ida Green Endowment. The authors would like to specifically thank Dr. Qingxiao Wang and Prof. Moon J. Kim from the University of Texas at Dallas for their general supports in taking the high‐resolution electron microscopy images.
Ye H., Nowak C., Liu Y., Li Y., Zhang T., Bleris L., Qin Z., Plasmonic LAMP: Improving the Detection Specificity and Sensitivity for SARS‐CoV‐2 by Plasmonic Sensing of Isothermally Amplified Nucleic Acids. Small 2022, 18, 2107832. 10.1002/smll.202107832
Contributor Information
Leonidas Bleris, Email: bleris@utdallas.edu.
Zhenpeng Qin, Email: zhenpeng.qin@utdallas.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Weiss C., Carriere M., Fusco L., Capua I., Regla‐Nava J. A., Pasquali M., Scott J. A., Vitale F., Unal M. A., Mattevi C., Bedognetti D., Merkoci A., Tasciotti E., Yilmazer A., Gogotsi Y., Stellacci F., Delogu L. G., ACS Nano 2020, 14, 6383. [DOI] [PubMed] [Google Scholar]
- 2. Bhalla N., Pan Y., Yang Z., Payam A. F., ACS Nano 2020, 14, 7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moitra P., Alafeef M., Dighe K., Frieman M., Pan D., ACS Nano 2020, 14, 7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seo G., Lee G., Kim M. J., Baek S. H., Choi M., Ku K. B., Lee C. S., Jun S., Park D., Kim H. G., Kim S. J., Lee J. O., Kim B. T., Park E. C., Kim S. Il., ACS Nano 2020, 14, 5135. [DOI] [PubMed] [Google Scholar]
- 5. Khodakov D., Wang C., Zhang D. Y., Adv. Drug Delivery Rev. 2016, 105, 3. [DOI] [PubMed] [Google Scholar]
- 6. Sidstedt M., Hedman J., Romsos E. L., Waitara L., Wadsö L., Steffen C. R., Vallone P. M., Rådström P., Anal. Bioanal. Chem. 2018, 410, 2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Opota O., Jaton K., Greub G., Clin. Microbiol. Infect. 2015, 21, 323. [DOI] [PubMed] [Google Scholar]
- 8. Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N., Hase T., Nucleic Acids Res. 2000, 28, 63e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanner N. A., Zhang Y., Evans T. C. Jr., BioTechniques 2015, 58, 59. [DOI] [PubMed] [Google Scholar]
- 10. Bokelmann L., Nickel O., Maricic T., Pääbo S., Meyer M., Borte S., Riesenberg S., Nat. Commun. 2021, 12, 1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thi V. L. D., Herbst K., Boerner K., Meurer M., Kremer L. P., Kirrmaier D., Freistaedter A., Papagiannidis D., Galmozzi C., Stanifer M. L., Boulant S., Sci. Transl. Med. 2020, 12, eabc7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hardinge P., Murray J. A., Sci. Rep. 2019, 9, 7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanner N. A., Evans T. C., in Current Protocol in Molecular Biology, John Wiley & Sons, New York: 2014. [Google Scholar]
- 14. Safavieh M., Kanakasabapathy M. K., Tarlan F., Ahmed M. U., Zourob M., Asghar W., Shafiee H., ACS Biomater. Sci. Eng. 2016, 2, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang D. G., Brewster J. D., Paul M., Tomasula P. M., Molecules 2015, 20, 6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Varona M., Anderson J. L., Anal. Chem. 2019, 91, 6991. [DOI] [PubMed] [Google Scholar]
- 17. Cai S., Jung C., Bhadra S., Ellington A. D., Anal. Chem. 2018, 90, 8290. [DOI] [PubMed] [Google Scholar]
- 18. Du Y., Hughes R., Bhadra S., Jiang Y. S., Ellington A. D., Li B., Sci. Rep. 2015, 5, 11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardinge P., Murray J. A., BMC Biotechnol. 2019, 19, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaminski M. M., Abudayyeh O. O., Gootenberg J. S., Zhang F., Collins J. J., Nat. Biomed. Eng. 2021, 5, 643. [DOI] [PubMed] [Google Scholar]
- 21. Zhou W., Hu L., Ying L., Zhao Z., Chu P. K., Yu X. F., Nat. Commun. 2018, 9, 5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J. S., Ma E., Harrington L. B., Da Costa M., Tian X., Palefsky J. M., Doudna J. A., Science 2018, 360, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gootenberg J. S., Abudayyeh O. O., Lee J. W., Essletzbichler P., Dy A. J., Joung J., Verdine V., Donghia N., Daringer N. M., Freije C. A., Myhrvold C., Science 2017, 356, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou R., Li Y., Dong T., Tang Y., Li F., Chem. Commun. 2020, 56, 3536. [DOI] [PubMed] [Google Scholar]
- 25. Katalani C., Boone H. A., Hajizade A., Sijercic A., Ahmadian G., Biol. Proced. Online 2020, 22, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khambhati K., Bhattacharjee G., Singh V., J. Cell. Biochem. 2019, 120, 2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hogan C. A., Garamani N., Lee A. S., Tung J. K., Sahoo M. K., Huang C., Stevens B., Zehnder J., Pinsky B. A., J. Clin. Microbiol. 2020, 58, e01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donato L. J., Trivedi V. A., Stransky A. M., Misra A., Pritt B. S., Binnicker M. J., Karon B. S., Diagn. Microbiol. Infect. Dis. 2021, 100, 115307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roberts R. J., Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crannell Z., Castellanos‐Gonzalez A., Nair G., Mejia R., White A. C., Richards‐Kortum R., Anal. Chem. 2016, 88, 1610. [DOI] [PubMed] [Google Scholar]
- 31. Kumvongpin R., Jearanaikool P., Wilailuckana C., Sae‐Ung N., Prasongdee P., Daduang S., Wongsena M., Boonsiri P., Kiatpathomchai W., Swangvaree S. S., Sandee A., J. Virol. Methods 2016, 234, 90. [DOI] [PubMed] [Google Scholar]
- 32. Wong J. K., Yip S. P., Lee T. M. H., Small 2014, 10, 1495. [DOI] [PubMed] [Google Scholar]
- 33. Arunrut N., Tondee B., Khumwan P., Kampeera J., Kiatpathomchai W., Aquaculture 2021, 533, 736206. [Google Scholar]
- 34. Suebsing R., Prombun P., Kiatpathomchai W., Lett. Appl. Microbiol. 2013, 56, 428. [DOI] [PubMed] [Google Scholar]
- 35. Seo G., Lee G., Kim M. J., Baek S. H., Choi M., Ku K. B., Lee C. S., Jun S., Park D., Kim H. G., Kim S. J., ACS Nano 2020, 14, 5135. [DOI] [PubMed] [Google Scholar]
- 36. Nakatsuka N., Yang K. A., Abendroth J. M., Cheung K. M., Xu X., Yang H., Zhao C., Zhu B., Rim Y. S., Yang Y., Weiss P. S., Science 2018, 362, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Au L., Lu X., Xia Y., Adv. Mater. 2008, 20, 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar A., Dixit C. K., in Advances in Nanomedicine for the Delivery of Therapeutic Nucleic Acids, Woodhead Publishing, Sawston: 2017, p. 43. [Google Scholar]
- 39. Loiseau A., Asila V., Boitel‐Aullen G., Lam M., Salmain M., Boujday S., Biosensors 2019, 9, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ye H., Wang Q., Catalano M., Lu N., Vermeylen J., Kim M. J., Liu Y., Sun Y., Xia X., Nano Lett. 2016, 16, 2812. [DOI] [PubMed] [Google Scholar]
- 41. Xia X., Wang Y., Ruditskiy A., Xia Y., Adv. Mater. 2013, 25, 6313. [DOI] [PubMed] [Google Scholar]
- 42. Hohenester U., Trügler A., Comput. Phys. Commun. 2012, 183, 370. [Google Scholar]
- 43. Rioux D., Vallières S., Besner S., Muñoz P., Mazur E., Meunier M., Adv. Opt. Mater. 2014, 2, 176. [Google Scholar]
- 44. Shiju E., Abhijith T., Chandrasekharan K., J. Mol. Liq. 2021, 333, 115935. [Google Scholar]
- 45. Zhu J., Chen J. K., Li J. J., Zhao J. W., Appl. Phys. A 2019, 125, 62. [Google Scholar]
- 46. Genç A., Patarroyo J., Sancho‐Parramon J., Arenal R., Duchamp M., Gonzalez E. E., Henrard L., Bastús N. G., Dunin‐Borkowski R. E., Puntes V. F., Arbiol J., ACS Photonics 2016, 3, 770. [Google Scholar]
- 47. Gao Z., Ye H., Wang Q., Kim M. J., Tang D., Xi Z., Wei Z., Shao S., Xia X., ACS Nano 2020, 14, 791. [DOI] [PubMed] [Google Scholar]
- 48. Godakhindi V. S., Kang P., Serre M., Revuru N. A., Zou J. M., Roner M. R., Levitz R., Kahn J. S., Randrianalisoa J., Qin Z., ACS Sens. 2017, 2, 1627. [DOI] [PubMed] [Google Scholar]
- 49. Armbruster D. A., Pry T., Clin. Biochem. Rev. 2008, 29, S49. [PMC free article] [PubMed] [Google Scholar]
- 50. Wang X., Lim H. J., Son A., Environ. Health. Toxicol. 2014, 29, e2014007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suwannin P., Polpanich D., Leelayoova S., Mungthin M., Tangboriboonrat P., Elaissari A., Jangpatarapongsa K., Ruang‐Areerate T., Tangchaikeeree T., J. Pharm. Biomed. Anal. 2021, 203, 114178. [DOI] [PubMed] [Google Scholar]
- 52. Sivakumar R., Dinh V. P., Lee N. Y., Lab Chip 2021, 21, 700. [DOI] [PubMed] [Google Scholar]
- 53. Ruang‐Areerate T., Sukphattanaudomchoke C., Thita T., Leelayoova S., Piyaraj P., Mungthin M., Suwannin P., Polpanich D., Tangchaikeeree T., Jangpatarapongsa K., Choowongkomon K., Sci. Rep. 2021, 11, 12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wan Y., Guo Z., Jiang X., Fang K., Lu X., Zhang Y., Gu N., J. Colloid Interface Sci. 2013, 394, 263. [DOI] [PubMed] [Google Scholar]
- 55. Wang Y., Gao Z., Liu B., Xia X., J. Mater. Chem. C 2019, 7, 15179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.