

Germline mutations in the oxygen-sensing pathway (VHL-HIF2A-PHD2) or erythropoietin (EPO) signaling (EPOR) are relatively rare but may result in erythrocytosis with normal p50 measurement (oxygen tension at which hemoglobin is 50% saturated) accompanied by either an elevated or inappropriately normal EPO (VHL-HIF2APHD2) or subnormal EPO (EPOR).1 On the other hand, a left shift of the oxygen dissociation curve, with venous p50 <24 mmHg may result from high-oxygen affinity (HOA) hemoglobin variants, defective 2,3-bisphosphoglycerate mutase (BPGM) causing 2,3-BPG deficiency or methemoglobinemia.1 The incidence, clinical course and management of hereditary erythrocytosis has not been well-characterized due to its rare occurrence. In that regard, we recently reported on 41 patients with HOA variant associated erythrocytosis; over half of the patients manifested one or more symptoms thought to be related to increased hematocrit while thrombosis was documented in a quarter of the patients.2 Neither hematocrit level nor active phlebotomy showed significant correlation with either thrombotic or non-thrombotic symptoms, which might have resulted from the limited sample size.2 In a recent study which included 270 patients with idiopathic erythrocytosis, 1.1% harbored EPOR mutations, while pathogenic variants involving genes in the hypoxia pathway were identified in 23% of patients.3 Accordingly, we share the Mayo Clinic clinical and laboratory experience with hereditary erythrocytosis resulting from genetic alterations in the oxygen-sensing pathway (VHL-HIF2A-PHD2), EPOR or BPGM.
All patients that underwent hereditary erythrocytosis evaluation at the Mayo Clinic over the last 10 years (2012-2021), were retrospectively recruited after obtaining Institutional Review Board approval. Polycythemia vera was excluded with JAK2 exon 12-15 sequencing. Hereditary erythrocytosis testing was pursued at the Mayo Clinic laboratory utilizing an algorithmic approach which included p50 measurement, serum EPO level (Epo), and DNA sequencing by polymerase chain reaction (PCR) of EPOR (exon 8), hypoxia-inducible factor 2 a (HIF2A) encoded by endothelial PASS domain protein 1 (EPAS1) (exons 9 and 12), prolyl hydroxylase 2(PHD2) encoded by EGL-9 family hypoxia inducible factor 1(EGLN1)(exons 1-5), von Hippel Lindau (VHL) (three coding exons and intron/exon boundaries) and BPGM (exons 1-4) as detailed in our prior work.4
Of 592 patients tested at the Mayo Clinic for HIF2A/PHD2/EPOR alterations, 14 pathogenic variants were identified in HIF2A (n=6, 1%), PHD2 (n=3, 0.5%), EPOR (n=2, 0.3%), while two of 421 (0.5%) and one of 446 (0.2%) patients harbored BPGM and VHL variants, respectively. In addition, 22 variants of uncertain significance (VUS) were reported; EPOR (n=1), HIF2A (n=3), PHD2 (n=10), BPGM (n=2), VHL (n=6), resulting in combined (pathogenic + VUS) Mayo Clinic incidence rates of 0.5%, 1.5%, 2.2%, 1% and 1.6% for EPOR, HIF2A, PHD2, BPGM, and VHL aberrations, respectively.
Table 1 summarizes oxygen-sensing pathway (PHD2/HIF2A/VHL) pathogenic variants including clinical course of ten patients with median follow-up of 2 years, (range, 0.2-10 years). HIF2A pathogenic variants were noted in six patients; four harbored the heterozygous HIF2A c.1121T>A, p.(Phe374Tyr) alteration in exon 9, previously reported in association with neuroendocrine tumors with or without erythrocytosis.5 A 57- year-old male with heterozygous HIF2A c.1121T>A mutation presented with a hemoglobin (Hb)/hematocrit (Hct)/Epo of 17.9 g/dL/54.4%/93.4 mIU/mL, diabetes mellitus and prior cerebrovascular accident (CVA)/ left ventricular thrombus, was started on phlebotomy, continued aspirin with anticoagulation and did not experience additional thromboses. The second case was a 56- year-old female with heterozygous HIF2A c.1121T>A mutation. Hb/Hct/Epo at presentation; 19.1 g/dL/57%/40.8 mIU/mL, with hypertension and hyperlipidemia, developed multiple thromboses; myocardial infarction, followed by CVA, inferior vena caval thrombus post-diagnosis, the latter occurred despite ongoing phlebotomy and aspirin/clopidogrel. The remainder two patients with heterozygous HIF2A c.1121T>A mutations were 68- and 71-year-old males with hypertension and hyperlipidemia respectively, Hb/Hct/Epo at diagnosis were 19.1/57.2/20.7 and 17.2/52/7.7, both did not experience thrombosis with the former receiving phlebotomy and the latter low dose aspirin.
Additionally, a 61-year-old female harbored a heterozygous missense alteration in HIF2A c.1620C>A, resulting in amino acid substitution p.Phe540Leu (F540L) previously reported by our group.4 She had a history of hypertension, presented with Hb/Hct/Epo of 16.1/47.8/7.3 and did not experience thrombosis while on low-dose aspirin. On the other hand, a 69-year-old hypertensive male with heterozygous HIF2A c.1609G>A, mutation with Hb/Hct/Epo of 23/58.7/175 at diagnosis, developed a CVA with ongoing phlebotomy. An elevated Epo level (range, 20.7-175, reference range; 2.6-18.5 mIU/mL) was noted in four of six patients with HIF2A pathogenic variants, which in all instances was accompanied by phlebotomy. All patients had one or more cardiovascular risk factors, with three patients (50%) experiencing thrombosis, two of which occurred with ongoing phlebotomy, suggesting the lack of benefit of phlebotomy.
Of three patients with PHD2 pathogenic variants; a 35- year-old female with family history of erythrocytosis, current smoker, without history of prior thrombosis, and Hb/Hct/Epo 17.2/52.6/11.2, demonstrated a PHD2 c.1111C>T, p.(Arg371Cys) missense variant. This variant has been reported in the human gene mutation database,6,7 and involves a highly conserved amino acid in the Fe(2+) 2-oxoglutarate dioxygenase domain, critical for hydroxylation of HIF; functional studies have not been performed but studies involving (Arg371His) have shown decreased ability of PHD2 to bind and hydroxylate HIF. On the other hand, two patients harbored previously reported PHD2 c.461C>A, p.(S154*) and c.1030C>T, p.(Arg344*) nonsense variants predicted to result in a premature stop codon in exon 1 and 3, respectively, and expected to be loss of function mutations.4 This included a 67-year-old male with PHD2 c.461C>A and a 60-year-old female with PHD2 c.1030C>T mutation, Hb/Hct/Epo at diagnosis were 17.8/50.7/10.3 and 17/not available/30, both did not experience thrombosis, former had known coronary artery disease and was on low-dose aspirin while the latter was hypertensive and receiving phlebotomy along with aspirin.
A pathogenic variant in VHL was detected in a 19-yearold male, compound heterozygous (L188V and R200W) for the previously described VHL mutations,8 who presented with erythrocytosis (Hb/Hct 19/57) and a markedly elevated EPO level at 1465 mIU/mL. He was managed with phlebotomy every 4 weeks, in addition to aspirin and did not experience thrombosis.
Table 1.
Clinical features and management of ten patients with EGLN1(PHD2)/ EPAS1(HIF2A)/VHL pathogenic variant associated erythrocytosis.

Canonical exon 8 EPOR c.1316G>A mutations,9 occurred in two patients, 48- and 69-year-old females, with a family history of erythrocytosis, and Hb/Hct/Epo levels of 19.4/56.6/1.1 and 14.6/44.3/<1, respectively, underscoring the suppressed Epo levels with gain of function EPOR mutations (Table 2). Both patients underwent intermittent phlebotomy and had an uncomplicated course in terms of thrombosis and pregnancies.
Two patients harbored BPGM pathogenic variants (Table 2) which included a 25-year-old male with hypertension who presented with Hb/Hct/Epo/p50 of 20/58/17.7/31, found to have a heterozygous missense alteration in BPGM at c.184C>T resulting in amino acid substitution p.Arg62Trp (R62W). While this specific amino acid change is novel, (p.Arg62Gln) has been reported in association with erythrocytosis in patients homozygous for the variant10 and compound heterozygous for Arg62Gln and another BPGM pathogenic variant. 11 The second case was a 25-year-old male, current smoker with Hb/Hct/Epo of 17/49.1/5.1, who harbored a previously unreported BPGM c.258dup, p.(Leu87Serfs*3) frameshift variant in the first coding exon, predicted to result in a premature stop codon. Similar nonsense mutations leading to a predicted premature stop codon have been reported.10,12,13 Both patients had an uneventful clinical course, the first patient was receiving phlebotomy and aspirin while the second case was observed.
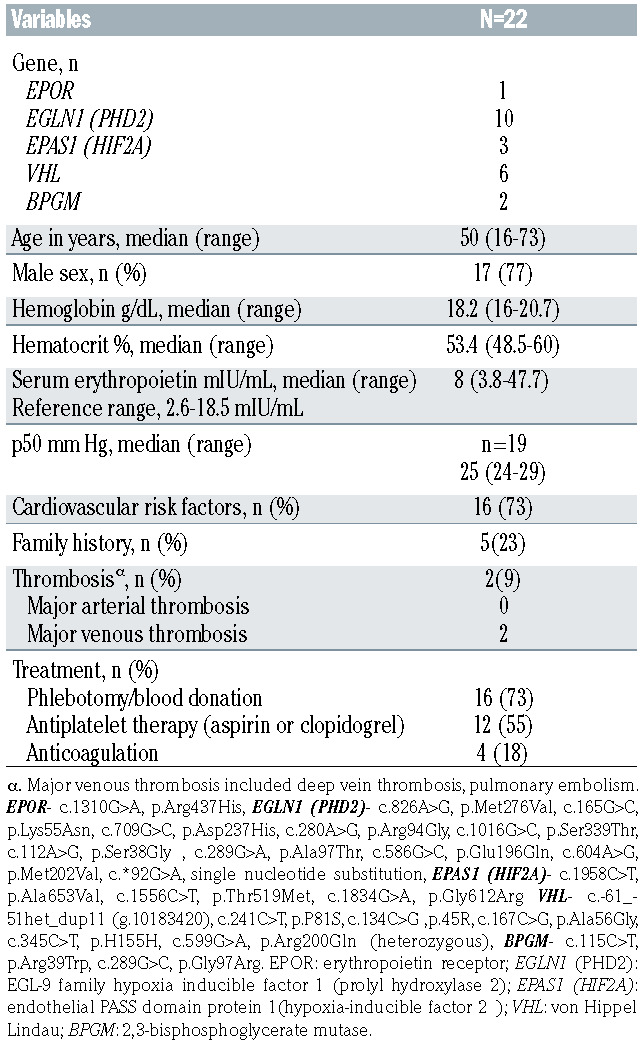
Among 22 VUS that were reported, PHD2 was most frequently involved (Table 3). The majority (n=17, 77%) of cases were males with median age at diagnosis of 50 years (range, 16-73 years). All patients had normal p50 testing, whereas EPO levels were highly variable, median 8 mIU/mL (range, 3.8-47.7 mIU/mL). A family history of erythrocytosis was known in five patients (23%) and thrombosis occurred in two (9%) of patients; the majority were managed with phlebotomy/blood donation (n=16, 73%) and/or antiplatelet therapy (n=12, 55%).
Table 2.
Clinical features and management of four patients with EPOR/BPGM pathogenic variant associated erythrocytosis.

In the current series, we share our decades worth of hereditary erythrocytosis testing experience from the Mayo Clinic in order to define the incidence of alterations involving the hypoxia sensing pathway, in addition to EPOR and BPGM, providing a clinical perspective on the likelihood of encountering such abnormalities during the course of erythrocytosis evaluation. We limited the above series to the hypoxia sensing pathway genes, EPOR, and BPGM, since we have recently published on HOA variant associated erythrocytosis. Of the hypoxia sensing pathway alterations, homozygous VHL (598C>T) mutation Chuvash polycythemia [CP] is phenotypically well-characterized by an unusual propensity for vascular events leading to early mortality.14 In a prospective, age, sex-matched controlled study on the subject matter, age and prior thrombotic events emerged as independent predictors of thrombosis; moreover, phlebotomy was associated with an increased incidence of thrombosis.15 Similarly, among eight patients harboring the HIF2A p.M535V variant, five experienced thrombotic events versus none in 17 HIF2A wild-type patients.15 Furthermore, thrombotic events occurred despite phlebotomy and in the absence of cardiovascular risks.15 In our series, all three thrombotic events occurred in patients harboring HIF2A pathogenic variants, two of which were receiving phlebotomy, in addition to dual antiplatelet therapy in one patient. Of note, HIF2A alterations may be associated with neuroendocrine tumors such as pheochromocytoma, paraganglioma, somatostatinoma; 16 however, none of our patients with HIF2A alterations developed tumors. Limitations of our study include the retrospective design, and heterogeneity in clinical practice in regard to diagnosis and management.
Table 3.
Clinical features and management of 22 patients with variants of uncertain significance involving EPOR/EGLN1(PHD2)/ EPAS1(HIF2A)/VHL/BPGM and associated erythrocytosis.
In summary, we confirm the infrequent (0.5-2.2%) occurrence of genetic alterations involving the hypoxia pathway, EPOR and BPGM among patients undergoing hereditary erythrocytosis evaluation at the Mayo Clinic which includes testing for all congenital mutations except recently described EPO and iron-responsive element binding protein 1 (IRP1) mutations.17,18 Additionally, phenotypic correlations and management details are provided which may serve as a useful guide for clinicians.
References
- 1.Gangat N, Szuber N, Pardanani A, Tefferi A. JAK2 unmutated erythrocytosis: current diagnostic approach and therapeutic views. Leukemia. 2021;35(8):2166-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gangat N, Oliveira JL, Hoyer JD, Patnaik MM, Pardanani A, Tefferi A. High-oxygen-affinity hemoglobinopathy-associated erythrocytosis: Clinical outcomes and impact of therapy in 41 cases. Am J Hematol. 2021;96(12):1647-1654. [DOI] [PubMed] [Google Scholar]
- 3.Filser M, Aral B, Airaud F, et al. Low incidence of EPOR mutations in idiopathic erythrocytosis. Haematologica. 2021;106(1):299-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliveira JL, Coon LM, Frederick LA, et al. Genotype-phenotype correlation of hereditary erythrocytosis mutations, a single center experience. Am J Hematol. 2018;93(8):1029-1041. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzo FR, Yang C, Ng Tang Fui M, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl). 2013;91(4):507-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson R, Syed N, Shah P. Erythrocytosis due to PHD2 mutations: a review of clinical presentation, diagnosis, and genetics. Case Rep Hematol. 2016;2016:6373706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardie B, Percy MJ, Hoogewijs D, et al. The role of PHD2 mutations in the pathogenesis of erythrocytosis. Hypoxia (Auckl). 2014;2:71-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bento C, Percy MJ, Gardie B, et al. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat. 2014;35(1):15-26. [DOI] [PubMed] [Google Scholar]
- 9.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90(10):4495-4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol. 2004;75(4):205-208. [DOI] [PubMed] [Google Scholar]
- 11.Lemarchandel V, Joulin V, Valentin C, et al. Compound heterozygosity in a complete erythrocyte bisphosphoglycerate mutase deficiency. Blood. 1992;80(10):2643-2649. [PubMed] [Google Scholar]
- 12.Petousi N, Copley RR, Lappin TR, et al. Erythrocytosis associated with a novel missense mutation in the BPGM gene. Haematologica. 2014;99(10):e201-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camps C, Petousi N, Bento C, et al. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica. 2016;101(11):1306-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103(10):3924-3932. [DOI] [PubMed] [Google Scholar]
- 15.Gordeuk VR, Miasnikova GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarade D, Robinson CM, Lee JE, Ohh M. HIF-2alpha-pVHL complex reveals broad genotype-phenotype correlations in HIF-2alpha-driven disease. Nat Commun. 2018;9(1):3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zmajkovic J, Lundberg P, Nienhold R, et al. A gain-of-function mutation in EPO in familial erythrocytosis. N Engl J Med. 2018;378(10):924-930. [DOI] [PubMed] [Google Scholar]
- 18.Oskarsson GR, Oddsson A, Magnusson MK, et al. Predicted loss and gain of function mutations in ACO1 are associated with erythropoiesis. Commun Biol. 2020;3(1):189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Percy MJ, Beer PA, Campbell G, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111(11):5400-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Percy MJ, Chung YJ, Harrison C, et al. Two new mutations in the HIF2A gene associated with erythrocytosis. Am J Hematol. 2012;87(4):439-442. [DOI] [PMC free article] [PubMed] [Google Scholar]