

Abstract
Background
Particulate matter (PM) exposure is related to mitochondria dysfunction and airway inflammation. Antioxidant drug edaravone (EDA) is reported to improve the occurrence and development of oxidative stress-related diseases. At present, there is no data on whether EDA can alleviate lung inflammation caused by PM.
Methods
The anti-inflammatory effects of EDA were investigated in urban PM-induced human bronchial epithelial cells (HBECs) and C57/BL6J mouse models. In vitro, its effects on the production of intracellular reactive oxygen species (ROS), mitochondrial membrane potential (MMP), and inflammatory cytokines were assessed by DCFH-DA staining, JC-1 assay, and real-time PCR, respectively. In vivo, the oxidant stress in lung tissues was assessed by dihydroethidium (DHE) staining and malondialdehyde (MDA) activity, and inflammatory cytokines in bronchoalveolar lavage fluid (BALF) were assessed by ELISA, respectively. Furthermore, the potential signaling pathways were studied by siRNA transfection and western blot.
Results
PM increased the expression of inflammatory cytokines and protein, including IL-6, IL-1α, IL-1β, and COX-2, while these alternations were significantly alleviated following EDA treatment in a dose-dependent manner. EDA treatment also alleviated the inflammatory responses in lung tissues of PM-exposed mice. We further showed mitochondrial dysfunction in PM-exposed HBECs and mice, which were reversed by EDA treatment. Moreover, the phosphorylation of NF-κB p65 in PM-exposed HBECs and mice was weakened by EDA. Transfection with NF-κB p65 siRNA further inhibited PM-induced inflammation in HBECs.
Conclusion
We demonstrated that EDA treatment had a protective role in PM-induced lung inflammation through maintaining mitochondrial balance and regulating the ROS-NF-κB p65 signaling pathway. This provided a new therapeutic method for PM-induced lung inflammation in the future.
1. Introduction
Ambient particulate matter (PM) exposure is a severe public health problem worldwide, and it is the fourth leading risk factor for global death in 2019 [1]. It was reported that 92% of the world's population were exposed to PM2.5 levels far exceeding the average PM2.5 transition level of the World Health Organization's Air Quality Guidelines. Furthermore, PM was reported to induce adverse effects on human health after short-term and long-term exposure. Short-term exposure generally causes acute inflammatory responses in airways and peripheral blood [2], while long-term exposure is actively related to the mortality of cardiovascular diseases, respiratory diseases, and cancer [3–5]. The increasing burden of PM exposure makes it urgent to control airway diseases caused by PM exposure. Accumulating epidemiological data have proved that short-term PM exposure contributed to the occurrence and development of airway inflammation and mitochondria dysfunction. However, the specific target of interventions is still unclarified.
Mitochondria are sensitive targets of PM and the primary source of oxidative stress that leads to inflammatory responses. Dysfunction mitochondria display decreased mitochondria membrane potential (MMP) and increased ROS production. Once ROS led to a decrease in MMP and an increase in mitochondrial membrane permeability, mitochondrial proapoptotic factors would be released into the cytoplasm, triggering mitochondrial-dependent apoptosis [6]. Cumulative studies suggested that airway inflammation acted as the product of mitochondrial dysfunction [7–9]. Thus, our study attempted to explore an effective therapy to reduce PM-related mitochondrial oxidative damage and protect against lung injury.
Edaravone (3-methyl-1 phenyl-2-pyrazoline-5-one, EDA) is a powerful scavenger of hydroxyl radicals, eliminating the hydroxyl radicals that induce lipid peroxidation [10–12]. Emerging research showed that it had antioxidation and antiapoptosis effects and pointed out that EDA had a beneficial effect on diseases caused by redox imbalance in inflammatory diseases [13–16]. Zhang et al. showed that EDA protected against liver and lung injury by reducing the expression of inflammatory cytokines in animal sepsis models [17]. However, the effect of EDA on PM-induced lung injury has not been studied.
Therefore, the present study for the first time demonstrated that EDA protected PM-induced lung inflammation via regulating the ROS-NF-κB signaling pathway in vivo and in vitro.
2. Methods
2.1. Reagents and Antibodies
Urban PM 1649b was purchased from NIST (Gaithersburg, MD, USA). EDA was obtained from Selleck (Houston, Texas, USA). Dihydroethidium (DHE) was obtained from Sigma-Aldrich (Saint Louis, MO, USA). 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) was purchased from Beyotime (Shanghai, China). Antibodies against cyclooxygenase- (COX-) 2, phospho-NF-κB p65, and NF-κB p65 were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against GAPDH were purchased from Beyotime. Antibodies against β-actin were purchased from Abcam (Cambridge, MA, USA). The mRNA primers were synthesized by Shenggong Technologies (Shanghai, China). All kits for cDNA synthesis and real-time PCR (RT-PCR) detection were purchased from Yeasen Biotech (Shanghai, China). The reagents for western were purchased from Beyotime.
2.2. Animals and Cell Culture
Animal handling and experiments were conducted following the policies of the animal care facility of Zhongshan hospital, Fudan University. Male 8-12-week-old C57BL/6 mice were acquired from SLAC Laboratory Animals (Shanghai, China). Human bronchial epithelial cells (HBECs) were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI 1640 (Gibco, Waltham, MA, USA) containing 10% FBS (Gibco) and 50 U/mL penicillin and streptomycin (Gibco).
For in vivo experiments, the acute PM-exposed mouse models were established via intratracheal instillation with 100 μg PM in 30 μL PBS per day for 2 consecutive days, according to our previous studies [18–20]. EDA (2, 10, or 20 mg/kg) was intraperitoneally injected 1 h prior to PM stimulation every time. PBS was administered following the same protocol to obtain the control group. For in vitro experiments, HBECs were treated with PM at 300 μg/mL for 24 h, according to our previous studies [21, 22]. EDA (300, 400, or 500 μM) were used to treat HBECs 1 h before PM stimulation. The dose of EDA for in vitro and in vivo experiments was identified according to the previously reported studies [11, 17, 23–27]. To make our results accurate, the concentration gradient of EDA treatment was selected for our study.
2.3. Small Interfering RNA (siRNA) Transfection
NF-κB p65 siRNA was purchased from GenePharma (Shanghai, China). HBECs were transfected with NF-κB p65 siRNA or control siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at a concentration of 50 nM according to the manufacturer's protocol. After transfection, cells were treated with PM for 24 h. Subsequently, certain genes were analyzed by RT-PCR and western blot.
2.4. Real-Time PCR
RT-PCR analysis was carried out to detect the mRNA expression of COX-2, IL-6, IL-1α, and IL-1β. Total RNA extraction from HBECs was performed with the TRIzol Reagent (Invitrogen). Then, RNA was reverse transcribed and quantified by the 7500 RT-PCR system (Applied Biosystems) using the SYBR Green PCR kit (Takara, Japan). All protocols were used as we previously reported [18]. The primers used in our research are listed in Table 1.
Table 1.
Primers used in the study.
| Genes | Forward | Reverse |
|---|---|---|
| GAPDH | 5′-AAGGTGAAGGTCGGAGTCAAC-3′ | 5′-GGGGTCATTGATGGCAACAATA-3′ |
| COX-2 | 5′-TAAGTGCGATTGTACCCGGAC-3′ | 5′-TTTGTAGCCATAGTCAGCATTGT-3′ |
| IL-6 | 5′-ACTCACCTCTTCAGAACGAATTG-3′ | 5′-CCATCTTTGGAAGGTTCAGGTTG-3′ |
| IL-1α | 5′-TGGTAGTAGCAACCAACGGGA-3′ | 5′-ACTTTGATTGAGGGCGTCATTC-3′ |
| IL-1β | 5′-ATGATGGCTTATTACAGTGGCAA-3′ | 5′-GTCGGAGATTCGTAGCTGGA-3′ |
2.5. Western Blot
Western blot analyses were performed as previously described [18]. Briefly, the cells and the right lung upper lobe of mice were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer containing 1% phenylmethanesulfonyl fluoride (PMSF) and 1% phosphatase inhibitors (Biotool, Houston, TX, USA). Subsequently, the homogenate was centrifuged at 12,000 g for 30 min at 4°C, and the supernatant was collected.
After cell homogenates were boiled, the proteins (30 μg) were separated by SDS-PAGE and transferred to the PVDF membranes. After blocking with western blocking buffer for 1 h at room temperature, membranes were incubated with primary antibody against phospho-NF-κB p65, NF-κB p65, COX-2, GAPDH, or β-actin overnight at 4°C, respectively. Then, the membranes were washed with TBST thrice and incubated with goat anti-rabbit or goat anti-mouse horseradish peroxidase- (HRP-) conjugated secondary antibodies. The targeted proteins were detected by ECL reagents. The intensity of protein bands was quantified by Image J software (v1.8.0).
2.6. ROS Measurement
The DCFH-DA fluorescent dye probe was used to measure intracellular ROS production. One day after seeding, cells were pretreated with EDA (300, 400, or 500 μM) for 1 h, then treated with 300 μg/mL PM for another 1 h. Then, cells were washed with PBS and incubated with 10 μM DCFH-DA at 37°C for 15 min in the dark. Cells were washed with PBS twice, and images were obtained using the fluorescence microscope (Olympus, Tokyo, Japan) equipped with a digital camera. For in vivo studies, fresh lung tissues were embedded in OCT, sectioned, and stained with DHE to detect ROS, as described previously [18]. DHE entered cells and was oxidized by superoxide to emit red fluorescence detected by the fluorescence microscope. The value of relative fluorescence intensity was measured in selected areas that contain cells and was analyzed by Image J software (v1.8.0).
2.7. JC-1 Assay for Mitochondrial Membrane Potential
MMP was measured using a mitochondrial-specific dual-fluorescence probe, JC-1 (Beyotime). Briefly, HBECs were seeded into 12-well plates, treated with or without EDA (300, 400, or 500 μM) for 1 h, and then treated with 300 μg/mL PM for another 24 h. Next, cells were washed with PBS and incubated with 10 μg/mL JC-1 at 37°C in the dark for 30 min. The fluorescence was measured by a microscope. The red fluorescence represents the JC-1 monomers, while the green fluorescence represents JC-1 aggregates. The ratio of red fluorescence to green fluorescence was calculated to indicate the level of MMP.
2.8. Malondialdehyde (MDA) Activity
The right lung middle lobe and right lung lower lobe of mice were harvested. The tissue samples were homogenized and sonicated in 300 μL RIPA buffer on ice. Then, tissue lysates were centrifuged at 12,000 g for 15 min at 4°C, and the supernatant was collected. The proteins were diluted to the appropriate concentration and subjected to the MDA assay as described in the Lipid Peroxidation MDA assay kit (Beyotime). The MDA levels were detected by Multi-Mode Microplate Readers (BioTek, Santa Barbara, CA, USA) at 532 nm.
2.9. H&E Staining and Immunohistochemistry (IHC)
Mice from different groups were intraperitoneally injected with avertin (Sigma-Aldrich) for euthanasia at the dose of 25 mg/kg body weight, and the left lung lobe of mice was harvested and fixed in 4% paraformaldehyde. Then, the left lung was sliced and stained with hematoxylin and eosin (H&E) staining according to the manufacturer's instructions [18]. Pathologic changes were imaged and scored as reported previously [28].
Paraffin-embedded left lung tissue sections were stained by IHC. The slices were incubated with 3% H2O2 and then were incubated with a primary antibody against COX-2 at 4°C for 12 h. The second antibody labeled with horseradish peroxidase was incubated at 37°C for 30 min, and the immunohistochemical reaction was observed according to the manufacturer's instructions with 3,3′-diaminobenzidine (DAB) as a chromogenic agent [28]. IHC staining was evaluated by ImageJ software (v1.8.0) according to the MOD value.
2.10. Bronchoalveolar Lavage Fluid (BALF) Collection and Inflammatory Cell Counts
Mice were intraperitoneally injected with avertin for euthanasia at the dose of 25 mg/kg body weight. After euthanasia, the chest of each mouse was opened to collect the BALF. The trachea and the whole lung were cannulated and perfused with 1 mL normal saline. A three-in and three-out pattern of main bronchial instillation was adopted, and the BALF was collected with a high recovery. The BALF was centrifuged at 800 g for 5 min at room temperature. The supernatant was collected and stored at -80°C for further analysis. Cell pellets were resuspended with 0.5 mL of PBS, and the total cell number was counted. Then, 100 μL of the resuspended cells was spun to the slide, air-dried, and then fixed with 4% paraformaldehyde overnight. Cells were stained with H&E, and the number of macrophages and neutrophils in 200 cells was counted. The data were shown as 105/mL BALF.
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
The protein expression levels of IL-6, IL-1α, and IL-1β in the BALF were measured using ELISA kits (4A Biotech, Beijing, China) according to the manufacturer's instructions [18]. Values were expressed as pg/mL and deduced from standard curves of recombinant cytokines.
2.12. Statistical Analyses
The GraphPad Prism 5 software (GraphPad, Inc., La Jolla, CA, USA) was used to analyze all data. The significance of differences between control and experimental values was assessed using the unpaired t-test or one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant. All values were expressed as the mean ± standard error of the mean (SEM).
3. Results
3.1. EDA Attenuated PM-Induced Inflammation in HBECs
To investigate the anti-inflammatory effect of EDA on HBECs, the cells were treated with 300, 400, or 500 μM EDA for 1 h, respectively, and then stimulated with 300 μg/mL PM for an additional 24 h. The mRNA levels of inflammatory cytokines, including IL-6, IL-1α, and IL-1β, were significantly increased in HBECs with PM stimulation, and the increasing level of inflammatory cytokines was significantly decreased by EDA treatment in a dose-dependent manner in HBECs with PM stimulation (Figure 1(a)). COX-2 was induced by several inflammatory stimuli, and its expression was increased significantly by PM exposure in this study, as we previously reported [18]. EDA treatment could significantly decrease the expression of COX-2 in HBECs exposed to PM at the gene and protein levels (Figures 1(b)–1(d)). These results suggested that EDA could inhibit PM-induced inflammation in HBECs.
Figure 1.
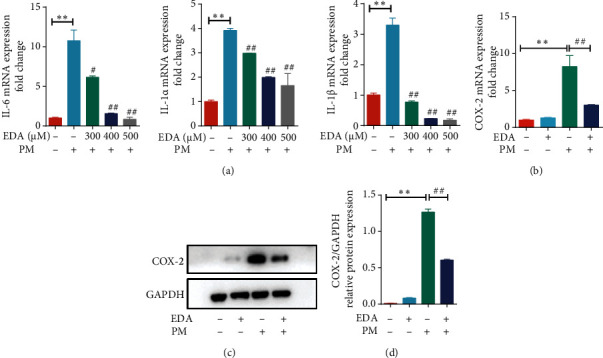
EDA attenuated PM-induced inflammation in HBECs. (a) HBECs were treated with EDA in a dose-dependent manner (300, 400, or 500 μM) and then stimulated with 300 μg/mL PM for 24 h. The mRNA expression of IL-6, IL-1α, and IL-1β was detected by RT-PCR. (b) The mRNA of COX-2 was detected by RT-PCR. (c) Western blot showed the expression of COX-2 in HBECs upon PM exposure after EDA treatment. The optical densities of COX-2 are shown in (d). Values are the mean ± SEM; ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3. EDA: edaravone; PM: particulate matter; HBECs: human bronchial epithelial cells.
3.2. EDA Attenuated PM-Induced ROS Generation and Reversed PM-Induced MMP Dysfunction in HBECs
To further investigate the antioxidant role of EDA in PM-induced inflammatory responses, the ROS level of HBECs was detected in PM-treated HBECs. The ROS level in HBECs was significantly increased after PM exposure, and EDA treatment markedly reduced PM-induced oxidant stress in HBECs (Figures 2(a) and 2(b)). To address the effect of EDA on mitochondria protection, we assayed MMP using a fluorescent mitochondria dye, JC-1. Red fluorescence defines normal MMP, whereas green fluorescence defines the loss of MMP. Our results showed that MMP was significantly degraded by PM, and EDA treatment significantly restored the MMP dysfunction (Figures 2(c) and 2(d)). These results suggested that EDA attenuated oxidative stress and treated mitochondrial imbalance in HBECs exposed to PM.
Figure 2.
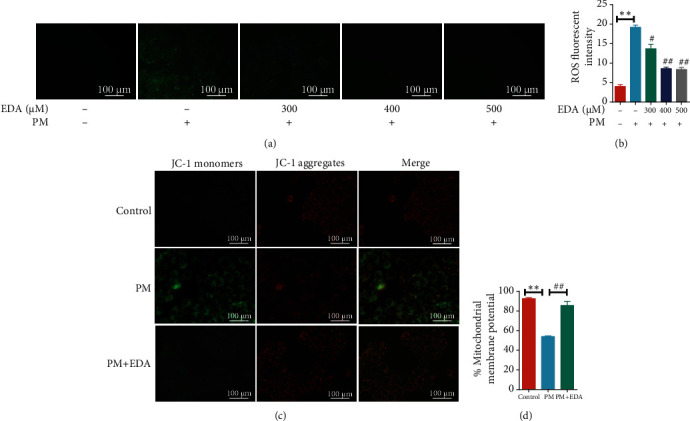
EDA inhibited PM-induced oxidative stress and mitochondrial dysfunction in HBECs. (a) HBECs were treated with EDA in a dose-dependent manner (300, 400, or 500 μM) and then stimulated with 300 μg/mL PM for 24 h. The ROS level was measured through the fluorescent probe, DCFH-DA, and fluorescence intensity was measured and shown in (b). (c) Mitochondrial membrane potential (ΔΨm) was detected using JC-1 staining, and data are presented as the ratio of red fluorescence (JC-1 aggregates) to green fluorescence (JC-1 monomers) in (d). Values are the mean ± SEM; ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3. EDA: edaravone; PM: particulate matter; HBECs: human bronchial epithelial cells.
3.3. EDA Inhibited PM-Induced Inflammation In Vivo
To clarify the anti-inflammation and antioxidative effect of EDA on PM-induced mice, mice were treated with EDA (2, 10, or 20 mg/kg) before PM exposure every time. Our results showed that EDA could dose-dependently attenuate the infiltration of inflammatory cells in lung tissues of mice exposed to PM (Figures 3(a) and 3(b)). Furthermore, the increasing number of the total cell, neutrophils, and macrophages in BALF in the PM group was significantly inhibited by EDA pretreatment in a dose-dependent manner (Figure 3(c)). Besides, we found that alone EDA treatment in different concentrations has no toxic effect on the lung tissues and on blood cell numbers (Figure S1).
Figure 3.
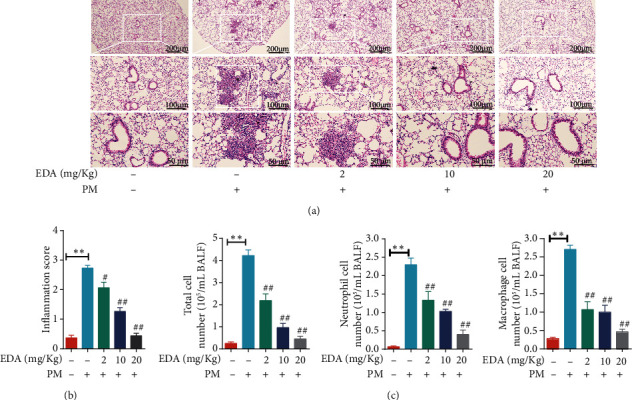
EDA alleviated the PM-induced inflammatory responses in vivo. Mice were intratracheally instilled with PM at 100 μg/day/mouse for 2 consecutive days. EDA (2, 10, or 20 mg/kg) was intraperitoneally injected prior to PM exposure for 1 h. (a) Representative images of lung sections stained with H&E. (b) The inflammation score for images of lung sections stained with H&E. (c) The number of the total cells, neutrophils, and macrophages in BALF. Values are the mean ± SEM; ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 5. EDA: edaravone; PM: particulate matter; BALF: bronchoalveolar lavage fluid.
ELISA showed that EDA could attenuate PM-induced production of inflammatory cytokines IL-6, IL-1α, and IL-1β (Figure 4(a)). Meanwhile, the expression of COX-2 was analyzed using IHC, and we found that EDA significantly decreased PM-induced expression of COX-2 in bronchial epithelium in a dose-dependent manner (Figures 4(b) and 4(c)). Similarly, we tested the expression of COX-2 in lung tissues using western blot and showed that EDA treatment inhibited the expression of COX-2 in lung tissues of mice exposed to PM (Figures 4(d) and 4(e)). Besides, we found alone EDA treatment had no effect on inflammatory and COX-2 expression in normal lung tissues (Figure S2). These results indicated that EDA could significantly inhibit PM-induced lung inflammation in vivo.
Figure 4.
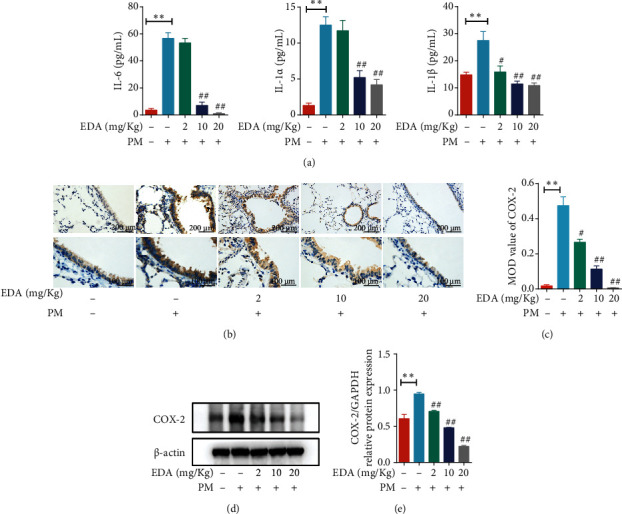
EDA attenuated the PM-induced inflammatory cytokines and protein expression in lung tissues. Mice were intratracheally instilled with PM at 100 μg/day/mouse for 2 consecutive days. EDA (2, 10, or 20 mg/kg) was intraperitoneally injected prior to PM exposure for 1 h. (a) The levels of IL-6, IL-1α, and IL-1β in the BALF were measured by ELISA. (b) The level of COX-2 expression in lung tissues was measured by IHC staining. (c) The semiquantitative analysis was applied to compare the relative COX-2 protein expression. (d) COX-2 expression was determined by western blot. The optical densities of COX-2 are shown in (e). Values are the mean ± SEM; ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 5. EDA: edaravone; PM: particulate matter; BALF: bronchoalveolar lavage fluid.
3.4. EDA Decreased PM-Induced Oxidative Stress In Vivo
The ROS level in lung tissues was detected with DHE staining and MDA activity. The ROS level in lung tissues of the PM-induced mouse model was significantly increased, and EDA treatment significantly reduced the PM-induced ROS generation in a dose-dependent manner (Figures 5(a) and 5(b)). Furthermore, the levels of MDA in lung tissues were also significantly increased in the PM group compared with those in the control group. EDA dose-dependent treatment significantly attenuated the levels of MDA induced by PM exposure (Figure 5(c)). These results showed that the levels of ROS in lung tissues were inhibited by EDA treatment in PM-exposed mice.
Figure 5.
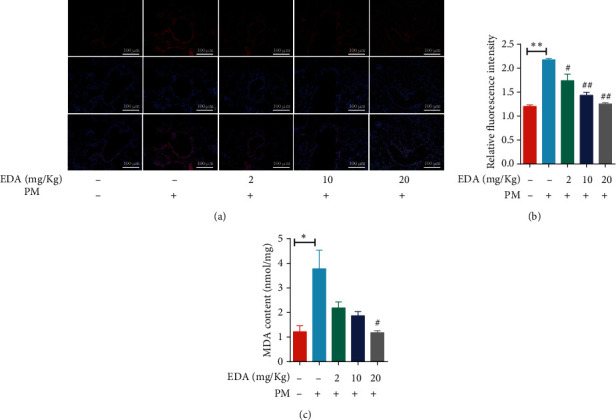
EDA inhibited the PM-induced oxidative stress in lung tissues. Mice were intratracheally instilled with PM at 100 μg/day/mouse for 2 consecutive days. EDA (2, 10, or 20 mg/kg) was intraperitoneally injected prior to PM exposure for 1 h. (a) ROS levels in lung tissues were examined by DHE staining. Relative DHE fluorescence is shown in (b). (c) MDA levels were measured in the lung tissues. Values are the mean ± SEM; ∗P < 0.05 or ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3 or 5. EDA: edaravone; PM: particulate matter.
3.5. EDA Inhibited the Activation of the NF-κB p65 Pathway in PM-Induced Inflammatory Responses
To investigate the effect of EDA on the NF-κB p65 pathway in PM-induced inflammatory responses, western blot was used the test the activation of the NF-κB p65 pathway. We found that pretreatment with EDA blocked PM-induced activation of the NF-κB p65 pathway in HBECs (Figures 6(a) and 6(b)). We also found that EDA treatment significantly decreased the expression of phospho-NF-κB p65 in PM-induced lung inflammation in a dose-dependent manner (Figures 6(c) and 6(d)). Besides, we performed a knockdown of NF-κB p65 by transfection with NF-κB p65 siRNA in HBECs to further clarify the key role of the NF-κB p65 signaling pathway in PM-induced inflammation. The efficiency of NF-κB p65 siRNA was examined by RT-PCR and western blot. We found a decrease in the expression of NF-κB p65 in HBECs after transfection with NF-κB p65 siRNA with or without PM exposure (Figures 6(e)–6(g)). Furthermore, we found that knockdown of NF-κB p65 significantly attenuated the production of inflammatory cytokines, including IL-6, IL-1α, and IL-1β in PM-exposed HBECs (Figure 6(h)). These results suggested that NF-κB p65 signaling pathway activation played a vital role in PM-induced inflammation and EDA treatment could inhibit the activation of the NF-κB p65 signaling pathway to alleviate PM-induced inflammation.
Figure 6.
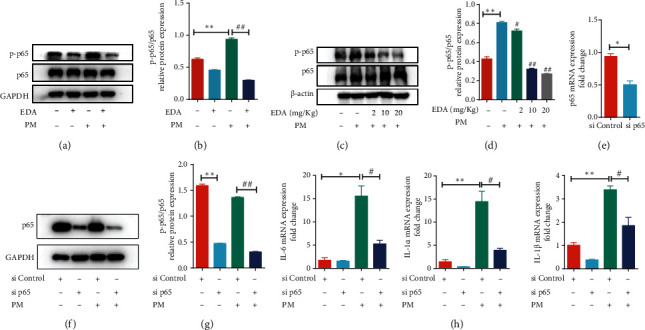
EDA attenuated PM-induced activation of NF-κB p65 signaling pathway. (a) HBECs were treated with 500 μM EDA and then stimulated with 300 μg/mL PM for 24 h. The phosphorylation of the NF-κB p65 signaling pathway was detected by western blot. The optical densities of phospho-NF-κB p65 are shown in (b). (c) Mice were intratracheally instilled with PM at 100 μg/day/mouse for 2 consecutive days. EDA (2, 10, or 20 mg/kg) was intraperitoneally injected prior to PM exposure for 1 h. The phosphorylation of NF-κB p65 in lung tissues was detected by western blot. The optical densities of phospho-NF-κB p65 are shown in (d). (e) The transfection efficiency of NF-κB siRNA was detected by RT-PCR. (f) The protein level of NF-κB was tested by western blot in HBECs which were transfected with NF-κB siRNA with or without PM exposure. The optical densities of phospho-NF-κB p65 are shown in (g). (h) The mRNA level of inflammatory factors IL-6, IL-1α, and IL-1β was detected by RT-PCR. ∗P < 0.05 or ∗∗P < 0.01, compared with the control group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3 or 5. EDA: edaravone; PM: particulate matter; HBECs: human bronchial epithelial cells.
4. Discussion
Inflammation is the primary biological mechanism of PM-induced lung damage and is responsible for the PM-induced development and exacerbation of chronic respiratory diseases. In this study, we showed that PM exposure triggered mitochondrial damage with the rapid increase in ROS and loss of MMP and thus promoted inflammatory cytokines and COX-2 expression in HBECs. EDA as the antioxidative drug decreased ROS production, reversed the MMP, and further inhibited the activation of the NF-κB p65 signaling pathway in PM-induced HBECs and lung tissues (Figure 7). To our knowledge, this is the first study to demonstrate the protective effect of EDA on PM-induced lung inflammation.
Figure 7.
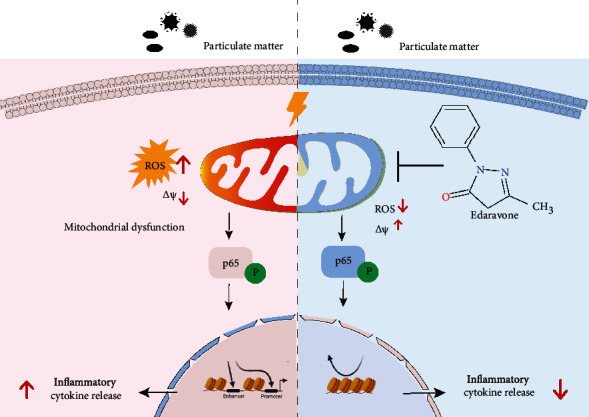
Schematic of the protective effect of EDA on PM-induced inflammation.
EDA is clinically approved for treating stroke and amyotrophic lateral sclerosis (ALS) in previous researches by removing hydroxyl radicals. It can also modulate inflammatory processes, matrix metalloproteinase levels, nitric oxide production, apoptotic cell death, and necrotic cell death [11, 16, 17]. Tajima et al. found that EDA prevented lung injury and attenuated inflammatory cell activation and the release of inflammatory cytokines (IL-6 and TNF-α) in the BALF of the LPS-induced ALI mouse model [29]. It was also reported that EDA attenuated LPS-mediated inflammatory response in astrocytes [30]. Thus, EDA might be a powerful drug for inhibiting inflammatory responses. However, the protective effects of EDA on PM-induced inflammatory responses in the lung were unknown so far. In this study, we used PM-induced HBECs and mice to investigate the anti-inflammatory role of EDA. Our results provided direct evidence that EDA protected against PM-induced inflammation in HBECs and lung tissues of mice in a dose-dependent manner.
The signs of mitochondrial metabolic dysfunction included the increased ROS and loss of the membrane potential difference between the inner and outer membranes, which were also the hallmark of apoptotic mitochondrial damage [31]. Mitochondria constitute a significant source of ROS, affecting inflammatory response processes [32]. Previous studies showed that oxidative stress participated in regulating PM-induced inflammatory response, cell apoptosis, and even lung injury [33, 34]. Ning et al. found that the expression of antioxidant enzymes, superoxide dismutase 2 (SOD2), SOD3, and peroxiredoxin 4 (PRDX4) was decreased in PM-exposed lung tissues of mice [35]. Similar to these previous studies, we detected that PM exposure decreased MMP and further promoted ROS production in HBECs. EDA exhibits beneficial effects on free radical scavenging and maintaining the stability of MMP. Li et al. found that EDA treatment could increase ATP and mitochondrial membrane potential levels by augmenting the expression of Nrf2 and the target genes in excessive evaporation-induced dry eyes [36]. Reyes et al. found that EDA inhibited ischemia-reperfusion injury-induced pulmonary dysfunction by suppressing MDA levels and the expression of inflammatory cytokines [37]. Similarly, we firstly found that EDA could inhibit PM-induced mitochondrial imbalance and ROS production in vitro and in vivo. Elimination of oxidative stress may be a strategy to prevent PM-induced inflammation.
The NF-κB pathway plays a crucial role in inflammatory responses by regulating the transcription of inflammatory cytokine genes [38]. Hassanein et al. demonstrated that EDA prevented cyclophosphamide-induced hemorrhagic cystitis and increased MDA by suppressing the NF-κB signaling pathway in rats [39]. We also reported that PM exposure induced the activation of the NF-κB pathway and increased the production of IL-6, IL-8, IL-1β, and COX-2 in HBECs [18]. In this study, we further demonstrated that the NF-κB pathway was also significantly activated in PM-induced lung tissues of mice. Knockdown of NF-κB p65 significantly inhibited the expression of inflammatory cytokines induced by PM exposure in HBECs.
Besides, the dose of PM exposure and EDA treatment in our studies was identified according to the previously reported studies [11, 17, 18, 23–27]. Based on the human exposure dose equivalent to the mouse-aspirated dose covert algorithm [40], we found that 100 μg PM2.5 exposure for mice via intratracheal instillation was equivalent to 141 μg/m3 PM2.5 exposure for humans in 24 hours. According to the World Health Organization (WHO) air quality guidelines, the recommended short-term (24-hour) AQG level of interim target-1 of PM2.5 is 75 μg/m3. Thus, the dose of PM exposure for mice in our study could contribute to lung damage. To make our results more accurate, we selected the concentration gradient of EDA for our study. EDA has been widely used in clinical treatment of acute cerebral infarction, and the recommended dose of EDA was 30 mg twice a day according to the drug instruction. According to the drug dose convert algorithm between humans and mice based on the body surface area [41], the concentration of EDA in our study was equivalent to 0.163, 0.813, and 1.626 mg/kg for humans. Thus, the concentration of EDA for the clinical treatment was included in our concentration range in this study. Furthermore, we demonstrated that alone EDA treatment with different concentrations had no toxic effect in our study.
5. Conclusion
Taken together, we firstly found that EDA inhibited the expression of IL-6, IL-1α, IL-1β, and COX-2 production in HBECs and mice exposed to PM. Meanwhile, EDA attenuated PM-induced mitochondrial dysfunction, ROS overproduction, and activation of the NF-κB signaling pathway. We demonstrated that EDA could significantly alleviate PM-induced lung inflammation by inhibiting the ROS-NF-κB p65 signaling pathway in vitro and in vivo. These data provided evidence that EDA would be a beneficial therapeutic choice in preventing and treating the harmful effects of PM exposure in the future.
Acknowledgments
This study was sponsored by the Shanghai Sailing Program (20YF1405700), National Natural Science Foundation of China (82000013, 81970060, 81770075, 81630001, and 82041003), Science and Technology Commission of Shanghai Municipality (20DZ2261200, 20Z1190100, and 20XD1401200), National Key R&D Plan (2020YFC2003700), and Shanghai Municipal Key Clinical Specialty (shslczdzk02201).
Contributor Information
Jian Wang, Email: 251328610@qq.com.
Meiling Jin, Email: mljin118@163.com.
Data Availability
All supporting the findings of this report are included in this article. Source raw data for all quantitations is fully available upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Yingying Zeng, Guiping Zhu, and Mengchan Zhu contributed equally to this work.
Supplementary Materials
Figure S1: alone EDA treatment had no toxic effect on lung tissues. Figure S2: alone EDA treatment had no effect on the expression of inflammatory cytokines and protein expression.
References
- 1.2018. http://www.who.int/mediacentre/factsheets/fs292/en/ http://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health .
- 2.Wooding D. J., Ryu M. H., Li H., et al. Acute air pollution exposure alters neutrophils in never-smokers and at-risk humans. The European Respiratory Journal . 2020;55(4, article 1901495) doi: 10.1183/13993003.01495-2019. [DOI] [PubMed] [Google Scholar]
- 3.Laden F., Schwartz J., Speizer F. E., Dockery D. W. Reduction in fine particulate air pollution and mortality: Extended follow-up of the Harvard Six Cities study. American Journal of Respiratory and Critical Care Medicine . 2006;173(6):667–672. doi: 10.1164/rccm.200503-443OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dockery D. W., Pope C. A., 3rd, Xu X., et al. An association between air pollution and mortality in six U.S. cities. The New England Journal of Medicine . 1993;329(24):1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 5.Miller K. A., Siscovick D. S., Sheppard L., et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. The New England Journal of Medicine . 2007;356(5):447–458. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 6.Chen H., Chomyn A., Chan D. C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction∗. The Journal of Biological Chemistry . 2005;280(28):26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 7.Mabalirajan U., Dinda A. K., Kumar S., et al. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. Journal of Immunology . 2008;181(5):3540–3548. doi: 10.4049/jimmunol.181.5.3540. [DOI] [PubMed] [Google Scholar]
- 8.Hock M. B., Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annual Review of Physiology . 2009;71(1):177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 9.Aguilera-Aguirre L., Bacsi A., Saavedra-Molina A., Kurosky A., Sur S., Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. Journal of Immunology . 2009;183(8):5379–5387. doi: 10.4049/jimmunol.0900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikuchi K., Uchikado H., Miyagi N., et al. Beyond neurological disease: new targets for edaravone (review) International Journal of Molecular Medicine . 2011;28(6):899–906. doi: 10.3892/ijmm.2011.795. [DOI] [PubMed] [Google Scholar]
- 11.Roh M. I., Murakami Y., Thanos A., Vavvas D. G., Miller J. W. Edaravone, an ROS scavenger, ameliorates photoreceptor cell death after experimental retinal detachment. Investigative Ophthalmology & Visual Science . 2011;52(6):3825–3831. doi: 10.1167/iovs.10-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C., Li M., Lin L., Chen S., Chen Y., Hong L. Clinical effects and safety of edaravone in treatment of acute ischaemic stroke: a meta-analysis of randomized controlled trials. Journal of Clinical Pharmacy and Therapeutics . 2021;46(4):907–917. doi: 10.1111/jcpt.13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bayrak B. B., Yilmaz S., Hacihasanoglu Cakmak N., Yanardag R. The effects of edaravone, a free-radical scavenger in lung injury induced by valproic acid demonstrated via different biochemical parameters. Journal of Biochemical and Molecular Toxicology . 2021;35(9, article e22847) doi: 10.1002/jbt.22847. [DOI] [PubMed] [Google Scholar]
- 14.Fan S. R., Ren T. T., Yun M. Y., Lan R., Qin X. Y. Edaravone attenuates cadmium-induced toxicity by inhibiting oxidative stress and inflammation in ICR mice. Neurotoxicology . 2021;86:1–9. doi: 10.1016/j.neuro.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Kim Y. S., Lee H. Y., Jang J. Y., Lee H. R., Shin Y. S., Kim C. H. Redox treatment ameliorates diabetes mellitus-induced skin flap necrosis via inhibiting apoptosis and promoting neoangiogenesis. Experimental Biology and Medicine . 2021;246(6):718–728. doi: 10.1177/1535370220974269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terada T., Kulkarni J. A., Huynh A., Tam Y. Y. C., Cullis P. Protective effect of edaravone against cationic lipid-mediated oxidative stress and apoptosis. Biological & Pharmaceutical Bulletin . 2021;44(1):144–149. doi: 10.1248/bpb.b20-00679. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z., Luo Z., Bi A., et al. Compound edaravone alleviates lipopolysaccharide (LPS)-induced acute lung injury in mice. European Journal of Pharmacology . 2017;811:1–11. doi: 10.1016/j.ejphar.2017.05.047. [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Huang J., Wang L., et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-κB signaling pathway. Journal of Thoracic Disease . 2017;9(11):4398–4412. doi: 10.21037/jtd.2017.09.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J., Huang J., Zhu M., et al. Osteopontin potentiates PM-induced IL-1α and IL-1β production via the ERK/JNK signaling pathway. Ecotoxicology and Environmental Safety . 2019;171:467–474. doi: 10.1016/j.ecoenv.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Wang J., Zhu M., Ye L., Chen C., She J., Song Y. miR-29b-3p promotes particulate matter-induced inflammatory responses by regulating the C1QTNF6/AMPK pathway. Aging . 2020;12(2):1141–1158. doi: 10.18632/aging.102672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Zhu M., Wang L., Chen C., Song Y. Amphiregulin potentiates airway inflammation and mucus hypersecretion induced by urban particulate matter via the EGFR-PI3Kα-AKT/ERK pathway. Cellular Signalling . 2019;53:122–131. doi: 10.1016/j.cellsig.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Wang J., Zhu M., Song J., et al. The circular RNA circTXNRD1 promoted ambient particulate matter-induced inflammation in human bronchial epithelial cells by regulating miR-892a/COX-2 axis. Chemosphere . 2022;286, Part 1, article 131614 doi: 10.1016/j.chemosphere.2021.131614. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Z. Y., Luan P., Huang S. X., et al. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neuroscience & Therapeutics . 2013;19(3):163–169. doi: 10.1111/cns.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai X., Zhang C., Chen A., et al. Protective effect of edaravone on glutamate-induced neurotoxicity in spiral ganglion neurons. Neural Plasticity . 2016;2016:10. doi: 10.1155/2016/4034218.4034218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan Y., Zha H., Rangarajan P., Ling E.-A., Wu C. Anti-inflammatory effects of edaravone and scutellarin in activated microglia in experimentally induced ischemia injury in rats and in BV-2 microglia. BMC Neuroscience . 2014;15(1) doi: 10.1186/s12868-014-0125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito H., Wate R., Fau-Zhang J., et al. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Experimental Neurology . 2008;213(2):448–455. doi: 10.1016/j.expneurol.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y., Zhu W., Kong W., Li T., Zou P., Chen H. Edaravone attenuates neuronal apoptosis in hippocampus of rat traumatic brain injury model via activation of BDNF/TrkB signaling pathway. Archives of Medical Science . 2021;17(2):514–522. doi: 10.5114/aoms.2019.89849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Y., Tong C., Wang Y., et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein & Cell . 2018;9(10):867–878. doi: 10.1007/s13238-017-0384-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tajima S., Soda M., Bando M., et al. Preventive effects of edaravone, a free radical scavenger, on lipopolysaccharide-induced lung injury in mice. Respirology . 2008;13(5):646–653. doi: 10.1111/j.1440-1843.2008.01322.x. [DOI] [PubMed] [Google Scholar]
- 30.Guo Z., Wu H. T., Li X. X., et al. Edaravone protects rat astrocytes from oxidative or neurotoxic inflammatory insults by restoring Akt/Bcl-2/Caspase-3 signaling axis. IBRO Reports . 2020;8:122–128. doi: 10.1016/j.ibror.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinvalet D., Zhu P., Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity . 2005;22(3):355–370. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., Liu Q., Yin H., Li S. Cadmium exposure induces pyroptosis of lymphocytes in carp pronephros and spleens by activating NLRP3. Ecotoxicology and Environmental Safety . 2020;202, article 110903 doi: 10.1016/j.ecoenv.2020.110903. [DOI] [PubMed] [Google Scholar]
- 33.Li N., Hao M., Phalen R. F., Hinds W. C., Nel A. E. Particulate air pollutants and asthma: a paradigm for the role of oxidative stress in PM-induced adverse health effects. Clinical Immunology . 2003;109(3):250–265. doi: 10.1016/j.clim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Riva D. R., Magalhaes C. B., Lopes A. A., et al. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhalation Toxicology . 2011;23(5):257–267. doi: 10.3109/08958378.2011.566290. [DOI] [PubMed] [Google Scholar]
- 35.Ning R., Li Y., Du Z., et al. The mitochondria-targeted antioxidant MitoQ attenuated PM2.5-induced vascular fibrosis via regulating mitophagy. Redox Biology . 2021;46, article 102113 doi: 10.1016/j.redox.2021.102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y., Liu H., Zeng W., Wei J. Edaravone protects against hyperosmolarity-induced oxidative stress and apoptosis in primary human corneal epithelial cells. PLoS One . 2017;12(3, article e0174437) doi: 10.1371/journal.pone.0174437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes Y. A., Shimoyama T., Akamatsu H., Sunamori M. MCI-186 (edaravone), a free radical scavenger, attenuates ischemia-reperfusion injury and activation of phospholipase A (2) in an isolated rat lung model after 18 h of cold preservation. European Journal of Cardio-Thoracic Surgery . 2006;29(3):304–311. doi: 10.1016/j.ejcts.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Yao L., Kan E. M., Kaur C., et al. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PLoS One . 2013;8(11, article e78439) doi: 10.1371/journal.pone.0078439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassanein E. H. M., Ahmed M. A., Sayed A. M., Rashwan E. K., Abd El-Ghafar O. A. M., Mahmoud A. M. Edaravone mitigates hemorrhagic cystitis by modulating Nrf2, TLR-4/NF-κB, and JAK1/STAT3 signaling in cyclophosphamide-intoxicated rats. Journal of Biochemical and Molecular Toxicology . 2021;35(11, article e22889) doi: 10.1002/jbt.22889. [DOI] [PubMed] [Google Scholar]
- 40.Gavett S. H., Haykal-Coates N., Highfill J. W., et al. World Trade Center fine particulate matter causes respiratory tract hyperresponsiveness in mice. Environmental Health Perspectives . 2003;111(7):981–991. doi: 10.1289/ehp.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nair A. B., Jacob S. A simple practice guide for dose conversion between animals and human. Journal of Basic and Clinical Pharmacy . 2016;7(2):27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: alone EDA treatment had no toxic effect on lung tissues. Figure S2: alone EDA treatment had no effect on the expression of inflammatory cytokines and protein expression.
Data Availability Statement
All supporting the findings of this report are included in this article. Source raw data for all quantitations is fully available upon request.