

Abstract
High volume blast furnace slag (BFS) resulting from iron-making activities has long been considered a burden for the environment. Despite considerable research efforts, attempts to convert BFS into high value-added products for environmental remediation are still challenging. In this study, calcium–magnesium–aluminium layered double hydroxides (CaMgAl-LDHs) and ordered mesoporous silica material (MCM-41) sorbents were simultaneously synthesized from BFS, and their CO2 adsorption performance was evaluated. Calcium (Ca), magnesium (Mg) and aluminium (Al) were selectively extracted from BFS using hydrochloric acid. Leaching conditions consisting of 2 mol L−1 acid concentration, 100 °C leaching temperature, 90 min leaching time and a solid-to-liquid ratio of 40 g L−1 achieved a high leaching ratio of Ca, Mg and Al at 88.08%, 88.59% and 82.27%, respectively. The silica-rich residue (SiO2 > 98.6 wt%) generated from the leaching process could be used as a precursor for MCM-41 preparation. Chemical composition, surface chemical bonds, morphology and textural properties of the as-synthesized CaMgAl-LDHs and MCM-41 sorbents were determined. Both the CaMgAl-LDHs and MCM-41 sorbents were found to be thermally stable and exhibited comparable adsorption uptake and rates over 20 CO2 adsorption/desorption cycles. This work demonstrated that a total solution for the utilisation of BFS can be achieved and the resulting valuable products, i.e. CaMgAl-LDHs and MCM-41 are promising sorbents for CO2 capture.
A total solution for the utilisation of BFS can be achieved and the resulting valuable products CaMgAl-LDHs and MCM-41 are promising sorbents for CO2 capture.
1. Introduction
One of the most significant environmental challenges faced by humans in the 21st century is global warming caused by the increase in carbon dioxide levels.1 In response to the issue of greenhouse gases, many countries have proposed a series of carbon emission reduction plans. Notably, research into options for the capture, utilisation and long-term storage of carbon dioxide has become a hot topic in the field of low-carbon and zero-carbon technologies, which include renewable energy, clean energy and carbon capture & storage (CCS).1–5 (CCS) is being investigated as a potential option to capture CO2 at the source of generation from various industrial facilities, and to transport and separate the captured CO2 in storage areas.5–9
Iron and steel manufacturing are not only major energy-consuming industries but also major emitters of greenhouse gases. The generation of large amounts of solid waste including blast furnace slag (BFS) and steel slag (SS) has caused severe environmental problems involving water and soil contamination, and local air pollution.10 To reduce or resolve these growing environmental concerns, many current research activities are focusing on the development of new waste management strategies such as the preparation of useful materials from waste. In the past ten years, mineral carbonation is the most dynamic and the fastest growing CO2 sequestration technology. In mineral carbonation, CO2 gas is stored by promoting the reaction of magnesium or calcium oxides in silicate minerals with carbon dioxide to form carbonates;4 BFS and SS from the steel industry contain a large number of alkaline oxides, such as CaO and MgO, which can be used for CO2 capture.11
In Finland, Eloneva et al.2–5 studied the possibility of utilizing SS for the production of pure calcium carbonate. They found that dilute acetic acid only selectively dissolved calcium from the steel slag. Tian et al.12 proposed a process for simultaneously producing CaO-based CO2 sorbents and high-quality iron ore from waste SS. The results showed that the extraction of calcium was always accompanied by the undesired dissolution of elemental impurities such as Al, Mg and Fe, resulting in decreased CO2 adsorption capacity.
As well as the intensive research activity on SS, BFS is also considered as valuable raw material in CO2 sequestration. Compared with SS, BFS is more greatly enriched in elemental Ca, Mg, Si and Al, which together account for around 90% of its total mass; this means that the aqueous extraction of BFS could potentially leads to less contamination. Seonhye Lee et al.1 studied the indirect sequestration of CO2 using BFS as the raw material and achieved 49.76% extraction efficiency of Ca and 95.69% carbonation efficiency using NH4NO3 under typical conditions (2 mol L−1 solvent concentration, 30 °C reaction temperature, 60 min reaction time, 10 g L−1 solid-to-liquid ratio).
In their early research, Yasutaka Kuwahara et al.13 successfully synthesised a Ca-based layered double hydroxide (LDH) compound from BFS, and applied it in CO2 fixation. In their methodology, the hydrochloric acid leaching resulted in a precursor solution suited for LDH preparation and hydrated silica gel. Inspired by this result, in this paper, a facile method for the simultaneous preparation of (LDHs) and ordered mesoporous silica (MCM-41) materials from BFS is reported. The objectives of this research are to focus on: (i) clarifying the influence of acid leaching conditions on the dissolution behavior of BFS, and seeking optimised conditions for the selective and highly efficient dissolution of elemental calcium, magnesium and aluminium; (ii) making best use of the BFS through synthesising LDHs and MCM-41 sorbents by recovering the leachate and residue after solvent extraction and subsequent structural characterisation, (iii) the evaluation of CO2 sequestration ability for both sorbents through adsorption–desorption measurements.
2. Experimental
2.1. Materials and test facilities
Water granulated blast furnace slag was received from the Shagang Steel Co., Ltd., China. The slag was crashed and ball milled to a particle size of less than 74 μm. The chemical composition of the BFS was analysed using X-ray fluorescence (XRF, SHIMADZU-EDX800, Japan), and the concentrations of Ca, Si, Mg and Al in the solution were measured using inductively coupled plasma-optic emission spectrometry (ICP-OES, Vista-MPX, Varian, USA). The compositionA results from XRF quantitative measurements are shown in Table 1, revealing that the BFS was mainly composed of 36.84 wt% CaO, 34.27 wt% SiO2, 10.14 wt% MgO and 14.47 wt% Al2O3, which together accounted for 95% of the total mass. Results of the ICP-OES analysis gave the compositionB of the solution, with these values used for estimating the leaching ratio of metal ions from the BFS.
Chemical composition of blast furnace slag A: measured by XRF analysis, B: by ICP-OES analysis.
| Al2O3 | SiO2 | CaO | MgO | K2O | Fe2O3 | Na2O | MnO | |
|---|---|---|---|---|---|---|---|---|
| Composition A | ||||||||
| wt% | 14.47 | 34.27 | 36.84 | 10.14 | 0.52 | 0.53 | 0.43 | 0.52 |
| Composition B | ||||||||
| wt% | 14.66 | 32.77 | 36.14 | 10.79 | NA | 0.26 | NA | NA |
The mineralogical phases were identified by X-ray diffraction (XRD, UltimaI VX, Rigaku, Japan) with CuKα radiation, utilising a generator voltage of 40 kV and tube current of 40 mA. Small-angle X-ray scattering (SAXS) measurements were carried out at room temperature using a SAXSess mc2 (Anton Paar) instrument.
Surface functional groups were determined with Fourier transform infrared spectroscopy (Jasco FT-IR-4100) with KBr in the 1800–400 cm−1 region.
Morphologies were characterized by scanning electron microscope (SEM, SU5000, Hitachi, Japan) and a transmission electron microscope (TEM, TECNAI G2 F20, FEI, USA). Textural properties were investigated by N2 adsorption–desorption measurements using an ASAP 2000 instrument (Micromeritics, USA). Thermogravimetric analysis (TGA) was performed to evaluate thermal stability using a TGA-50/50H analyser (Shimadzu, Kyoto, Japan).
2.2. Experimental methods
2.2.1. Hydrochloric acid dissolution experiments
As illustrated in Fig. 1, extraction experiments were carried out in a 500 mL flat-bottomed flask glass reactor with a working volume of 200 mL. A mechanical stirrer (Dragonlab® MS7-H550-S, Dragon Laboratory Instruments Co. Ltd., Beijing, China) was installed at the center of the reactor and used to mix the slurry at 500 rpm. The glass reactor was placed in a temperature-controlled water bath to maintain a consistent reaction temperature. To avoid evaporation losses, the reactor was equipped with a condenser cooled with tap water.
Fig. 1. Employed methodology for simultaneously producing LDHs and MCM-41 from blast furnace slag.

BFS (10 g) was added to the reactor containing a specific concentration (0.5 M, 1 M, 2 M or 3 M) of hydrochloric acid (11.74 M, Enox) at 100 °C. To monitor the progression of the dissolution, 5 mL of aqueous solution was sampled using quantitative pipette at 5, 10, 20, 40, 60, 80, 100, 120, 140, 160 and 180 min after the slag was added.
The leaching ratio of a specific element was calculated by the following equation:
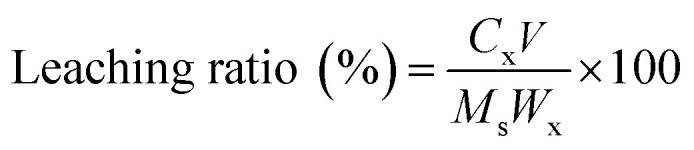 |
1 |
Here, Cx (mg L−1) represents the concentration of a selected element in the extracted aqueous solution, V (L) is the volume of aqueous solution, Ms (mg) denotes the mass of the slag and Wx (wt%) is the elemental content in the slag.
2.2.2. Synthesis of LDHs
After the leaching process, the obtained leachate containing Ca2+, Mg2+, Al3+ can be used as precursor for LDHs synthesis. NaOH aqueous solution (2 M) was added dropwise into the leachate (150 mL) and the final pH adjusted to 12. The resulting mixture was then aged at 80 °C for 18 h to enable crystallisation. The aged mixture was filtered and washed with deionised water several times until the pH was 7. Finally, the obtained product was dried at 100 °C overnight.
2.2.3. Synthesis of MCM-41
The solid residue resulting from acid leaching was subjected to secondary acid leaching (2 M acid concentration, 1 h of leaching at 100 °C) to remove impurities. The XRF results in Table 2 show that the secondary acid leaching yielded solid residue containing around 98.6 wt% of silica and could be utilised as a precursor for mesoporous silica preparation.
Chemical composition of the solid residue obtained from secondary acid leaching.
| Composition | SiO2 | Al2O3 | MgO | TiO2 | Fe2O3 | SO3 | MnO | Cl |
|---|---|---|---|---|---|---|---|---|
| wt% | 98.581 | 0.094 | 0.048 | 0.017 | 0.013 | <0.01 | <0.01 | 0.689 |
In a typical procedure, n-cetyltrimethylammonium bromide (CTAB, 1.457 g) was dissolved in of deionised water (60 mL) at room temperature. Then, acid leaching residue (1.2 g) and NaOH (1 g) were added to the CTAB aqueous solution. The mixture was stirred until a clear solution was obtained, following which NaOH or HCl solution was added dropwise to adjust the pH to 11. The resulting solution was further stirred for another one hour, then placed in a Teflon-lined autoclave for hydrothermal treatment at 80 °C for 48 h. After that, the slurry was filtered, washed repeatedly using deionised water and air-dried overnight at 100 °C. Finally, the as-synthesized silica powder was calcined at 550 °C in air for 6 h.
2.2.4. Measurements of CO2 adsorption capacity
CO2 adsorption–desorption measurements of synthesized LDHs and MCM-41 materials were performed on a thermogravimetric analyser (CP-2000, Beijing). Precisely 10 mg sample was activated by heating it to a desired temperature and maintaining the temperature for 30 min under N2 (50 mL min−1) before CO2 adsorption. The temperature was then adjusted to the adsorption temperature and the gas feed switched from N2 to CO2 with a constant flow of 50 mL min−1. After adsorption, the flow was switched from CO2 to N2 and the temperature adjusted to and maintained at the activated temperature for 30 min for completion of one desorption. This experiment was repeated twenty times to investigate the stability of CO2 adsorption.
3. Results and discussion
3.1. Leaching of BFS
3.1.1. Effect of hydrochloric acid concentration
According to the results in Fig. 2, the dissolution of elemental Ca, Mg, Al and Si in BFS was strongly dependent on acid concentration. The leaching ratios of Ca, Mg, Al significantly improved when the acid concentration exceeded 1 M. Surprisingly the rate of dissolution of BFS in acid solution was extremely fast and the leaching ratios for these elements reached a within 5–10 min of initial contact; this suggested that the water-granulated BFS exhibits outstanding reactivity. In contrast, the dissolution behaviour of Si followed a different pattern. When higher concentrations of 2 M and 3 M were employed, the leaching ratio of Si was drastically reduced from 65% to 1.27% after 80 min and continuously decreased to nearly zero.
Fig. 2. Effect of the variation in the hydrochloric acid concentrations (0.5 M, 1 M, 2 M, 3 M) on the leaching ratio of Ca, Mg, Al, Si at condition: temperature of 100 °C, solid to liquid ratio of 50 g L−1.

3.1.2. Effect of leaching temperature
Fig. 3 shows that increasing the temperature from 70 to 100 °C did not enhance the dissolution of Ca, Mg, Al and Si; on the contrary, the leaching ratios of these elements all decreased. In particular, the leaching ratio of Si dropped dramatically to 1.27% at 100 °C, which suggested that high temperature favours the formation of silica gel and consequently leads to decreased dissolution of Si. The newly forming silica gel with high surface area has the capability of caging metal ions, which may account for lower leaching ratios of Ca, Mg and Al at higher temperatures.
Fig. 3. Effect of leaching temperature (70 °C, 90 °C, 100 °C) on the leaching ratio of Ca, Mg, Al, Si at condition: acid concentration of 2 M, the solid to liquid ratio of 50 g L−1, leaching time of 90 min.

3.1.3. Effects of the solid-to-liquid ratio
To study the effect of the solid-to-liquid (S/L) ratio on the leaching behavior, the acid solution (concentration and volume) was kept constant and the amount of BFS doped in the solution was varied, i.e. 6, 8, 10, 12, 14 gram. As shown in Fig. 4, the general trend was that the leaching ratio of Ca, Mg, and Al increased as the S/L ratio decreased. At a S/L ratio of 40 g L−1, the highest leaching ratio of Ca, Mg, Al was achieved at 88.08%, 88.59% and 82.27%, respectively. Meanwhile, the S/L ratio had little effect on the dissolution behaviour of Si.
Fig. 4. Effect of the S/L ratio (30, 40, 50, 60, 70 g L−1) on the leaching ratio of Ca, Mg, Al, Si at condition: temperature of 100 °C, acid concentration of 2 M, leaching time of 90 min.

3.1.4. Leaching selectivity
The degree of elemental selectivity was used to identify the optimised extraction conditions for subsequent synthesis of high purity LDHs and MCM-41 materials. As can be seen from Table 3, a leaching temperature of 100 °C, acid concentration of 2 M and S/L ratio of 40 g L−1, yielded a satisfied elemental selectivity, that is the elemental selectivity for silicon was a minimum and high leaching ratio. Increase in the acid concentration resulted in the considerable decomposition of the amorphous silica phase within only a few minutes of leaching. Dissolved silicon presents as Si(OH)4, which is monomeric silica in an acidic solution. Once the monomeric concentration exceeds the saturation level, the discrete silica particles start to polymerise to into a gel. The above experimental results indicated that high temperature and long leaching times can promote the polymerisation of Si according to eqn (2) and (3):14
| Si(OH)4 + Si(OH)4 → (HO)3Si-O-Si(OH)3 + H2O | 2 |
| (HO)3Si-O-Si(OH)3 → (SiO2)2 + 3H2O | 3 |
Elemental selectivity of four elements and n(M2+)/n(M3+) in the leachate solution from different leaching conditionsa.
| Concentration of HCl (M) | Temperature (°C) | S/L ratios (g L−1) | Elemental selectivity (%) | n(M2+)/n(M3+) | |||
|---|---|---|---|---|---|---|---|
| Ca2+ (%) | Mg2+ (%) | Al3+ (%) | Si4+ (%) | ||||
| 0.5 | 100 | 50 | 67.94 | 22.36 | 8.52 | 1.17 | 10.60 |
| 1 | 100 | 50 | 69.36 | 23.59 | 6.81 | 0.24 | 13.65 |
| 2 | 100 | 50 | 58.99 | 20.05 | 20.73 | 0.23 | 3.81 |
| 3 | 100 | 50 | 59.55 | 19.67 | 20.47 | 0.31 | 3.87 |
| 2 | 70 | 50 | 35.90 | 15.45 | 17.13 | 31.52 | 3.00 |
| 2 | 90 | 50 | 36.45 | 15.63 | 17.16 | 30.77 | 3.04 |
| 2 | 100 | 30 | 52.70 | 21.01 | 24.11 | 2.17 | 3.06 |
| 2 | 100 | 40 | 50.89 | 20.38 | 22.91 | 5.82 | 3.11 |
| 2 | 100 | 60 | 50.54 | 20.96 | 23.47 | 5.03 | 3.05 |
| 2 | 100 | 70 | 52.33 | 22.08 | 24.10 | 1.49 | 3.09 |
Elemental selectivity (%) = nx/(nCa + nMg + nAl + nSi) × 100%, x = Ca, Mg, Al, Si.
Another important index that also needs to be considered is the molar ratio of divalent cations to trivalent cations in the leachate solution: the molar ratio should meet the general requirements (between 2 and 4)15 for the preparation of LDHs. The value of n(M2+)/n(M3+) is strongly related to the hydrochloric acid concentration. With increased acid concentration, n(M2+)/n(M3+) decreased, which suggests that the trivalent metal (Al3+) shows stronger resistance against acid attack compared with divalent alkali metals (Ca2+, Mg2+).
In summary, leaching conditions need to be rationally chosen for obtaining a high leaching ratio of elemental Ca, Mg and Al, low elemental selectivity for Si and a suitable n(M2+)/n(M3+). Leaching conditions comprising 2 M acid concentration, 100 °C leaching temperature, 90 min leaching time and a S/L ratio of 40 g L−1 allowed a high leaching ratio of Ca, Mg and Al to be achieved, at 88.08%, 88.59% and 82.27%, respectively, 5.82% of Si and a value of 3.11 for n(M2+)/n(M3+).
3.2. Characterization of CaMgAl-LDHs sorbent
Fig. 5(a) shows the XRD pattern of as-synthesized CaMgAl-LDHs. All the sharp reflections with a high-degree of crystallinity were attributed to the successful formation of LDHs. The LDHs showed obvious crystallographic changes as a result of calcination at different temperatures in air. The sample maintained hydrotalcite structure when the calcination temperature was 200 °C. Once calcined at 400 °C, the sample showed weak reflections characteristic of hydrotalcite with a small amount of periclase, indicating decomposition of the layered structure. Calcination above 800 °C generated new crystalline phases assignable to CaO, MgO and mayenite (Ca12Al4O33). The thermal events of as-synthesized CaMgAl-LDHs from BFS was further studied by TGA analysis. The thermal decomposition of the CaMgAl-LDHs occurred in three main weight loss steps (ESI, Fig. S1†). In addition, this LDHs had a nominal structural formula of Ca1.89Mg0.89Al1.0(OH)7.5Cl0.83(CO3)0.11·yH2O, and most of the intercalated anions were Cl− (ESI, Table S1†).
Fig. 5. (a) X-ray diffraction patterns of LDHs calcined at different temperatures for 4 h: (A) as-synthesized, (B) 200 °C, (C) 400 °C and (D) 800 °C (b) BET results of LDHs calcined at different temperatures.

The textural properties obtained from BET analysis are presented in Fig. 5(b). The specific surface area increased with increase in calcination temperature from 200 °C to 400 °C. The sample after calcination at 400 °C for 4 h had a specific surface area of 38.55 m2 g−1, which was twice as large as that of the LDHs. A slight increase in average pore diameter and pore volume were also observed. When the temperature further increased to 800 °C, the crystallisation of resulting oxides resulted in a decreased surface area and porosity.
SEM micrographs of as-synthesized LDHs calcined at different temperatures are shown in Fig. 6. Fig. 6(a) shows the formation of a typical plate-like structure with well-defined hexagonal shape.16 The particle size of LDHs was about 1 μm and the thickness was less than 40 nm. Fig. 6(b) shows that the plate-like particles stacked together after calcination at 200 °C. When the temperature further increased, calcination resulted in the disappearance of characteristic LDHs features with the formation of amorphous mixed oxides (Fig. 6(c)), which further developed into mineral crystallites as a consequence of sintering events (Fig. 6(d)).17
Fig. 6. SEM images of LDHs sorbents calcined at different temperatures: (a) as-synthesized, (b) 200 °C, (c) 400 °C and (d) 800 °C.

3.3. Characterization of MCM-41 sorbent
The SEM image in Fig. 7(a) shows that the as-synthesized mesoporous silica nanoparticles on a scale of less than 200 nm had a good dispersity, with some coalescence into aggregates. The TEM micrograph in Fig. 7(b) reveals a highly ordered 2D hexagonal pore structure array of the MCM-41 type silica; this feature was also confirmed by the presence of (100), (110) and (200) plane reflections in Fig. 7(c).18 The diameter between the fringes of the pore in the mesoporous structure was estimated to be around 3 nm, which is in good agreement with the BJH pore size distribution analysis in Fig. 7(d), having a primary distribution in the range of 2.6–2.8 nm. The sorption isotherm in Fig. 7(d) shows a type IV isotherm according to the IUPAC classification with a sharp increase step in the relative pressure region of 0.31–0.35. This was attributed to capillary condensation within the narrow distribution of mesopores.19
Fig. 7. (a) SEM image and (b) TEM image of MCM-41 sorbent prepared from BFS; (c) SAXs scattering pattern; (d) N2 adsorption–desorption isotherm and pore size distribution.

Textural properties of MCM-41 including BET surface area (SBET), total pore volume (Vp), average pore radius (DBJH) and the wall thickness (bp) are shown in Table 4. This material had a surface area of 1270 m2 g−1, much higher than when synthesized from fly ash (732 m2 g−1),20 iron ore tailings (527 m2 g−1),21 sodium silicate (874 m2 g−1)22 and pure TEOS (1100 m2 g−1).20 The thickness of pore walls was comparable at 1.04 nm with results in the previous literature.23
Textural properties of MCM-41 derived from BFSa.
| Sample | S BET (m2 g−1) | V p (cm3 g−1) | D BJH (nm) | d 100 (nm) | b p (nm) |
|---|---|---|---|---|---|
| MCM-41 | 1270 | 1.342 | 3.034 | 3.531 | 1.04 |
S BET, specific surface area calculated using BET equation. Vp, the total pore volumes were determined at a P/P0 value of 0.995. DBJH, the mean pore size distributions were determined by BJH mode applied to the desorption branches of the isotherms. bp, the pore wall thickness, calculated from equation.24bp = 2d100/31/2 − DBJH, where d100 refers to the interplanar spacing of the (100) reflection by Bragg's equation (2d100 sin θ = λ).
3.4. Evaluation of CO2 capture performance
The CO2 adsorption behaviour of as-synthesized Ca1.89Mg0.89Al1.0-LDHs and after calcination at 200 °C, 400 °C, 800 °C, denoted as CaMgAl-LDHs-200, CaMgAl-LDHs-400, CaMgAl-LDHs-800, respectively, was measured at 400 °C. As seen from Fig. 8(a), after calcination at 400 °C for 4 h, CaMgAl-LDHs-400 exhibited a first contact capacity of 1.58 mmol g−1, almost 2 times greater than that of CaMgAl-LDHs without thermal activation. However, calcination at 800 °C caused a great decline in the CO2 adsorption. These results were in agreement with those of Ram Reddy et al.,25 indicating that there exists an optimised thermal activation temperature for CaMgAl-LDHs. The events of dehydroxylation, decomposition of intercalated anions, followed by crystallization upon thermal calcination (confirmed by the XRD and TGA analyses) gave rise to the variation in surface area and a number of active basic sites, thus having a great influence on the CO2 uptake.
Fig. 8. (a) The effect of calcination temperature on the first CO2 contact capacities of CaMgAl-LDHs sorbents adsorption temperature was set at 400 °C; (b) adsorption/desorption cycles for CaMgAl-LDHs-400 at 400 °C.

The thermal stability of CaMgAl-LDHs-400 was evaluated through continuous CO2 adsorption/desorption cycles. As shown in Fig. 8(b), CaMgAl-LDHs-400 demonstrates good reusability and cyclic performance throughout the 20 adsorption/desorption cycles. After 20 times cycles, the CaMgAl-LDHs-400 sorbent still adsorbs around 1.2 mmol g−1 of CO2 at 400 °C and 1 atm, CO2 adsorption capacity reduces 24%.
As depicted in Fig. 9(a), for MCM-41, the highest CO2 adsorption capacity increases from 0.95 to 1.24 mmol g−1 when the adsorption temperature increases from 25 to 75 °C, which is a characteristic feature of physisorption processes.26 However, the adsorption capacity decreased slightly to 1.13 mmol g−1 when the temperature exceeded 100 °C, which could be related to the dominant CO2 desorption reaction at high temperature. The adsorption/desorption cycles for MCM-41 were conducted at 75 °C. As shown in Fig. 9(b), there was about 20% deterioration in the adsorption capacity after 20 adsorption/desorption cycles, indicating that the MCM-41 sorbents had a good thermal stability.
Fig. 9. (a) The first CO2 contact capacities of MCM-41 sorbent at different temperatures (b) adsorption/desorption cycles for MCM-41(adsorbed at 75 °C, desorbed at 105 °C).

3.5. Adsorptive mechanism for the two prepared composites for CO2 capture
The composite stability for the prepared two composites was studied using the thermogravimetric analysis with results shown in Fig. 10. LDHs-400 sample was heated to 400 °C and maintained for a while, the weight loss was small. Similarly, MCM-41 sample was heated to 700 °C and maintained for a while, the weight loss was due to the adsorbed gas and water at the beginning and did not change as the temperature rised to 700 °C. This confirmed that both composites had good stability.
Fig. 10. Thermogravimetric analysis of CaMgAl-LDHs-400 and MCM-41.

As depicted Fig. 8(a), the LDHs calcined at 400 °C has a maximum adsorption capacity, which was most likely due to the optimum balance between the surface area and enough basic sites.25 The BET analysis for LDHs calcined at different temperatures in Fig. 5(b) supported this theory. The LDHs-400 sample, which is an amorphous mixed oxide phase, had the maximum BET surface area compared to the other samples at 38.55 m2 g−1. The LDHs-200 sample had the minimum BET surface area because its layered structure was maintained, with CO32− ions occupying the basic sites. Calcination at higher temperature (800 °C) decomposed most CO32− ions to release some basic sites for CO2 adsorption but resulted in a low BET surface area due to the phase transformation of amorphous mixed oxide into crystalline spinel, CaO and MgO phases. So, the LDHs-400 sample had not only the highest surface area but also enough basic sites for improving the CO2 adsorption performance.
FTIR spectra patterns of CaMgAl-LDHs-400 and MCM-41 before and after adsorption are shown in Fig. 11. For LDHs-400, the reflection at 1633 cm−1 for LDHs material was the δH2O mode of interlayer water molecules.27 The relatively weak CO32− reflection at 1425–1475 cm−1 indicated a low level of contamination with CO2 before adsorption,28 while after CO2 adsorption the corresponding band located at around 1417 cm−1 was strong. The AlO6 stretching mode with the indicative reflection around 457 cm−1 was observed after calcination.26 The new reflection at 846 cm−1 indicated the presence of Ca–O bonding, which was consistent with previous results reported in the synthesis of hydrotalcite with varied (Ca + Mg)/Al molar ratios.29 After CO2 adsorption, the samples exhibited extremely weak vibration peaks at 1633 cm−1 and the disappearance of other vibration peaks. The disappearance of peaks related to metal–oxygen vibrations at low wavenumber of around 500 cm−1 was due to the chemical reaction between basic sites and CO2.
Fig. 11. FTIR spectra patterns of CaMgAl-LDHs-400 and MCM-41 before and after adsorption.

For MCM-41, the band around 3450 cm−1 attributed to hydrogen bonded surface silanols (Si–OH) and adsorbed water molecules were due to O–H stretching and the band at 1633 cm−1 was due to bending vibrations of adsorbed water molecules. The peaks centred at 1093 cm−1, 804 cm−1 and 460 cm−1 corresponded to the asymmetric stretching, symmetric stretching and rocking vibrations of the Si–O–Si bonds, respectively. The band appearing at 956 cm−1 was attributed to Si–O stretching in Si–OH. Silanol groups (Si–OH) on the surface of MCM-41 maybe the adsorption sites for capturing CO2 molecules.30 After CO2 adsorption, the band at 956 cm−1 disappeared and a very weak band at 1384 cm−1 was observed for the symmetric stretching mode of linear CO2 which verified that physisorbed CO2 on mesoporous silica was detected.31 The intensity of band corresponded to OH stretching of free silanol groups around 3450 cm−1 decreased after CO2 adsorption and a weakly bonded adduct between silanol groups and CO2: –OH⋯O C O formed which can easily break down as the temperature increases. As depicted Fig. 9(a), the CO2 adsorption capacity at higher temperature (100 °C) decreased because interaction between CO2 and silanol groups is van der Waals force, which does not favor the adsorption at higher temperatures.32
3.6. Comparison of the CO2 adsorption capacities of various materials
Different kinds of LDHs and mesoporous silica and their CO2 adsorption performances are compared in Table 5. The maximum CO2 adsorption capacity of CaMgAl-LDHs derived from BFS is 1.66 mmol g−1, which is higher than that of other LDHs synthesized using pure reagents. The maximum CO2 adsorption capacity of MCM-41 derived from BFS was 1.25 mmol g−1, which is comparable to other mesoporous silica synthesized from other materials. Comparing the results of this study with those reported in literature, BFS was found to be a promising raw material for CaMgAl-LDHs and MCM-41 production and their further use as efficient sorbents for capturing CO2.
Comparison of the CO2 adsorption capacities of various materials.
| Sorbents | Materials | CO2 uptake | Ref. |
|---|---|---|---|
| LDHs | Mg3Al-stearate-LDH-400 | 1.30 mmol g−1 at 300 °C, 1 bar | 33 |
| Ca3Al-LDH-550 | 0.87 mmol g−1 at 350 °C, 1 bar | 34 | |
| Mg3Al-LDH-550 | 1.18 mmol g−1 at 350 °C, 1 bar | 34 | |
| CaMg2Al-LDH-550 | 1.28 mmol g−1 at 350 °C, 1 bar | 34 | |
| Nano-sized 20 wt% K2CO3 impregnated Mg3Al-LDH-400 | 1.21 mmol g−1 at 200 °C, 1 bar | 35 | |
| Ca1.89Mg0.89Al1.0-LDHs-400 | 1.66 mmol g−1 at 400 °C, 1 bar | In this study | |
| Mesoporous silica | MCM-41/CPA | 1.5 mmol g−1 at 25 °C, 1 bar | 36 |
| MCM-48/CPA | 1.1 mmol g−1 at 25 °C, 1 bar | 36 | |
| MCM-41-NH2 | 1.15 mmol g−1 at 30 °C, 2.1 bar | 37 | |
| TRI-PE-MCM-41 | 2.21 mmol g−1 at 55 °C, 1 bar | 38 | |
| MCM-41 | 1.25 mmol g−1 at 75 °C, 1 bar | In this study |
4. Conclusions
In this work, the conversion of blast furnace slag into two different kinds of promising sorbents of CO2 has been presented. The calcium, magnesium, aluminium in BFS were selectively extracted by hydrochloric acid and used for preparation of layered double hydroxides (LDHs). The leaching residue which contained a high amount of silica was utilised for MCM-41 material synthesis. Several operating variables including initial hydrochloric acid concentration, leaching temperature, solid-to-liquid ratio, and leaching time were optimised. It was found that higher hydrochloric acid concentration, higher leaching temperature, and longer leaching time could promote silica gel formation; this provided a great advantage for the selective leaching out of calcium, magnesium, aluminium with high efficiency. Characterisation of the as-synthesized CaMgAl-LDHs and MCM-41 sorbents was performed to determine chemical composition, morphology and textural properties. The structural formula of CaMgAl-LDHs was determined as Ca1.89Mg0.89Al1.0(OH)7.5Cl0.83(CO3)0.11·yH2O, with Cl− as the predominate intercalated anion. Thermal calcination at 400 °C enhanced the specific surface area, thus leading to improved CO2 uptake and a maximum CO2 adsorption capacity of 1.58 mmol g−1 at 400 °C. The MCM-41 sorbents derived from BFS exhibited a high surface area of 1270 m2 g−1 and showed comparable CO2 adsorption capacity with a value of 1.24 mmol g−1 at 75 °C. In the CO2 adsorption/desorption cycles, both sorbents showed good reusability and cyclic performance. With all these results, the CaMgAl-LDHs and MCM-41 sorbents derived from blast furnace slag are promising sorbents for practical applications in the field of CO2 capture.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The National Natural Science Foundation of China (No. 51574169, No. 51604178, No. 51704202, No. 51604202, No. 51774209), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 17KJB450002), are duly acknowledged for their financial support. The authors also are grateful for the analysis support from Soochow University.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08458k
References
- Lee S. Kim J. W. Chae S. Bang J. H. Lee S. W. J. CO2 Util. 2016;16:336–345. doi: 10.1016/j.jcou.2016.09.003. [DOI] [Google Scholar]
- Teir S. Eloneva S. Fogelholm C. J. Zevenhoven R. Energy. 2007;32:528–539. doi: 10.1016/j.energy.2006.06.023. [DOI] [Google Scholar]
- Eloneva S. Teir S. Salminen J. Fogelholm C. J. Zevenhoven R. Energy. 2008;33:1461–1467. doi: 10.1016/j.energy.2008.05.003. [DOI] [Google Scholar]
- Eloneva S. Teir S. Salminen J. Fogelholm C. J. Zevenhoven R. Ind. Eng. Chem. Res. 2008;47:7104–7111. doi: 10.1021/ie8004034. [DOI] [Google Scholar]
- Eloneva S., Teir S., Savolahti J., Fogelholm C. and Zevenhoven R., Proceeding ECOS 2007, Padova, Italy, 2007, vol. II, pp. 1389–1396 [Google Scholar]
- Hall C. Large D. J. Adderley B. West H. M. Miner. Eng. 2014;65:156–162. doi: 10.1016/j.mineng.2014.06.002. [DOI] [Google Scholar]
- jun Xu A. ning Zhang H. Yang Y. Cui J. feng He D. yuan Tian N. J. Iron Steel Res. Int. 2012;19:34–38. doi: 10.1016/S1006-706X(12)60084-3. [DOI] [Google Scholar]
- Bang J. H. Lee S. W. Jeon C. Park S. Song K. Jo W. J. Chae S. Energies. 2016;9(12):996. doi: 10.3390/en9120996. [DOI] [Google Scholar]
- Bao W. Li H. Yi Z. Ind. Eng. Chem. Res. 2010;49:2055–2063. doi: 10.1021/ie801850s. [DOI] [Google Scholar]
- Wang J. Huang L. Yang R. Zhang Z. Wu J. Gao Y. Wang Q. O'Hare D. Zhong Z. Energy Environ. Sci. 2014;7:3478–3518. doi: 10.1039/C4EE01647E. [DOI] [Google Scholar]
- Wang S. H. Bin Wang Y. Dai Y. M. Jehng J. M. Appl. Catal., A. 2012;439–440:135–141. doi: 10.1016/j.apcata.2012.06.049. [DOI] [Google Scholar]
- Tian S. Jiang J. Yan F. Li K. Chen X. Manovic V. Green Chem. 2016;18:4022–4031. doi: 10.1039/C6GC00400H. [DOI] [Google Scholar]
- Kuwahara Y. Yamashita H. ISIJ Int. 2015;55:1531–1537. doi: 10.2355/isijinternational.55.1531. [DOI] [Google Scholar]
- Song K. Park S. Kim W. Jeon C. Ahn J.-W. Metals. 2017;7:199. doi: 10.3390/met7060199. [DOI] [Google Scholar]
- Wang Q. Ohare D. Chem. Rev. 2012;112:4124–4155. doi: 10.1021/cr200434v. [DOI] [PubMed] [Google Scholar]
- Tathod A. P. Gazit O. M. Cryst. Growth Des. 2016;16:6709–6713. doi: 10.1021/acs.cgd.6b01272. [DOI] [Google Scholar]
- Vieira A. C. Moreira R. L. Dias A. J. Phys. Chem. C. 2009;113:13358–13368. doi: 10.1021/jp902566r. [DOI] [Google Scholar]
- Flodström K. Alfredsson V. Microporous Mesoporous Mater. 2003;59:167–176. doi: 10.1016/S1387-1811(03)00308-1. [DOI] [Google Scholar]
- Selvam P. Bhatia S. K. Sonwane C. G. Ind. Eng. Chem. Res. 2001;40:3237–3261. doi: 10.1021/ie0010666. [DOI] [Google Scholar]
- Misran H. Singh R. Begum S. Yarmo M. A. J. Mater. Process. Technol. 2007;186:8–13. doi: 10.1016/j.jmatprotec.2006.10.032. [DOI] [Google Scholar]
- Yu H. Xue X. Huang D. Mater. Res. Bull. 2009;44:2112–2115. doi: 10.1016/j.materresbull.2009.07.003. [DOI] [Google Scholar]
- Zhou Y. Y. xuan Li X. xing Chen Z. Powder Technol. 2012;226:239–245. doi: 10.1016/j.powtec.2012.04.054. [DOI] [Google Scholar]
- Kumar P. Mal N. Oumi Y. Yamana K. Sano T. J. Mater. Chem. 2001;11:3285–3290. doi: 10.1039/B104810B. [DOI] [Google Scholar]
- Zhao D. Huo Q. Feng J. Chmelka B. F. Stucky G. D. J. Am. Chem. Soc. 1998;120:6024–6036. doi: 10.1021/ja974025i. [DOI] [Google Scholar]
- Ram Reddy M. K. Xu Z. P. Lu G. Q. Da Costa J. C. D. Ind. Eng. Chem. Res. 2006;45:7504–7509. doi: 10.1021/ie060757k. [DOI] [Google Scholar]
- Frost R. L. Spratt H. J. Palmer S. J. Spectrochim. Acta, Part A. 2009;72:984–988. doi: 10.1016/j.saa.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Galindo R. López-Delgado A. Padilla I. Yates M. Appl. Clay Sci. 2014;95:41–49. doi: 10.1016/j.clay.2014.03.022. [DOI] [Google Scholar]
- Boumaza A. Favaro L. Lédion J. Sattonnay G. Brubach J. B. Berthet P. Huntz A. M. Roy P. Tétot R. J. Solid State Chem. 2009;182:1171–1176. doi: 10.1016/j.jssc.2009.02.006. [DOI] [Google Scholar]
- Zhou J. Xu Z. P. Qiao S. Liu Q. Xu Y. Qian G. J. Hazard. Mater. 2011;189:586–594. doi: 10.1016/j.jhazmat.2011.02.078. [DOI] [PubMed] [Google Scholar]
- Wang W. Li J. Wei X. Ding J. Feng H. Yan J. Yang J. Appl. Energy. 2015;142:221–228. doi: 10.1016/j.apenergy.2014.12.072. [DOI] [Google Scholar]
- Bacsik Z. Ahlsten N. Ziadi A. Zhao G. Garcia-Bennett A. E. Martín-Matute B. Hedin N. Langmuir. 2011;27:11118–11128. doi: 10.1021/la202033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witoon T. Chareonpanich M. Songklanakarin J. Sci. Technol. 2012;34:403–407. [Google Scholar]
- Wang Q. Tay H. H. Zhong Z. Luo J. Borgna A. Energy Environ. Sci. 2012;5:7526–7530. doi: 10.1039/C2EE21409A. [DOI] [Google Scholar]
- Xiao P. W. Jun J. Y. Cheng J. Zheng P. H. Zhi P. X. Environ. Sci. Technol. 2008;42:614–618. doi: 10.1021/es072085a. [DOI] [PubMed] [Google Scholar]
- Wang Q. Gao Y. Luo J. Zhong Z. Borgna A. Guo Z. O'Hare D. RSC Adv. 2013;3:3414–3420. doi: 10.1039/C2RA22607C. [DOI] [Google Scholar]
- Bhagiyalakshmi M. Yun L. J. Anuradha R. Jang H. T. J. Porous Mater. 2010;17:475–484. doi: 10.1007/s10934-009-9310-7. [DOI] [PubMed] [Google Scholar]
- Mello M. R. Phanon D. Silveira G. Q. Llewellyn P. L. Ronconi C. M. Microporous Mesoporous Mater. 2011;143:174–179. doi: 10.1016/j.micromeso.2011.02.022. [DOI] [Google Scholar]
- Belmabkhout Y. Serna-Guerrero R. Sayari A. Adsorption. 2011;17:395–401. doi: 10.1007/s10450-011-9348-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.