

Abstract
Autologous hematopoietic stem cell transplantation using genome-edited cells can become a definitive therapy for hematological and non-hematological disorders with neurological involvement. Proof-of-concept studies using human genome-edited hematopoietic stem cells have been hindered by the low efficiency of engraftment of the edited cells in the bone marrow and their modest efficacy in the CNS. To address these challenges, we tested a myeloablative conditioning regimen based on Busulfan in an immunocompromised model of mucopolysaccharidosis type 1. Compared with sub-lethal irradiation, Busulfan conditioning enhanced the engraftment of edited CD34+ cells in the bone marrow, as well the long-term homing and survival of bone-marrow-derived cells in viscera, and in the CNS, resulting in higher transgene expression and biochemical correction in these organs. Edited cell selection using a clinically compatible marker resulted in a population with low engraftment potential. We conclude that conditioning can impact the engraftment of edited hematopoietic stem cells. Furthermore, Busulfan-conditioned recipients have a higher expression of therapeutic proteins in target organs, particularly in the CNS, constituting a better conditioning approach for non-hematological diseases with neurological involvement.
Keywords: genome editing, CRISPR-Cas9, hematopoietic stem cell, Busulfan, total body irradiation, engraftment, conditioning, lysosomal storage disease, mucopolysaccharidosis type I, iduronidase, tNGFR
Graphical abstract
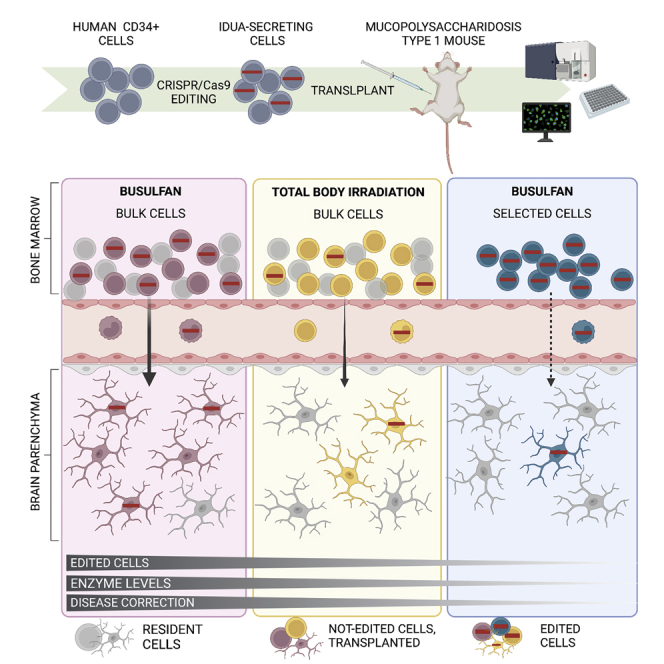
Busulfan conditioning enhances engraftment of human genome-edited CD34+ cells in the bone marrow and long-term homing of bone-marrow-derived cells in viscera and the CNS, resulting in higher transgene expression and phenotypic correction in these organs. Cell selection of edited cells results in a population with low engraftment potential.
Introduction
Autologous hematopoietic stem cell transplantation using genetically modified cells holds immense promise for the one-off treatment of hematological and non-hematological disorders. Hence, genome-editing tools have been extensively applied to hematopoietic stem and progenitor cells (HSPCs).1 Among these tools, CRISPR-Cas9 has been widely adopted as it uses an RNA molecule (gRNA) to target the nuclease (Cas9) to the desired genomic locus. Cas9 binding and the subsequent double-stranded break (DSB) generation recruit two main DNA-damage response pathways: non-homologous end joining (NHEJ), which can create insertions or deletions (indels), and homology-directed repair (HDR), which can generate specific modifications when an exogenous template containing the desired changes is also included.2, 3, 4 NHEJ is often more efficient than HDR in HSPCs and, for this reason, has advanced faster to non-human primate models5,6 and into the clinic for diseases like sickle cell disease and beta thalassemia.7 Several groups, including ours, have achieved efficient HDR in human HSPCs for several disease models of hematological diseases such as sickle cell disease,8, 9, 10, 11 beta thalassemia,12 X-linked severe combined immunodeficiency,13,14 X-linked chronic granulomatous disease,15,16 Fanconi anemia,17 and Wiskott-Aldrich syndrome,18,19 as well as non-hematological diseases such as the lysosomal storage diseases (LSDs), mucopolysaccharidosis type I (MPSI),20 and Gaucher disease.21 Despite significant progress, approaches based on HDR have been hindered by the decreased engraftment potential observed in HSPCs that have undergone HDR. This is in addition to the geno- and cytotoxicity associated with DSB-triggered DNA damage responses that can result in apoptosis, arrest, or differentiation.22 Across multiple loci, studies reported a 3- to 9-fold reduction in the number of modified cells that engrafted compared with the transplanted HSPC population.11,20,21,23, 24, 25
Conditioning refers to a group of treatments used to suppress the immune system and clear out stem cell niches prior to hematopoietic stem cell transplantation (HSCT). Treatment regimens include chemotherapy such as the alkylating agent busulfan (BU; 1,4-butanediol dimethanesulfonate), radiation, and monoclonal antibodies, and can be administered at different intensities based on the clinical indication.26, 27, 28 BU induces cell senescence29,30 and apoptosis31 in the host’s myeloid compartment, thereby conferring an engraftment and growth advantage for transplanted cells. In mouse models, including immunocompromised NOD scid gamma (NSG) mice, bone marrow engraftment of human CD34+ HSPCs is equivalent using both BU and irradiation,29,32,33 and donor chimerism is often dose dependent for both conditioning regimens until complete myeloablation is achieved.29,32,34 Despite the importance of the conditioning regimen in HSCT, a comparison of the effect of conditioning on the engraftment of human genome-edited HPSCs has not been performed.
For multi-systemic and neurological diseases such as MPSI, Gaucher, and potentially other LSDs,28 how HSCT addresses disease pathology in non-hematopoietic tissues is not entirely understood. The effect is mediated, in part, by bone-marrow-derived cells that migrate to become tissue-resident myeloid cells where they can cross-correct other cells.35 Several studies have shown that this migration is determined in large part by the conditioning regimen.36,37 In the clinic, this is traditionally achieved with high-intensity BU myeloablation, though high-dose radiation can be used in the setting of malignancy. Specifically, conditioning with BU has been shown to improve the homing to the central nervous system (CNS) by lentiviral-vector-modified mouse and human HSPCs in animal models when compared with radiation,36,37 and it is currently used in clinical settings.38,39 How human genome-edited HPSCs and their progeny engraft in the bone marrow and migrate into the CNS following BU-based myeloablation remains poorly understood.
Herein, we examined the effect of BU conditioning on the engraftment of human genome-edited CD34+ cells and their therapeutic efficacy in a mouse model of a multi-systemic disease. We used a previously developed HDR-based platform to target expression cassettes of the lysosomal enzyme alpha-L-iduronidase (IDUA) and a human cell-engraftable mouse model of MPSI, a lysosomal storage disorder resulting from IDUA deficiency.20 Using a targeted gene-addition approach, the IDUA cassettes are inserted into CCR5, a well-established safe harbor locus as germ deletions in this gene are common (up to 10% in the Northern European population), have no overt developmental phenotype, and provide protection against HIV.40,41 We examined the edited CD34+ cells’ ability to engraft long term, express the transgene, and correct biochemical abnormalities in visceral organs and the CNS. Furthermore, to establish the optimal cell product for transplantation, we compared engraftment and biochemical correction with either non-selected cell preparations (bulk samples with modified and unmodified cells) or purified CD34+ cells expressing a clinically compatible selection marker.
Results
Improved engraftment of genome-edited human CD34+ HSPCs in BU-conditioned mice
To examine the effect of the pre-transplant conditioning regimen, we used a donor template for repair previously optimized in pre-clinical studies for the expression of the IDUA enzyme for the treatment of the lysosomal disorder MPSI. We edited CD34+ cells from healthy cord blood 48 h after CD34+ isolation using the single guide RNA (sgRNA)/Cas9 ribonucleoprotein (RNP) and adeno-associated virus type 6 (AAV6) system and an sgRNA targeting exon 3 of the CCR5 locus, which has been extensively characterized for having high on-target and low off-target activity.20 The template for HDR contained the IDUA cDNA sequence under the control of the phosphoglycerate kinase (PGK) promoter and the bovine growth hormone polyadenylation (BGH-PolyA) signal flanked by 500 bp homology arms (Figure 1A).
Figure 1.

Improved engraftment of genome-edited human HSPCs in TBI- and busulfan-conditioned mice
(A) Schematic depicting the AAV6 vector genome used for HDR, the experimental design, and timeline. (B) Human cell engraftment measured by human CD45 and HLA double-positive cells and presented as the percentage of total CD45 cells in the bone marrow, peripheral blood, and spleen of NSG-MPSI mice 20 weeks post-transplantation. Each dot/square represents a mouse. Three different human HSPCs donors were transplanted into n = 14 mice for total body irradiation (TBI) and n = 12 for busulfan (BU); two-tailed Mann-Whitney test. (C) Percentage of indels in human cells that did not undergo HDR engrafted in the bone marrow of primary recipients. Dashed line represents the average of indels found in the transplanted cells. (D) Fraction of edited alleles in human cells engrafted in the bone marrow of primary recipient mice. Dashed line represents the fraction of edited alleles in the transplanted HSPC population (input) as measured by ddPCR; two-tailed unpaired t test. (E) Fraction of edited alleles in the bone marrow of secondary recipient mice, after 36 weeks in vivo (20 weeks in primary and 16 weeks in secondary recipient mice), measured by ddPCR. Each dot represents a mouse (TBI, n = 5; BU, n = 4); two-tailed unpaired t test. (F and G) Percentage of edited alleles in the input HSPC population and after transplant in primary and secondary recipient mice conditioned with BU or TBI. Each dot represents a mouse, and each shade of pink or yellow represents a different human cell donor. Lines link recipients of cells with their respective donors. Input: independent donor HSPCs before transplantation (n = 3). Primary: human cells in the bone marrow after 20 weeks in vivo (TBI, n = 13; BU, n = 12). Secondary: edited cells in the bone marrow of serially transplanted recipient mice, 36 weeks total in vivo (TBI, n = 5; BU, n = 4). All data are shown as median. (F) Ordinary one-way ANOVA with post-hoc Tukey. (G) Kruskal-Wallis with Dunn’s post-hoc test. ns, not significant.
Two days after editing, CD34+ cells were transplanted in bulk into 6- to 8-week-old mice via intrafemoral injection. For each transplant experiment and human cell donor, an equal number of cells were transplanted per mice, but cell doses ranged between experiments depending on the degree of cell expansion during the culturing period (0.8–2.7 × 106 cells/mouse). The recipient mice are in the NSG background and carry a nonsense mutation in the Idua gene (NSG-MPSI), constituting a human cell engraftable model of the disease.20 Before transplantation, mice underwent either sublethal total body irradiation (TBI), the most common protocol used in engraftment studies using human CD34+ HSPCs, or complete myeloablation using BU. Myeloablation with BU was completed 24 h before transplant and consisted of four daily intraperitoneal injections of 17 mg/kg, followed by a single support dose of genotype-matched bone marrow (e.g., IDUA-deficient cells for homozygous NSG-MSPI mice) injected intravenously 4 days post-transplantation (2–5 × 106 cells/mouse).42 TBI was administered 2 h before transplant at a dose of 2.1 Gy (Figure 1A).
Human cell engraftment, measured by double-positive human CD45+ and human leukocyte antigen-ABC (HLA-ABC+) cells (Figure S1), was statistically similar for both regimens in the bone marrow, peripheral blood, and spleen 20 weeks post-transplantation (Figure 1B). Median (min, max) human chimerism in bone marrow was BU 73% (25, 91) and TBI 45% (2.1, 97). Although the frequency of human cells in the bone marrow did not differ between BU and TBI, the proportion of cells with targeted integration was significantly higher in BU-conditioned mice, while indel frequency at CCR5 in cells that did not undergo HDR did not change (Figures 1C and 1D). Twenty weeks post-transplantation, BU-conditioned mice had 20% ± 13% of edited alleles in the bone marrow as measured by droplet digital PCR (ddPCR), compared with 5.7% ± 4.1% in the TBI group (p = 0.0002) (Figure 1D). Compared with an edited allele fraction in the input HSPC population before transplantation of 30%, the drop in the fraction of edited alleles was 5.3-fold for TBI and 1.5-fold for BU. The TBI versus BU effect did not appear to be transgene or NSG-MPSI model specific. A similar construct targeting CCR5 but encoding for a different lysosomal enzyme (glucocerebrosidase) used to target CD34+ cells transplanted in NSG mice showed a similar effect (8- to 1.8-fold; Figure S2).
The engraftment of edited CD34+ cells with serial transplantation did not change with either regimen (Figure 1E), consistent with the idea that the initially manipulated cell product differs from the HSPC population engrafted in vivo. Median percentages of edited alleles in secondary transplants were comparable to those observed in the primary engraftment studies for both groups (BU 22% and TBI 4.7%). Notably, the BU group had mice where the engrafted cells had a higher edited allele fraction in primary and secondary transplantation than the initially transplanted cells (Figure S1F), a phenomenon observed only in secondary transplants in the TBI group (Figure 1G). The in vivo multi-lineage differentiation of the edited CD34+ cells was comparable in both conditioning regimens in primary and secondary transplants as measured by the frequency of edited myeloid (CD33+), B cells (CD19+), and T cells (CD3+) in hematopoietic organs (Figure S3). These data confirm that edited HSCPs engraft long term in BU- and TBI-conditioned mice and suggest that myeloablation with BU results in a more conducive niche for the engraftment of human CD34+ HSPCs that have undergone HDR via the CRISPR-AAV6 platform.
The bone marrow niche is less pro-inflammatory in BU-conditioned mice
Previous studies in wild-type immunocompetent mice have reported that TBI triggers a more pro-inflammatory niche compared with BU.43,44 Analysis of 48 cytokines in the plasma of NSG mice at 2 and 24 h after TBI or BU conditioning showed increased levels of pro-inflammatory cytokines (e.g., MCP-1, tumor necrosis factor alpha [TNFα], interleukin 6 [IL-6], IL1-β) only in mice treated with TBI (Figures 2A and S4). Other significant changes in cytokine levels were found at these early time points in both groups but were more pronounced with TBI (Figures 2A and S4). Analyses of cytokine production in the bone marrow 24 h post-conditioning showed more prominent changes with TBI including significant decreases in eight cytokines. This marked reduction in cytokine production could reflect greater depletion of cytokine-producing cells in TBI-conditioned mice (Figures 2B and S5). To examine the cellular changes in the bone marrow associated with each regimen, we measured erythrocytes (TER119) and monocyte/macrophages (CD11b, Ly6C) using flow cytometry (NSG mice lack lymphocytes; Figure S6). Decreases in the Ly6C staining by population and median fluorescence intensity (MFI), as well as decreases in CD11b MFI, were more significant in TBI (Figures 2C and 2D). While both conditions resulted in lower expression of the erythrocyte marker TER119, TBI resulted in an apparent increase in this population (Figures 2E and 2F). These data suggest that at the time of CD34+ cell transplantation, the BU-conditioned bone marrow is less pro-inflammatory and likely more amenable to cell engraftment compared with TBI.
Figure 2.

The bone-marrow niche is less pro-inflammatory in BU-conditioned NSG mice
(A) Cytokine secretion measured using the mouse 48-plex Luminex assay in the plasma of NSG mice at 2 and 24 h (H) following TBI (2.1 Gy, n = 5) or BU (66 mg/kg, n = 5) conditioning. The 2 and 24 h time points correspond to the time of transplantation after conditioning, respectively. (B) Cytokine secretion in bone marrow 24 h (24H) post-conditioning. (A and B) Fold changes were calculated relative to untreated NSG control mice (n = 4). Statistically significant changes are depicted in bold. No statistically significant cytokine changes were found other than those depicted. (C–F) Frequency and median fluorescence intensity (MFI) monocyte and macrophage progenitors (CD11b+, Ly6C+) and erythrocytes (TER119+) measured in the bone marrow by flow cytometry. (A–F) Statistical analysis: one-way ANOVA with Tukey post-hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Improved enzyme expression and biochemical correction in visceral tissues of MPSI mice
Humans and mice affected with MPSI present with hepatosplenomegaly due to the cumulative storage of glycosaminoglycans (GAGs), IDUA’s substrates, inside cells. To examine the ability of engrafted cells to migrate to visceral tissues and secrete IDUA enzyme, we analyzed IDUA activity and GAG storage in NSG-MPSI mice. We measured IDUA enzyme activity in serum, liver, and spleen and expressed the results as the percentage of littermate heterozygous mice (Figures 3A–3C). Compared with TBI, BU-conditioned mice exhibited higher IDUA enzyme activity in liver (771% versus 50%; p = 0.0409; Figure 3B) and spleen (4,460% versus 167%; p = 0.0037; Figure 3C). A consequence of increased IDUA with BU conditioning was that metabolic correction was also improved. We first used the modified dimethylmethylene blue (DMB) assay45 to assess total GAG levels in urine samples, which report on visceral GAG storage including in the kidney. Mice conditioned with TBI showed a significant reduction in GAG excretion (p < 0.0001) but still increased compared with normal mice (p = 0.0073), while BU-conditioned mice had GAG levels in the normal ranges (p = 0.46; Figure 3D). Because this assay provided a non-specific measurement of total GAGs, we also measured urinary GAG excretion in BU-conditioned mice using liquid chromatography tandem mass spectrometry (LC-MS/MS), which enables characterization of different sulfated GAG species.46 Compared with sham-treated mice, mice transplanted with IDUA-expressing CD34+ cells showed significant reduction in dermatan sulfate (Di-0S; p = 0.0004), HS-NS, and HS-0S disaccharides of heparan sulfate (p = 0.0007 and p < 0.0001; Figure 3E). In tissues, storage of GAG was completely normalized in the liver (medians 1.6, 6.2, and 1.6 for normal, MPSI, and transplanted mice, respectively; p < 0.0001 versus sham-treated mice; Figure 3F). Similarly, dermatan and heparan levels were normalized in the spleen of BU-conditioned mice (Figure 3G). Mono- and di-sulfated keratan sulfate showed no differences between groups in urine or spleen, which is expected since keratan sulfate is not a substrate of IDUA (Figure S7).
Figure 3.

BU conditioning enhances biochemical correction of genome-edited HSPCs in visceral organs of NSG-MPSI mice
(A–G) White: normal; green: sham-transplanted MPSI; pink: BU-conditioned and transplanted MPSI; yellow: TBI-conditioned and transplanted MPSI. (A–C) Enzyme activity of IDUA in (A) serum, (B) liver, and (C) spleen of BU- (pink, n = 8) and TBI- (yellow, n = 5) conditioned and transplanted mice, represented as percentage of the average of normal heterozygous mice. (D) Urinary GAG excretion in 28-week-old mice measured by modified DMB assay. Results are presented as μg of GAGs normalized by mg of creatinine. (E) Disaccharides of dermatan (Di-0S) and heparan (HS-NS, HS-0S) sulfate measured by LC-MS/MS. Results are presented as ng of GAGs normalized by mg of creatinine. Normal, n = 6; MPSI, n = 7; BU, n = 8. (F) Storage of GAGs in the liver measured by DMB assay. Results are presented as μg of GAGs normalized by mg of protein. Normal, n = 6; MPSI, n = 5; BU, n = 9. (G) GAG disaccharides in the spleen. Results are presented as ng of GAGs normalized by mg of protein. Normal, n = 7; MPSI, n = 3; BU, n = 9. For all graphs, each symbol represents a mouse, and all data are represented as mean ± SD. Statistical analysis: (A) Mann-Whitney test; (B and C) two-tailed unpaired t test; (D–G) ordinary one-way ANOVA with post-hoc Tukey test. ns, not significant.
BU conditioning enhances therapeutic efficacy in the CNS
Given that neurodegeneration is a key feature of severe MPSI, we also compared the effect of BU conditioning versus TBI in the CNS. Twenty weeks post-transplantation, IDUA activity was significantly higher in the BU group compared with TBI, despite considerable variability in the levels of enzymatic activity in each mouse (medians 17% versus 6.8%; p = 0.0451; Figure 4A). Consistent with enhanced enzymatic correction, BU-conditioned mice showed significant reductions of GAG accumulation in the brain, whereas TBI-conditioned mice did not (p = 0.0118; Figures 4B and S8A). The statistically significant decrease in GAG storage in BU-conditioned mice was also corroborated with toluidine blue staining in Purkinje cells (p = 0.0015; Figures 4C and 4D). These data suggest that conditioning with BU is superior to sub-lethal TBI at targeting the CNS.
Figure 4.

BU conditioning enhances therapeutic efficacy of edited HSPCs in the CNS
(A) Enzyme activity of IDUA in the brain of BU- (n = 8) and TBI- (n = 5) treated mice, represented as the percentage of normal heterozygous mice (n = 5). MPSI mice have undetectable levels of enzyme, represented in this log scale as 0.01%. (B) Storage of GAGs in the brain measured by the DMB assay. Results are presented as μg of GAGs normalized by mg of protein. Normal, n = 6; MPS I, n = 5; BU, n = 9. (C) Percentage of Purkinje cells with visible vacuoles, indicative of substrate storage within the cell. Ten histological sections were analyzed per mouse. Normal, n = 2; MPS I, n = 3 and BU, n = 4. (D) Representative histological sections from cerebellum, stained with toluidine blue for visualization of storage vacuoles. (E) Representative flow cytometry plots from microglia-enriched populations from mouse brains. (F) Percentage of human CD45+ cells among live cells (propidium iodide negative) and among total CD45 cells (mouse and human CD45+) in the microglia isolated from BU-treated mice. (G) Percentage of human DNA measured by ddPCR in microglia cells from BU-treated mice, normalized by total mouse and human DNA. BU, n = 5, BU-2ary, n = 4. (H) Correlation between percentage of human DNA in microglia cells and IDUA activity measured in brains of BU-treated mice. n = 5; p = 0.0070; R2 = 0.9362. (I) Representative brain sections of BU-conditioned HSPC transplanted mice, stained for a human-specific Ku80 marker and the microglial marker IsolectinB. Scale bar is 50 μm. (J) Percentage of human cells (given by Ku80 positivity) normalized by total nuclei (given by DAPI+ cells) in the meninges and in the parenchyma of BU- and TBI-conditioned mice. (K) Percentage of human microglia (Ku80+IB4+) normalized by total human cells (Ku80+) in the meninges and parenchyma. (A–C and F–H) Medians shown; each dot represents a mouse. (J and K) Medians; each symbol represents a section. (A, J, and K) Two-tailed Mann-Whitney test. (B and C) Ordinary one-way ANOVA with post-hoc Tukey test.
Several groups have demonstrated the presence of bone-marrow-derived cells in the brains of mice and non-human primates following syngeneic HSCT after BU conditioning in normal37,47 and disease models.36,48 To examine this, we isolated microglia from well-perfused mouse brains using density gradients and analyzed them with flow cytometry. Mouse microglia cells (mCD45+/mCD11b+) in affected MPSI had higher light-scattering behavior consistent with larger cells with greater internal complexity (granularity), likely reflecting storage and morphological changes due to activation. This observation provided an additional quantitative tool to phenotype microglia cells in this disease model. Specifically, we found that microglia from BU-conditioned and transplanted MPSI mice had lower side scatter (SSC) compared with sham-treated MPSI mice, though the light-scattering behavior did not normalize (Figures S8B and S8C). To specifically detect human cells in the CNS, we also measured the fraction of human CD45+ cells in these microglia-enriched preparations and found a mean frequency of 1.5% hCD45+ in all live cells or 3.9% in the human and mouse CD45+ subset (Figures 4E, 4F, and S9). When examined for hCD11b and HLA-DR expression, the hCD45+ population had an almost equal distribution of single- and double-positive cells reflecting abnormal marker expression by these human cells in this xenograft model (Figure S9). As an orthogonal approach, we used ddPCR to measure the ratio of human DNA to mouse DNA in the microglia fraction of transplanted mice using probes targeting the human and mouse CCRL2/Ccrl2 genes. In the microglia population of transplanted mice, the median value of human DNA was 3.5% (Figure 4G). Similarly, a median of 2.6% human to mouse DNA fraction was observed in secondary recipient mice (Figure 4G). The levels of IDUA activity in the brain showed a strong positive correlation with the percentage of human DNA in the mixed microglia population (p = 0.007; R2 value of 0.9362; Figure 4H), supporting that increased enzyme activity observed in the brain is the result of human cells in the brain and not from blood contamination.
To provide independent evidence for the presence of human cells in the mouse CNS, we tested a panel of human-specific antibodies in immunohistochemical studies of transplanted NSG-MPSI brains. Among these, the anti-human Ku80 antibody showed the best signal to noise and highest specificity when tested in positive control sections of mouse brains injected with human CD34+ cells edited at CCR5 locus to express yellow fluorescent protein (YFP; Figure S10). We found human Ku80+ cells in multiple regions of the brain but primarily enriched in the leptomeninges, making up as high as 4.9% of all nuclei, and approximately half the cells were also positive for Isolectin B4 (Figures 4I–4K). This was markedly different from TBI-conditioned brains where minimal Ku80+ cells were identified (only 0.57% in the meninges; Figure 4J). Notably, despite multiple attempts, several anti-IDUA antibodies failed to detect any signal above background in immunohistochemistry, including in positive control sections with edited human cells injected intracerebroventricularly.
Together, these data suggest that conditioning with BU before transplantation with edited CD34+ cells is more effective than TBI at achieving metabolic correction in the CNS. The observed homing of BM-derived cells to the CNS could be the result of more efficient engraftment in this organ or increased cell penetration due to damage to the blood-brain barrier (BBB). Previous studies have shown that BU even at myeloablative doses does not damage the BBB while irradiation at high doses (10 Gy) does.43 However, given that these studies used immunocompetent mice and different doses of TBI and BU, we examined this question in NSG mice and with our conditioning regimens. To measure BBB permeability, we looked for immunoglobulin G (IgG) leakage into the CNS parenchyma using an anti-IgG stain. Neither TBI nor BU resulted in detectable IgG staining in the parenchyma in NSG when measured 7 and 14 days post-conditioning (Figure S11). These data suggest that BBB disruption is not a major contributor to the observed increased homing of cells from the bone marrow.
Homing to the CNS by human HSPC-derived cells in SGM3 mice
To better determine the number, marker, and transgene expression of CNS-engrafted human genome-edited cells in the brain following BU-based myeloablation, we sought to increase the sensitivity of our system by using a reporter gene under the control of a strong ubiquitous promoter and an immunodeficient, humanized mouse model. The NSG-SGM3 transgenic mouse expresses the human cytokines IL3, SCF, and CSF2 and has been validated to improve human cell engraftment and myeloid differentiation in the context of HSPC xenotransplants.49,50 The insertion of the YFP-expression cassette at the CCR5 locus as well as the conditioning and experimental timeline were the same as in prior experiments, except that the mice were analyzed at 15 weeks instead of 20 due to morbidity. We achieved efficient isolation of mouse CD45+ CD11b+ microglia with clear detection of HSPC-derived human cells (Figure 5A). Mouse microglia were approximately 80% of the total CD45+ cell fraction isolated from the brain (Figure 5B). Human cells engrafted in the brain expressed the CD45, CD11b, and HLA-DR markers (Figures 5C and 5D). Notably, the median engraftment of human CD45+ cells over the total CD45+ fraction was 16% (Figure 5C). Sensitive ddPCR analyses using the DNA extracted from the total cell population isolated from the brain showed a 4% median human engraftment (Figure 5E). Genome-edited YFP+ cells were detected in the brain by flow cytometry (Figures 5F and 5G), ddPCR (Figure 4H), and histological analyses (Figure 5I). The YFP+ cells co-expressed all tested human markers (CD45, CD11b, and HLA-DR markers) and represented 1%–2% of the total (mouse plus human) CD45+ fraction or 13%–19% of the human cells, consistent with the fraction of edited alleles detected by ddPCR (Figures 5F and 5G). These results confirm that the progeny of human genome-edited HSPCs can engraft and maintain long-term transgene expression in the brain following BU conditioning.
Figure 5.

Engraftment of human genome-edited HSPCs in the CNS of NSG-SGM3 mice
(A) Representative flow cytometry plots showing mouse (mCD45+ CD11b+), human (hCD45+ CD11b+), and edited (YFP+) human microglia-like cells isolated from brain tissue of transplanted NSG-SGM3 mice. (B) Purity of murine microglia (mCD45+ CD11b+) among total myeloid CD45+ cells. (C and D) Characterization of human cells among (C) total myeloid CD45+ cells and (D) human CD45+ cell populations. (E) Proportion of human alleles measured by ddPCR in the microglia preparations, normalized by mouse genomic DNA. (F and G) Characterization of edited YFP+ human HSPCs among (F) total CD45+ and (G) human CD45+ cell populations. (H) Fraction of edited human CCR5 alleles in cells isolated from microglia preparations. Dashed line refers to the fraction of edited alleles in the transplanted HSPC population (input). (I) Representative brain sections of transplanted mice showing YFP expression (natural fluorescence) from edited HSPCs and staining for nuclei (DAPI) and for the specific human marker Ku80. Scale bar is 100 μm. Medians shown; each dot represents a mouse, n = 4.
Human cell chimerism in hematopoietic organs in the NSG-SGM3 mouse was robust but less so than in the NSG model, likely due to constant CSF2 transgene-mediated mobilization from the bone marrow (Figure S12). As expected, constitutive expression of human IL3, SCF, and CSF2 resulted in robust generation of edited myeloid and T cells in bone marrow, peripheral blood, and spleen, and the editing frequencies persisted with differentiation, confirming their potential to differentiate along these lineages (Figures S12C and S12D).
Cell selection decreases engraftment potential and therapeutic efficacy
To establish a potential transplantation protocol that includes BU as conditioning for genome-edited CD34+ cells, we next tested the best cell product for transplantation. We compared engraftment and biochemical correction between non-selected cell preparations (bulk samples with modified and unmodified cells) to purified cells expressing a clinically compatible selection marker. We designed a new donor template containing the sequence for the truncated nerve growth factor receptor (tNGFR) after the IDUA cDNA and the self-cleaving peptide P2A (Figure 6A). tNGFR is a signaling-impaired version of the endogenous receptor, which is not expected to be antigenic, and is not expressed in hematopoietic cells.51 Furthermore, tNGFR has already been used in clinical studies and has available good manufacturing practice (GMP)-compatible reagents, facilitating its translation to the clinic. Human CD34+ cells were cultured and edited as in previous experiments using the IDUA-only targeting vector, and the cells were analyzed, purified, and transplanted 4 days post-editing to allow for marker expression. CD34+ cells modified in this manner showed normal rates of expansion and distribution of clonogenic progenitors, confirming that expression of tNGFR did not affect proliferation or multi-lineage potential in vitro (Figures S13A and S13B).
Figure 6.

Selection of modified cells decreases engraftability and therapeutic efficacy of genome-edited HSPCs in NSG-MPS I mice
(A) Schematic depicting experimental design, timeline, and the AAV6 vector used to express truncated nerve growth factor receptor (tNGFR) as a selectable marker downstream of IDUA. (B) Representative flow cytometry plots showing expression of tNGFR in edited HSPCs. (C) Fraction of edited alleles measured by ddPCR in bulk HSPCs edited with the IDUA (n = 3) or IDUA-tNGFR (n = 8) AAV6 donor templates. Each symbol represents a different human cell donor. (D) Engraftment of human cells (CD45+HLA+) in different hematopoietic tissues of mice transplanted with bulk cells edited with IDUA alone (n = 12), selected IDUA-tNGFR+ (n = 10, 10 and 8), and IDUA-tNGFR- (n = 5) cells. Each symbol represents a mouse. (E) Fraction of edited alleles in the bone marrow of transplanted mice (input). Dashed lines represent the median of edited alleles in the transplanted cells. Each symbol represents a mouse. (F) Edited alleles fraction in input cells (n = 6 different human cell donors) and in bone-marrow-engrafted cells (n = 7 mice). Lines link transplant recipient mice with their respective donors. (G) Engraftability of edited cells when transplanted as bulk population (n = 3) or as selected IDUA-tNGFR+ cells (n = 6). Each symbol represents a different HSPC donor. (H) Percentage of long-term HSCs (LT-HSCs), given by CD34+CD90+CD38-CD45RA- in mock and selected cells; each dot represents a donor. (I) Enzyme activity of IDUA in serum, liver, spleen, and brain from NSG-MPS I mice transplanted with IDUA-bulk (n = 8), IDUA-tNGFR+ (n = 7), and IDUA-tNGFR-cells (n = 3). Each symbol represents a mouse. (J) GAG storage measured in the liver, spleen, and brain using the DMB method. Liver: Normal, n = 15; MPS I, n = 5; IDUA-tNGFR+, n = 6; IDUA-tNGFR-, n = 3. Spleen: Normal, n = 8; MPS I, n = 8; IDUA-tNGFR+, n = 8; IDUA-tNGFR-, n = 6. Brain: Normal, n = 15; MPS I, n = 7; IDUA-tNGFR+, n = 7; IDUA-tNGFR-, n = 3. All numerical data is presented as median. (C and E–G) Two-tailed unpaired t test; (D and H–J) (liver) Kruskal-Wallis test with post-hoc Dunn test; (J) (spleen and brain) ordinary one-way ANOVA with post-hoc Tukey test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
We first established a GMP-compatible method for immuno-purification of tNGFR+ cells. Four days after genome editing, CD34+ cells showed cell-surface expression of tNGFR by flow cytometry (Figure 6B). The median percentage of edited alleles in IDUA-tNGFR cells measured by ddPCR was 14.5%, significantly lower than the 31% observed with the IDUA-only construct (p = 0.0034) (Figure 6C). Positive selection of tNGFR+ cells using magnetic beads enriched the tNGFR-expressing cell population from 15.5% to 72% (or 12.5%–33% edited alleles), and a second round of selection did not increase yield (Figures S13C and S13D). The potency of the tNGFR+-selected HPSCs and their in-vitro-differentiated monocytes as measured by IDUA expression was comparable to cells expressing the YFP (IDUA-YFP) and selected using fluorescence-activated cell sorting (FACS)-based isolation, confirming that the inclusion of the tNGFR sequence did not affect enzyme production (Figure S14).
To examine their engraftment and therapeutic potency, tNGFR+ and tNGFR- purified populations were transplanted intrafemorally into BU-conditioned NSG-MPSI mice. Sixteen weeks after transplantation, engraftment of human cells was analyzed in bone marrow, peripheral blood, and spleen (Figure 6D). Selected IDUA-tNGFR+ cells engrafted poorly in the bone marrow (median 1.4%; min 0.01, max 19), peripheral blood (0.08%; 0.01, 1.3), and spleen (0.24%; 0.01, 8.3) compared with bulk IDUA cells (medians 73%, 8.6%, and 56%) or IDUA-tNGFR- cells (74%, 14%, and 52%). Although IDUA-tNGFR+ cells had a higher fraction of edited alleles prior to transplantation due to enrichment (median 41% tNGFR+ versus 31% IDUA), this fraction dropped in the bone marrow, resulting in a lower fraction of targeted alleles in the engrafted cells compared with cells lacking the selectable marker (13% tNGFR+ versus 20% IDUA) (Figures 6E and 6F).
Because cell selection resulted in a smaller cell dose at transplantation, to better quantify and compare the engraftment potential in IDUA+, tNGFR+, and tNGFR- cells, we calculated the total number of edited cells in the bone marrow and normalized it by the number of cells that were initially transplanted, resulting in a metric we named engraftability (Figures S15A–S15C). This analysis shows that the engraftability of selected cells is significantly lower than that of bulk cells (p = 0.0024; Figure 6G). Consistent with this, flow-based analysis of long-term repopulating HSCs (CD34+ CD38- CD45RA- CD90+ cells) in three independent cord blood donors after editing and tNGFR selection showed that the selected tNGFR+ fraction contained fewer HSCs compared with unedited cells and the tNGFR- fraction (fold decrease 1.4 ± 0.1; Figures 6H and S16). Together, cell selection seems to negatively impact the engraftment potential of the cell product, ultimately reducing the therapeutic cell dose.
The reduced engraftability and consequent lower therapeutic dose observed with tNGFR-selected cells predicted a worse performance at correcting biochemical features in the mouse model. Despite IDUA-tNGFR enzymatic activity in vitro being over 100-fold compared with unmodified cells (Figure S14C), only a few mice transplanted with IDUA-tNGFR+ CD34+ cells had detectable levels of IDUA activity in serum, liver, spleen, and brain (Figure 6I). Consequently, reduction in GAG storage was not pronounced in IDUA-tNGFR+ transplanted mice, where GAGs were only partially reduced in the spleen (p < 0.0001) but not in the brain or liver (Figure 6J).
Prior studies examining FACS-based sorted edited human CD34+ cells expressing IDUA and a fluorescent reporter (YFP) also suggested that selection negatively impacted engraftment and in vivo efficacy.20 Experiments comparing the engraftment and biochemical correction capacity of YFP+ (cells that underwent HDR and express supraphysiological IDUA levels) and YFP- (cells that did not undergo HDR and express endogenous IDUA levels) redemonstrated the decreased engraftability of selected cell populations that have undergone HDR (Figures S15D–S15F). Consistent with their supraphysiological IDUA expression in culture (∼50-fold; Figure S9C), YFP+ CD34+ cells were more potent at reconstituting IDUA activity in NSG-MPSI mice (Figure S15G), suggesting that the observed biochemical correction in vivo is in part due to the integrated cassette in addition to endogenous IDUA. Together, these data support that selection negatively impacts efficiency, engraftment, and phenotypic correction compared with bulk cell preparations.
Discussion
Autologous transplantation of genome-edited HSPCs has the potential to treat many hematological and non-hematological diseases including several lysosomal and peroxisomal disorders. Despite significant advances in genome-editing efficiencies in these cells, therapeutic approaches that depend on CRISPR-Cas9-mediated HDR for making specific modifications have been hindered by a reproducible disadvantage in the long-term engraftment of the edited cells in the initial cell product used for transplantation. While this challenge can be circumvented by newer technologies like base editors and prime editing, these tools are limited to small genetic modifications.52 Accordingly, finding ways to facilitate the engraftment of edited HSPCs is not only widely beneficial but necessary for making larger therapeutic modifications and leveraging the versatility of targeted gene-addition approaches into safe harbors, which are still mostly dependent on HDR.
HSPCs are a heterogeneous population that includes long-term repopulating stem cells and more committed progenitors. An accepted mechanism underlying the reduced engraftment potential in HDR-edited HSPCs is that, compared with the more quiescent stem cells (HSCs), proliferating progenitors preferentially undergo HDR.53,54 This loss of engraftment capacity is further supported by our experiments selecting HDR-edited HSPCs. Interestingly, this phenomenon is also observed in HSPCs modified by lentiviral vectors,55, 56, 57, 58 which transduce dividing and non-dividing cells, suggesting that other factors, such as ex vivo culturing conditions or the susceptibility of HSCs to modification, might also play a role. Notably, edited HSPCs produced in vivo after serial transplantation have normal long-term engraftment potential, suggesting that this phenomenon is specific to the transplanted population and not to genome editing or the transgene per se.54
Herein, we sought to determine the effect of the pre-conditioning regimen on the engraftment of edited human CD34+ cells and on phenotypic correction in a non-hematological disease model with prominent CNS involvement (MPSI). Across multiple human cell donors and compared with sub-lethal radiation in immunocompromised mice, BU-mediated myeloablation resulted in higher engraftment of edited cells in all organs tested. This implies that BU-based myeloablation produces a niche that is more conducive for the engraftment of edited hematopoietic stem cells. Comprehensive cytokine profiling in plasma and bone marrow suggests that niche amenability might be impacted by the cytokine milieu and that BU is less pro-inflammatory than TBI. Interestingly, human cell chimerism in the bone marrow was the same in both protocols, suggesting that this effect cannot be explained by different degrees of myelosuppression. In human diseases for which autologous transplantation of genome-edited HSPCs would be considered, BU will be the myeloablative clinical agent of choice. Compared with irradiation, BU is less toxic, produces fewer adverse events, and elicits less inflammatory reactions, which is important especially in the context of graft-versus-host disease.59, 60, 61 BU myeloablation has also been successfully combined with immunosuppression to ensure efficient engraftment of lentiviral-vector-modified CD34+ cells in non-human primates (NHPs) and in humans to prevent anti-transgene immune responses.28,62,63
Consistent with higher engraftment of edited cells, biochemical correction as measured by transgene expression (IDUA) and metabolite storage (GAG) was improved. We observed significantly increased biochemical correction in the brain of NSG-MPSI mice conditioned with BU compared with TBI. Enzyme levels were 2.5-fold higher in BU-treated mice, and a significant reduction in brain GAG storage and in Purkinje cells in the cerebellum was also observed. This improved tissue correction is likely due to a combination of improved engraftment of edited cells in the bone marrow (higher therapeutic cell dose) and enhanced tissue recruitment of bone marrow (BM)-derived cells. In the CNS in particular, prior studies have shown that BU-based myeloablation results in better infiltration of BM-derived cells in the CNS,36,37 and we saw, for the first time, the progeny of human-edited HSPCs in the mouse brain. Specifically, compared with sub-lethal TBI, BU likely results in a greater migration of BM-derived cells to macrophage/microglia niches. How BU enhances homing to the CNS is not well understood, but overt BBB disruption does not seem to be a major factor. BU is BBB permeable and achieves plasma-equivalent concentrations,64 exerting long-term effects in the CNS and suggesting that conditioning elicits changes in the CNS parenchyma that prompt the recruitment and long-term survival of cells from the BM. Several studies demonstrate that BU affects the microglia niche by reducing microglia density and inducing cell senescence, resulting in a loss of regenerative capacity that enables repopulation from BM-derived cells.36,65 It is likely that similar phenomena take place in macrophage niches in other organs. Other possible reasons for BU’s superiority compared with TBI are the long-term CNS production of MCP-1, an essential cytokine for monocyte recruitment,37 and the release of granulocyte colony-stimulating factor (G-CSF), which is responsible for the proliferation and mobilization of hematopoietic cells. At much higher doses than used here, TBI disrupts the BBB43 and provokes an intense release of pro-inflammatory cytokines, but this seems transitory without affecting long-term cell migration.37
To establish a potential transplantation protocol that includes BU as conditioning for genome-edited HSPCs, we also examined the long-term repopulation potential and therapeutic efficacy of selected and non-selected preparations of edited HSPCs that had undergone HDR. We determined that selection not only complicates the manufacturing protocol and reduces yield but results in a population with reduced engraftment potential. These results are consistent with the idea that HDR happens preferentially in the progenitor population but could also suggest that preparations containing unedited cells could support engraftment of edited cells. These data are also consistent with a previous study where the selection of edited cells was based on YFP expression and FACS. However, it was not clear if the lower efficiency observed was due to toxicity caused by cytoplasmic YFP.20
The study highlights the importance of the pre-conditioning regimen in studies using genome edited HSPCs. Increased engraftment of edited HSPCs will benefit the development of this approach for hematological and non-hematological diseases, but it is of particular interest in diseases with neurological involvement. Specifically, our data support the development of protocols that combine BU with unselected preparations of cells as a general strategy for several diseases. For MPSI, we provide convincing data of phenotypic correction in the CNS that further support the clinical development of human genome-edited HSPCs for this disease and other lysosomal storage disorders.
Materials and methods
Cell culture and isolation of CD34+ HSPCs
CD34+ HSPCs were collected from umbilical cord blood obtained from healthy donors under informed consent via the Binns Program for Cord Blood Research at Stanford University. The mononuclear cells were isolated by Ficoll Paque Plus density gradient centrifugation. CD34+ HSPCs were positively selected using CD34+ Microbead Kit Ultrapure (Miltenyi Biotec, San Diego, CA, USA) according to the manufacturer’s instructions. Cells were used fresh or immediately frozen after CD34+ isolation. Cells were cultured for 48 h prior to editing at 37 °C, 5% CO2, and 5% O2 in growth culture media consisting of StemSpan SFEM II (Stemcell Technologies, Vancouver, BC, Canada), SCF (100 ng/mL), TPO (100 ng/mL), Flt3-Ligand (100 ng/mL), IL-6 (100 ng/mL), UM171 (35 nM), and StemRegenin1 (0.75 μM).
AAV donor plasmids and viral vector construction
Human genomic DNA was used as template to PCR-amplify 500 bp of sequence upstream and downstream of the CCR5 sgRNA cut site to serve as left and right homology arms in the donor vectors. PGK, IDUA, tNGFR, spleen focus-forming virus (SFFV), and YFP sequences were amplified from plasmids. Fragments generated were assembled by Gibson Assembly into the pAAV-MCS plasmid (Agilent Technologies, Santa Clara, CA, USA). rAAV6 viruses encoding IDUA were produced and purified by Vigene Biosciences (Rockville, MD, USA), while SFFV-YFP was purified in house using iodixanol gradient ultracentrifugation to remove impurities and empty capsids. Titers were performed using ddPCR and the following ITR2 oligos: 5′-CGGCCTCAGTGAGCGA-3′, 5′-GGAACCCCTAGTGATGGAGTT-3′, 5′-FAM-/CACTCCCTC/ZEN/TCTGCGCGCTCG-3IABkFQ-3′.
Electroporation and transduction of cells
The CCR5 gRNA used was chemically modified and purchased from TriLink BioTechnologies (San Diego, CA, USA). The sequence was previously described:9 5′-GCAGCATAGTGAGCCCAGAA-3′. Cas9 protein was purchased from Aldevron (Fargo, ND, USA). For intracellular delivery, the RNP, Cas9, and gRNA were complexed by pre-mixing at room temperature at a molar ratio of 1:2.5 and were electroporated in HSPCs 48 h after CD34+ isolation using Lonza Nucleofector 4D, program DZ-100, in P3 primary cell solution. One million cells were electroporated in 100 μL with 30 μg Cas9 protein complexed with 15 μg gRNA. Immediately after electroporation, cells were rescued with warm growth culture media, and rAAV6 vectors were added as 10,000 viral genomes/cell (vg/cell) (IDUA, IDUA-tNGFR, IDUA-YFP) or 4–6,000 (YFP). Mock electroporated controls without RNP or with RNP only were included.
Measuring allele modification efficiency at the CCR5 locus
Genomic DNA was extracted from either bulk or sorted populations using QuickExtract DNA Extraction Solution. The frequency of indel formation was quantified using Tracking Indels by Decomposition (TIDE).66 For ddPCR, droplets were generated on a QX200 Droplet Generator (Bio-Rad) per the manufacturer’s protocol. A HEX reference assay detecting copy-number input of the CCRL2 gene was used to quantify the chromosome 3 input. The assay designed to detect insertions at CCR5 consisted of F:5′-GGG AGG ATT GGG AAG ACA -3′, R:5′-AGG TGT TCA GGA GAA GGA CA-3′, and labeled probe 5′− FAM/AGC AGG CAT/ZEN/GCT GGG GAT GCG GTG G/3IABkFQ-3′. The reference assay was designed to detect the CCRL2 genomic sequence F:5′-CCT CCT GGC TGA GAA AAA G-3′, R:5′-GCT GTA TGA ATC CAG GTC C-3′ and labeled probe 5′− HEX/TGT TTC CTC/ZEN/CAG GAT AAG GCA GCT GT/3IABkFQ-3′. The accuracy of this assay was established with genomic DNA from a mono-allelic colony (50% allele fraction) as a template. The final concentrations of primer and probes were 900 and 250 nM, respectively. Twenty μL of the PCR reaction was used for droplet generation, and 40 μL of the droplets was used in the following PCR conditions: 95° for 10 min, 45 cycles of 94° for 30 s, 57°C for 30 s, and 72° for 2 min, then finalize with 98° for 10 min and 4°C until droplet analysis. Droplets were analyzed on a QX200 Droplet Reader (Bio-Rad) detecting FAM- and HEX-positive droplets. Control samples with non-template control, genomic DNA, and mock-treated samples and 50% modification control were included. Data were analyzed using QuantaSoft (Bio-Rad).
Quantification of human/mouse DNA
DNA extraction was performed with Quick Extract in 25 μL reagent. Human DNA relative to mouse DNA was quantified by ddPCR, targeting the mouse and human CCRL2 gene. The reference mouse labelled probe was 5′-/56-FAM/CTCTGCGGC/ZEN/TGACAGAAGCT/3IABkFQ/-3′; primer F: 5′-GTTCTGAAAGCCTCTCTC-3′; primer R: 5′-CTGGCTAAGAGGTACAG-3′. The human assay used labeled probe 5′- HEX/TGTTTCCTC/ZEN/CAGGATAAGGCAGCT GT/3IABkFQ-3′, and primers F 5′-CCTCCTGGCTGAGAAAAAG-3′ and R: 5′-GCTGTATGAATCCAGGTCC-3′. Reaction conditions were the same as described in Measuring allele modification efficiency at the CCR5 locus with ddPCR.
Selection of IDUA-tNGFR+ cells and flow cytometry
HSPCs edited with IDUA-tNGFR were kept in culture for 4 days after editing for full expression of the cassette. The selection of tNGFR+ cells was optimized using the GMP-compatible reagents anti-CD271 (LNGFR)-biotin antibodies (ME20.4–1.H4, Miltenyi Biotec) and Anti-Biotin Microbeads (Miltenyi Biotec), following the manufacturer’s protocol with an antibody concentration of 1:50 dilution for up to 106 cells. To confirm tNGFR expression in edited and selected HSPCs, cell preparations were stained with anti-tNGFR fluorescein isothiocyanate (FITC; ME20.4, Thermo Fisher Scientific, Waltham, MA, USA) or anti-Biotin FITC (130-098-796, Miltenyi Biotec, North Rhine-Westphalia, Germany) antibodies for 30 min on ice and were protected from light. A BD FACSAria II flow cytometer was used, and analysis was made first by gating cells only and excluding debris, then subgating the tNGFR+ events. The staining of long-term repopulating HSCs using recognized cell surface markers67 was performed in unedited and edited HSPCs with IDUA-tNGFR kept in culture for 4 days (2 days after editing) using the following antibodies: CD34-APC (581, Biolegend, San Diego, CA, USA), CD38-PE-Cy7 (HIT2, Biolegend), CD90-PE (5E10, Biolegend) CD45RA-APC-Cy7 (HI100, Biolegend), and the eBioscience Fixable Viability Dye eFluor 450 (Thermo Fisher Scientific). Stained cells were acquired using Cytoflex (Beckman Coulter, Jersey City, NJ, USA) and analyzed using FlowJo software (FlowJo, Ashland, OR, USA).
Analysis of CD34+ HSPCs and derived myeloid cells in culture
Seven days after genome editing with either the PGK-IDUA-P2A-YFP or the PGK-IDUA-P2A-tNGFR donors, the cord-blood-derived HSPCs were FACS sorted to isolate YFP+, YFP-, NGFR+, and NGFR- cells. Mock HSPCs and HSPCs treated with the RNP only were used as controls. The sorted cells (5 × 104 cells/sample) were used to measure IDUA enzyme activity as described above. Sorted cells (1 × 106 cells/sample) were differentiated toward the myeloid/monocyte lineage for 8 days and collected to measure IDUA enzyme activity (5 × 104 cells/sample). To differentiate CD34+ cells toward the myeloid/monocyte lineage, the cells were cultured at 37 °C, 5% CO2, and 21% O2 on Cultrex Reduced Growth Factor Basement Membrane Matrix (R&D Systems, Minneapolis, MN, USA; 35-330-1002) in StemSpan SFEM II supplemented with human cytokines (PeproTech, East Windsor, NJ, USA): M-CSF (50 ng/mL), Flt3-Ligand (50 ng/mL), and GM-CSF (25 ng/mL). Control undifferentiated HSPCs and non-adherent myeloid/monocyte cells, collected 8 days after differentiation, were analyzed by flow cytometry upon staining with the following antibodies: anti-human CD45 PB (2D1, Biolegend), anti-human CD14 APC (61D3, Thermo Fisher Scientific), anti-mouse/human CD11b PE (M1/70, Biolegend), and anti-human CD271 (NGFR) FITC (ME20.4, Thermo Fisher Scientific). Propidium iodide (1 μg/mL) was used to detect dead cells, and cells were analyzed on Cytoflex (Beckman Coulter). Cells were identified by the specific markers and quantified by FlowJo software (FlowJo).
Analysis of myeloid cells in NSG BM
BM was collected by flushing femurs and tibiae using phosphate-buffered saline (PBS) 1X (no calcium/no magnesium). BM cells were stained for flow cytometry analyses using the following antibodies: CD45.1 PE-Cy7 (A20, eBioScience), TER119 PE-Cy5 (TER-119, BD Biosciences, Franklin Lakes, NJ, USA), CD11b PE (M1/70, Biolegend), Ly6C BV605 (AL-21, Fisher Scientific), CD3 APC (17A2, eBioScience), CD19-PB (6D5, Biolegend), and eFluor 780 viability Dye (eBioScience). Stained cells were acquired using Cytoflex (Beckman Coulter) and analyzed using FlowJo software (FlowJo).
Mice
NOD.C-PrkdcscidIL2rgtmlWjl/Sz [NSG] IDUA X/X [MPS I] (NSG-MPSI) mice were previously described,9 and NSG-SGM3 (SGM3) expressing human IL3, GM-CSF (CSF2), and SCF (KITLG) were obtained from Jackson Laboratory (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ; Jax stock no.: 013062). Mice were housed in the animal barrier facility at Stanford University in a 12 h dark/light cycle, temperature- and humidity-controlled environment. Sterile bedding, food, and water were used. Cages were sterile and ventilated individually. All experimental procedures were performed in accordance with National Institutes of Health institutional guidelines and were approved by the University Administrative Panel on Laboratory Animal Care (IACUC 33365).
Conditioning regimens
BU conditioning was done with four daily doses of 17 mg/kg, 24 h apart, in a total dose of 68 mg/kg.42 Myeloablation was confirmed, as BU-conditioned mice that did not receive support of blood and BM cells died up to 20 days post-treatment. BU was obtained from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in sterile DMSO at 40 mg/kg aliquots, stored at −20°C. Immediately before administration, BU stock solutions were thawed and diluted in PBS 1X to 2 mg/kg. BU was injected intraperitoneally in volumes not higher than 250 μL. TBI was performed 2 h before transplantation at a dose of 2.1 Gy.
CD34+ cell transplantation
Transplantations were performed 24 h after the last BU dose or 1 h after TBI. IDUA-edited bulk (800,000–2,700,000 cells/mouse), IDUA-tNGFR+ (100,000–1,450,000 cells/mouse) or IDUA-tNGFR- (550,000–1,600,000 cells/mouse) cells were suspended in 25 μL of PBS 1X and transplanted intrafemorally in anesthetized mice. This route was chosen to minimize hematopoietic stem cell loss in the peripheral organs and encourage engraftment by putting the cells directly into the BM environment.68 For the BU-TBI comparisons, the same number of total cells were transplanted per condition. Bulk preparations were transplanted 48 h after editing, while tNGFR cells were selected 96 h after editing and transplanted immediately after selection. Secondary transplants were performed after the collection of BM from primary recipient mice and immunomagnetic selection of CD34+ cells. Four days after transplantation, BM and blood cells collected from IDUA knockout mice were given intravenously to all BU-conditioned mice for support (5 × 106 cells/mouse). For supportive BM transplants, femurs and tibiae were flushed using PBS 1X (no calcium/no magnesium) supplemented with 4U/mL heparin (H3149-500KU, Sigma-Aldrich). The cells were filtered sequentially through 100 and 45 μm cell strainers and washed twice with PBS 1X (no calcium/no magnesium). 2–5 × 106 cells/mouse were injected intravenously in a total volume of 100 uL PBS 1X (no calcium/no magnesium).
SGM3 mice were transplanted with bulk HSPCs edited with the SFFV-YFP donor (1 × 106 cells/mouse for the intravenous study, Figures 4 and S7, or 1 × 105 cells/ventricle for the intracerebroventricular study, Figure S6). Support BM and blood cells collected from an untreated SGM3 mouse were given intravenously to all BU-conditioned SGM3 mice (1.5 × 106 cells/mouse).
Assessment of human engraftment in hematopoietic organs
Fifteen to 20 weeks after transplantation, mice were euthanized, and peripheral blood, BM, and spleen were collected. After several washes and treatment with ammonium chloride to remove erythrocytes, cells were treated with 10% vol/vol TruStain FcX (BioLegend, San Diego, CA, USA) to block non-specific antibody binding. Cells were stained in the dark for 30 min on ice using the following antibodies: anti-human HLA-ABC APC-Cy7 (W6/32, BioLegend), anti-mouse CD45.1 PE-Cy7 (A20, eBioScience, San Diego, CA, USA), anti-human CD45 PB (2D1, Biolegend), CD19 APC (HIB19, BD511 Biosciences), CD33 PE (WM53, BD Biosciences), anti-mouse/human TER-r119 PE-Cy5 (TER-119, BD Biosciences), and CD3 BV650 (HiT3A, BioLegend). After staining, cells were washed and resuspended in MACS buffer containing propidium iodide to detect dead cells. Samples were analyzed in a BD FACSAria II flow cytometer, and human engraftment was given by double-positive human HLA-ABC+/human CD45+ cells.
Microglia isolation and flow cytometry analysis
Brains were dissected from mice after transcardial perfusion, and approximately one quarter (for MPSI mice) or one sagittal half (for SGM3 mice) of the organ was used for microglia isolation using a modified version of a reported method.69 The piece of tissue was minced in ice-cold Hank’s balanced salt solution (HBSS) 1X, washed, and then incubated with enzyme mix 1 (containing enzyme P and buffer X from Neural Tissue Dissociation Kit P, Miltenyi Biotec) at 37°C for 15 min. After incubation, enzyme mix 2 (enzyme A + buffer Y) was added, and the sample was incubated for another 15 min, with eventual up-and-down pipetting during the total 30 min incubation. Digested samples were passed through a 70 μm cell strainer until no big pieces of tissue remained. After washing with HBSS 1X for 320 × g at 4°C, the pellet was resuspended in 33% Percoll (GE Healthcare, Chicago, IL, USA) and centrifuged for 15 min, 700 × g at 18°C, with brakes off and acceleration 7. The resulting gradient contained myelin on top and the microglia pelleted, which was collected, washed with FACS-BL buffer (PBS 1X, 5% fetal bovine serum, 1% bovine serum albumin), and pelleted again for DNA extraction and flow cytometry analyses. For flow cytometry antibody staining, the cell extracts were incubated in FACS-BL with 10% vol/vol Fc-receptor-blocking solutions (TruStain FcX, BioLegend, and Mouse BD Fc Block, Biosciences) to block non-specific antibody binding. Cell were then stained in the dark for 30 min on ice using the following antibodies: anti-mouse CD45.1 PE-Cy7 (A20, eBioScience, San Diego, CA, USA), anti-mouse/human TER-119 PE-Cy5 (TER-119, BD Biosciences), anti-human CD45 PB (2D1, Biolegend, San Diego, CA, USA), anti-mouse/human CD11b PE (M1/70, Biolegend, San Diego, CA, USA), and anti-human HLA-DR BV645 (LN3, Thermo Fisher Scientific). After staining, cells were washed and resuspended in FACS-BL buffer containing propidium iodide (1 μg/mL) to detect dead cells. Samples were analyzed in BD FACSAria II flow cytometer or Cytoflex (Beckman Coulter). Mouse microglia cells and human microglia-like/myeloid cells were identified by the specific marker labelling and quantified by FlowJo software (FlowJo).
IDUA activity assay
IDUA enzyme activity was measured by a fluorometric assay using the fluorescent specific substrate 4-methylumbelliferyl α-L-iduronide (4MU-iduronide) (LC Scientific, Concord, ON, Canada), as described previously.9 Briefly, the 4-methylumbelliferyl-iduronide substrate was diluted at 6.6 mM with sodium formate buffer (0.4 M, pH 3.5). Substrate and tissue homogenates were mixed 1:1 to a final volume of 40 μL and incubated at 37 °C for 60 min. To stop the reaction, 200 μL glycine carbonate buffer (pH 10.4) was added. The fluorescence was measured in a plate reader (SpectraMax iD5, Molecular Devices) with excitation at 355 nm and emission at 460 nm, and results were calculated using a 4-MU standard curve.
Analysis of GAGs using the DMB method
Urine and tissue GAGs were measured with the modified DMB assay and by LC/MS-MS, as described in detail previously.9 For DMB, tissue samples (10–30 mg) were incubated for 3 h, 2,000 RPM, at 65°C in papain digest solution (calcium- and magnesium-free PBS containing 1% papain suspension [Sigma], 5 mM cysteine, and 10 mM EDTA, pH 7.4) to a final concentration of 0.05 mg tissue/mL buffer. Urine samples were diluted in water accordingly in order to be within the linear range of the assay. Fifty μL of tissue extract or diluted urine were incubated with 200 μL DMB reagent (9:1 31 μM DMB stock in formiate buffer 55 nM:2 M Tris base). The samples were read on a microplate reader at 520 nm and compared with a heparan sulfate standard curve. GAGs in tissue or urine were normalized by protein and creatinine contents, respectively.
Analysis of GAGs by LC/MS-MS
Disaccharides were produced from polymer GAGs by digestion with chondroitinase B, heparitinase, and keratanase II, resulting in DS (di-0S), HS (diHS-NS, diHS-0S), and KS (mono-sulfated KS, di-sulfated KS). Chondrosine was used as an internal standard (IS). Unsaturated disaccharides, ΔDiHS-NS, ΔDiHS-0S, ΔDi-4S, mono-sulfated KS, and di-sulfated KS were obtained from Seikagaku (Tokyo, Japan) and used to make standard curves. Stock solutions ΔDiHS-NS (100 μg/mL), ΔDiHS-0S (100 μg/mL), and ΔDi-4S (250 μg/mL) and mono- and di-sulfated KS (1,000 μg/mL) and IS (5 mg/mL) were prepared separately in milliQ water. Standard working solutions of ΔDiHS-NS, ΔDiHS-0S, ΔDi-4S (7.8125, 15.625, 31.25, 62.5, 125, 250, 500, and 1,000 ng/mL) and mono- and di-sulfated KS (80, 160, 310, 630, 1,250, 2,500, 5,000, and 10,000 ng/mL), each mixed with IS solution (5 μg/mL), were prepared. Mass spectrometer apparatus, run condition, brain homogenate preparation, and disaccharide analysis were done as described in the supplemental methods.
Immunofluorescence
After bleeding, mice were transcardially perfused with PBS (pH 7.4) followed by 4% paraformaldehyde (PFA) in PBS. Brains were fixed overnight at 4°C. For frozen sections, brains were transferred to a 30% sucrose solution overnight for cryoprotection, embedded in Tissue-Tek optimal cutting temperature (OCT) compound, and cut (15–20 μm sections) on a freezing cryostat (Leica, Wetzlar, Germany, CM3050). All tissue was stored at −80°C until further use. For immunohistochemistry, slides were washed in PBS to remove excess OCT. Sections were blocked in 10% normal goat plasma (NGS; Gibco) containing 0.25–3% Triton X-100 for 1 h at 25°C. Primary antibody (anti-human Ku80, STEM101, Y4040, Takara Bio, San Jose, CA, USA) was applied overnight in 10% NGS with 0.1% Triton X-100 at 4°C followed by the appropriate fluorochrome-conjugated secondary antibody (Alexa conjugates; Molecular Probes) for 1 h at 25°C. For imaging microglia, Isolectin GS-IB4 From Griffonia simplicifolia, Alexa Fluor 568 Conjugate (Invitrogen - Molecular Probes, USA) was reconstituted as a 1 mg/mL stock in PBS with 0.5 mM CaCl2 and 0.01% sodium azide, and the slides were incubated with a working solution of 5 μg/mL in calcium-containing PBS for 1 h at 25°C. Slides were then washed in PBS with 0.1% BSA, counterstained with Hoechst 3342 (PI62249, Thermo Fisher Scientific), and mounted in Aqua Poly/Mount (Polysciences) for fluorescent microscopy. Slides were visualized by conventional epifluorescence microscopy using an all-in-one Fluorescence Microscope BZ-X800 (Keyence, Itasca, IL, USA). Human Ku80-positive cells were counted using ImageJ.
Histological assessment with toluidine blue
GAG quantification in cerebellum was performed as described previously.70 Briefly, PFA-fixed brains were embedded in paraffin and cut in thin cross sections (4 μm). Resulting slides were submitted to routine histological processing and stained with toluidine blue, where storage can be observed in Purkinje cells as white vacuoles in the cytoplasm. Slides were analyzed by a researcher blinded to the groups, counting vacuole-positive cells in 10 high-power fields (1,000 X).
Cytokine measurement and BM phenotype after conditioning
Ten-week-old female NSG mice were conditioned with TBI (2.1 Gy, n = 5) or BU (66 mg/kg, n = 5) or left untreated (Untreated, n = 4); samples were collected at 2 and 24 h after conditioning. Blood was collected in Safe-T-fill Dipotassium EDTA tubes (RAM Scientific, Nashville, TN, USA), and plasma was separated by centrifugation at 200 × g for 10 min. BM was collected by flushing femurs and tibiae using PBS 1X (no calcium/no magnesium). BM homogenates were prepared using RIPA Lysis buffer 1X (G Biosciences, St. Louis, MO, USA) and were supplemented with protease inhibitor cocktail (Complete, Sigma-Aldrich), and protein concentration was normalized using the BCA protein assay kit (Thermo Fisher Scientific). Mouse cytokines were quantified using the Immune Monitoring 48-Plex Mouse ProcartaPlex Panel (EPX480-20834-901, Thermo Fisher Scientific). Luminex assay was performed at the Human Immune Monitoring Center (HIMC, Stanford University School of Medicine, Stanford, CA, USA). Plasma samples were diluted 3 folds with PBS 1X and run in duplicates. BM homogenates, normalized to 5 mg/mL, were run undiluted, in duplicates. BM cells were stained for flow cytometry analyses using the following antibodies: CD45.1 PE-Cy7 (A20, eBioScience), TER119 PE-Cy5 (TER-119, BD Biosciences), CD11b PE (M1/70, Biolegend), Ly6C BV605 (AL-21, Fisher Scientific), CD3 APC (17A2, eBioScience), CD19-PB (6D5, Biolegend), and eFluor 780 viability Dye (eBioScience). Stained cells were acquired using Cytoflex (Beckman Coulter) and analyzed using FlowJo software (FlowJo).
IgG immunostaining
Thirty-five μm-thick sections were washed in 0.01 M PBS and then incubated in 3% hydrogen peroxide (methanol: 30% H2O2 = 1:1 in 0.01 M PBS) for 20 min and permeabilized with PBS containing 0.25% Triton X-100 (PBST, 3 × 10 min). The sections were blocked in 10% normal goat serum at room temperature (RT) for 1 h, followed by incubation with biotinylated-goat anti-mouse IgG (#BA9200, Vector Laboratories, Burlingame, CA, USA) overnight at 4°C. Free-floating sections were subsequently washed in permeabilizing buffer three times. For detection of mouse IgG, sections were incubated in avidin Vectastain ABC KIT (#sk4600, Vector Laboratories) for 30 min and revealed for 5 min using Vector NovaRed peroxidase substrate (#sk4800, Vector Laboratories). The reaction was stopped with PBS (3 × 5 min), and sections were mounted to a glass slide. Then samples were air dried in air overnight and dehydrated using isopropanol xylene before being cover slipped with Aqua/polymount (Polyscience, Philadelphia, PA, USA). Images were captured under a light microscope using a 2X lens (B-Z Series, Keyence, Osaka, Japan). Pixel intensity was calculated using Fiji software.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism v.8 for macOS X software (La Jolla, CA, USA). Normality tests were conducted for all groups, and means were presented when data passed D’Agostino & Pearson, Shapiro-Wilk, and Kolmogorov-Smirnov (KS) normality tests. ANOVA with Tukey post-hoc and two-way unpaired t tests were conducted in normally distributed data; Kruskal-Wallis and Mann-Whitney tests were performed in not normally distributed sets.
Acknowledgments
We thank Stanford’s Binns Program for Cord Blood Research for providing cells. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS; K08NS102398 to N.G.-O.) and MPSI Pilot Grant from the Orphan Disease Center, University of Pennsylvania. We also thank Yael Rosenberg-Hasson (Institute for Immunity, Transplantation, and Infection, Stanford University School of Medicine, Stanford, CA, USA), Tran Thi Thanh Nguyen, and the Stanford Human Immune Monitoring Center (HIMC) for running the Luminex assay.
Author contributions
E.P. and P.C. collected data, performed experiments, carried out the analyses, and participated in manuscript preparation and figure design; L.N.P.V. performed studies on BBB damage; S.K. and S.T. performed LC-MS/MS analysis of GAGs; G.B. performed histological analysis of GAG storage; N.G.-O. conceived and directed the project, collected data, designed and cloned vectors, performed experiments and analysis with E.P. and P.C., and assisted with manuscript preparation and figure design.
Declaration of interests
N.G.-O. is a consultant and has equity interest in Graphite Bio, but this company had no input or opinions on the subject matter described in this manuscript. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.04.009.
Supplemental information
Data availability
Data supporting the findings of this work are available within the paper and its supplemental information. A reporting summary for this article is available as supplemental information. The datasets generated and analyzed during the current study are also available from the corresponding author upon request.
References
- 1.Dever D.P., Porteus M.H. The changing landscape of gene editing in hematopoietic stem cells: a step towards Cas9 clinical translation. Curr. Opin. Hematol. 2017;24:481–488. doi: 10.1097/moh.0000000000000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/elife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bak R.O., Gomez-Ospina N., Porteus M.H. Gene editing on center stage. Trends Genet. 2018;34:600–611. doi: 10.1016/j.tig.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Demirci S., Zeng J., Wu Y., Uchida N., Shen A.H., Pellin D., Gamer J., Yapundich M., Drysdale C., Bonanno J., et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Invest. 2020;130:6677–6687. doi: 10.1172/jci140189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O., et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11:aaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [Google Scholar]
- 7.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/nejmoa2031054. [DOI] [PubMed] [Google Scholar]
- 8.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Munoz D.P., Boffelli D., et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [Google Scholar]
- 10.Hoban M.D., Lumaquin D., Kuo C.Y., Romero Z., Long J., Ho M., Young C.S., Mojadidi M., Fitz-Gibbon S., Cooper A.R., et al. CRISPR/Cas9-Mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 2016;24:1561–1569. doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H., et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cromer M.K., Camarena J., Martin R.M., Lesch B.J., Vakulskas C.A., Bode N.M., Kurgan G., Collingwood M.A., Rettig G.R., Behlke M.A., et al. Gene replacement of alpha-globin with beta-globin restores hemoglobin balance in beta-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021;27:677–687. doi: 10.1038/s41591-021-01284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N., et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R., et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9:eaan0820. doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- 15.De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M., et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 16.De Ravin S.S., Reik A., Liu P.Q., Li L., Wu X., Su L., Raley C., Theobald N., Choi U., Song A.H., et al. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016;34:424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diez B., Genovese P., Roman-Rodriguez F.J., Alvarez L., Schiroli G., Ugalde L., Rodriguez-Perales S., Sevilla J., Diaz de Heredia C., Holmes M.C., et al. Therapeutic gene editing in CD34(+) hematopoietic progenitors from Fanconi anemia patients. EMBO Mol. Med. 2017;9:1574–1588. doi: 10.15252/emmm.201707540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Guerrero A., Sanchez-Hernandez S., Galvani G., Pinedo-Gomez J., Martin-Guerra R., Sanchez-Gilabert A., Aguilar-Gonzalez A., Cobo M., Gregory P., Holmes M., et al. Comparison of zinc finger nucleases versus CRISPR-specific nucleases for genome editing of the Wiskott-Aldrich syndrome locus. Hum. Gene Ther. 2018;29:366–380. doi: 10.1089/hum.2017.047. [DOI] [PubMed] [Google Scholar]
- 19.Rai R., Romito M., Rivers E., Turchiano G., Blattner G., Vetharoy W., Ladon D., Andrieux G., Zhang F., Zinicola M., et al. Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott - Aldrich Syndrome. Nat. Commun. 2020;11:4034. doi: 10.1038/s41467-020-17626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J., et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scharenberg S.G., Poletto E., Lucot K.L., Colella P., Sheikali A., Montine T.J., Porteus M.H., Gomez-Ospina N. Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun. 2020;11:3327. doi: 10.1038/s41467-020-17148-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P., et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genovese P., Schiroli G., Escobar G., Di Tomaso T., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.V., Lill G.R., Kaufman M.L., et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 2018;23:2606–2616. doi: 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J., et al. Editing the sickle cell disease mutation in human hematopoietic stem cells: comparison of endonucleases and homologous donor templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drysdale C.M., Tisdale J.F., Uchida N. Immunoresponse to gene-modified hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2020;16:42–49. doi: 10.1016/j.omtm.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw P., Shizuru J., Hoenig M., Veys P., Iewp E. Conditioning perspectives for primary immunodeficiency stem cell transplants. Front. Pediatr. 2019;7:434. doi: 10.3389/fped.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucci F., Scaramuzza S., Aiuti A., Mortellaro A. Update on clinical ex vivo hematopoietic stem cell gene therapy for inherited monogenic diseases. Mol. Ther. 2021;29:489–504. doi: 10.1016/j.ymthe.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robert-Richard E., Ged C., Ortet J., Santarelli X., Lamrissi-Garcia I., de Verneuil H., Mazurier F. Human cell engraftment after busulfan or irradiation conditioning of NOD/SCID mice. Haematologica. 2006;91:1384. [PubMed] [Google Scholar]
- 30.Meng A., Wang Y., Van Zant G., Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 2003;63:5414–5419. [PubMed] [Google Scholar]
- 31.Qiao J., Wu Y., Liu Y., Li X., Wu X., Liu N., Zhu F., Qi K., Cheng H., Li D., et al. Busulfan triggers intrinsic mitochondrial-dependent platelet apoptosis independent of platelet activation. Biol. Blood Marrow Transplant. 2016;22:1565–1572. doi: 10.1016/j.bbmt.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Hayakawa J., Hsieh M.M., Uchida N., Phang O., Tisdale J.F. Busulfan produces efficient human cell engraftment in NOD/LtSz-Scid IL2Rγ null mice. Stem Cells. 2009;27:175–182. doi: 10.1634/stemcells.2008-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi B., Chun E., Kim M., Kim S.T., Yoon K., Lee K.Y., Kim S.J. Human B cell development and antibody production in humanized NOD/SCID/IL-2Rγnull (NSG) mice conditioned by busulfan. J. Clin. Immunol. 2011;31:253–264. doi: 10.1007/s10875-010-9478-2. [DOI] [PubMed] [Google Scholar]
- 34.Chevaleyre J., Duchez P., Rodriguez L., Vlaski M., Villacreces A., Conrad-Lapostolle V., Praloran V., Ivanovic Z., Brunet de la Grange P. Busulfan administration flexibility increases the applicability of scid repopulating cell assay in NSG mouse model. PLoS One. 2013;8:e74361. doi: 10.1371/journal.pone.0074361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 36.Capotondo A., Milazzo R., Politi L.S., Quattrini A., Palini A., Plati T., Merella S., Nonis A., di Serio C., Montini E., et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. U S A. 2012;109:15018–15023. doi: 10.1073/pnas.1205858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkinson F.L., Sergijenko A., Langford-Smith K.J., Malinowska M., Wynn R.F., Bigger B.W. Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol. Ther. 2013;21:868–876. doi: 10.1038/mt.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 39.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 40.Novembre J., Galvani A.P., Slatkin M. The geographic spread of the CCR5 Δ32 HIV-resistance allele. PLoS Biol. 2005;3:e339. doi: 10.1371/journal.pbio.0030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samson M., Libert F., Doranz B.J., Rucker J., Liesnard C., Farber C.M., Saragosti S., Lapoumeroulie C., Cognaux J., Forceille C., et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 42.Capotondo A., Milazzo R., Garcia-Manteiga J.M., Cavalca E., Montepeloso A., Garrison B.S., Peviani M., Rossi D.J., Biffi A. Intracerebroventricular delivery of hematopoietic progenitors results in rapid and robust engraftment of microglia-like cells. Sci. Adv. 2017;3:e1701211. doi: 10.1126/sciadv.1701211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kierdorf K., Katzmarski N., Haas C.A., Prinz M. Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS One. 2013;8:e58544. doi: 10.1371/journal.pone.0058544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youshani A.S., Rowlston S., O'Leary C., Forte G., Parker H., Liao A., Telfer B., Williams K., Kamaly-Asl I.D., Bigger B.W. Non-myeloablative busulfan chimeric mouse models are less pro-inflammatory than head-shielded irradiation for studying immune cell interactions in brain tumours. J. Neuroinflammation. 2019;16:25. doi: 10.1186/s12974-019-1410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Jong J.G., Wevers R.A., Liebrand-van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin. Chem. 1992;38:803–807. doi: 10.1093/clinchem/38.6.803. [DOI] [PubMed] [Google Scholar]
- 46.Tomatsu S., Shimada T., Mason R.W., Kelly J., LaMarr W.A., Yasuda E., Shibata Y., Futatsumori H., Montano A.M., Yamaguchi S., et al. Assay for glycosaminoglycans by tandem mass spectrometry and its applications. J. Anal. Bioanal. Tech. 2014;2014:006. doi: 10.4172/2155-9872.S2-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson C.W., Adair J.E., Wohlfahrt M.E., Deleage C., Radtke S., Rust B., Norman K.K., Norgaard Z.K., Schefter L.E., Sghia-Hughes G.M., et al. Autologous, gene-modified hematopoietic stem and progenitor cells repopulate the central nervous system with distinct clonal variants. Stem Cell Rep. 2019;13:91–104. doi: 10.1016/j.stemcr.2019.05.016. [DOI] [Google Scholar]
- 48.Sergijenko A., Langford-Smith A., Liao A.Y., Pickford C.E., McDermott J., Nowinski G., Langford-Smith K.J., Merry C.L., Jones S.A., Wraith J.E., et al. Myeloid/Microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol. Ther. 2013;21:1938–1949. doi: 10.1038/mt.2013.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coughlan A.M., Harmon C., Whelan S., O'Brien E.C., O'Reilly V.P., Crotty P., Kelly P., Ryan M., Hickey F.B., O'Farrelly C., Little M.A. Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cells Dev. 2016;25:530–541. doi: 10.1089/scd.2015.0289. [DOI] [PubMed] [Google Scholar]
- 50.Billerbeck E., Barry W.T., Mu K., Dorner M., Rice C.M., Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor–, granulocyte-macrophage colony-stimulating factor–, and interleukin-3–expressing NOD-SCID IL2Rγnull humanized mice. Blood. 2011;117:3076–3086. doi: 10.1182/blood-2010-08-301507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonini C., Grez M., Traversari C., Ciceri F., Marktel S., Ferrari G., Dinauer M., Sadat M., Aiuti A., Deola S., et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat. Med. 2003;9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 52.Newby G.A., Yen J.S., Woodard K.J., Mayuranathan T., Lazzarotto C.R., Li Y., Sheppard-Tillman H., Porter S.N., Yao Y., Mayberry K., et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595:295–302. doi: 10.1038/s41586-021-03609-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lomova A., Clark D.N., Campo-Fernandez B., Flores-Bjurstrom C., Kaufman M.L., Fitz-Gibbon S., Wang X., Miyahira E.Y., Brown D., DeWitt M.A., et al. Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells. 2019;37:284–294. doi: 10.1002/stem.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin J.J., Schroder M.S., Caiado F., Wyman S.K., Bray N.L., Bordi M., Dewitt M.A., Vu J.T., Kim W.T., Hockemeyer D., et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 2020;32:108093. doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrua F., Cicalese M.P., Galimberti S., Giannelli S., Dionisio F., Barzaghi F., Migliavacca M., Bernardo M.E., Calbi V., Assanelli A.A., et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239–e253. doi: 10.1016/s2352-3026(19)30021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hacein-Bey Abina S., Gaspar H.B., Blondeau J., Caccavelli L., Charrier S., Buckland K., Picard C., Six E., Himoudi N., Gilmour K., et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313:1550–1563. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marktel S., Scaramuzza S., Cicalese M.P., Giglio F., Galimberti S., Lidonnici M.R., Calbi V., Assanelli A., Bernardo M.E., Rossi C., et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019;25:234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 58.Dahl M., Smith E.M.K., Warsi S., Rothe M., Ferraz M.J., Schambach A., Mason C., Karlsson S., Aerts J.M., Aerts J., et al. Correction of pathology in mice displaying Gaucher disease type 1 by a clinically-applicable lentiviral vector. Mol. Ther. Methods Clin. Dev. 2021;20:312–323. doi: 10.1016/j.omtm.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flowers M.E.D., Inamoto Y., Carpenter P.A., Lee S.J., Kiem H.P., Petersdorf E.W., Pereira S.E., Nash R.A., Mielcarek M., Fero M.L., et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117:3214–3219. doi: 10.1182/blood-2010-08-302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gyurkocza B., Sandmaier B.M. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood. 2014;124:344–353. doi: 10.1182/blood-2014-02-514778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xun C.Q., Thompson J.S., Jennings C.D., Brown S.A., Widmer M.B. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–2367. doi: 10.1182/blood.v83.8.2360.bloodjournal8382360. [DOI] [PubMed] [Google Scholar]
- 62.Tarantal A.F., Giannoni F., I Lee C.C., Wherley J., Sumiyoshi T., Martinez M., Kahl C.A., Elashoff D., Louie S.G., Kohn D.B. Nonmyeloablative conditioning regimen to increase engraftment of gene-modified hematopoietic stem cells in young rhesus monkeys. Mol. Ther. 2012;20:1033–1045. doi: 10.1038/mt.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Bonifacino A.C., Krouse A.E., Linde N., Hsieh M.M., Donahue R.E., et al. Busulfan combined with immunosuppression allows efficient engraftment of gene-modified cells in a rhesus macaque model. Mol. Ther. 2019;27:1586–1596. doi: 10.1016/j.ymthe.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peake K., Manning J., Lewis C.A., Tran K., Rossi F., Krieger C. Bone marrow-derived cell accumulation in the spinal cord is independent of peripheral mobilization in a mouse model of amyotrophic lateral sclerosis. Front. Neurol. 2017;8:75. doi: 10.3389/fneur.2017.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sailor K.A., Agoranos G., Lopez-Manzaneda S., Tada S., Gillet-Legrand B., Guerinot C., Masson J.B., Vestergaard C.L., Bonner M., Gagnidze K., et al. Hematopoietic stem cell transplantation chemotherapy causes microglia senescence and peripheral macrophage engraftment in the brain. Nat. Med. 2022;28:517–527. doi: 10.1038/s41591-022-01691-9. [DOI] [PubMed] [Google Scholar]
- 66.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Majeti R., Park C.Y., Weissman I.L. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhan Y., Zhao Y. Hematopoietic stem cell transplant in mice by intra-femoral injection. Methods Mol. Biol. 2008;430:161–169. doi: 10.1007/978-1-59745-182-6_11. [DOI] [PubMed] [Google Scholar]
- 69.Molina Estevez F.J., Mathews T.D., Biffi A., Peviani M. Simultaneous flow cytometric characterization of multiple cell types retrieved from mouse brain/spinal cord through different homogenization methods. J. Vis. Exp. 2019 doi: 10.3791/60335. [DOI] [Google Scholar]
- 70.Baldo G., Mayer F.Q., Martinelli B., Dilda A., Meyer F., Ponder K.P., Giugliani R., Matte U. Evidence of a progressive motor dysfunction in Mucopolysaccharidosis type I mice. Behav. Brain Res. 2012;233:169–175. doi: 10.1016/j.bbr.2012.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this work are available within the paper and its supplemental information. A reporting summary for this article is available as supplemental information. The datasets generated and analyzed during the current study are also available from the corresponding author upon request.