

Abstract

A 3 × 3 isomer grid of nine N-(chlorophenyl)pyridinecarboxamides (NxxCl) is reported with physicochemical studies and single crystal structures (Nx = pyridinoyl moiety; xCl = aminochlorobenzene ring; x = para-/meta-/ortho-), as synthesized by the reaction of the substituted p-/m-/o-pyridinecarbonyl chlorides (Nx) with p-/m-/o-aminochlorobenzenes (xCl). Several of the nine NxxCl crystal structures display structural similarities with their halogenated NxxX and methylated NxxM relatives (x = p-/m-/o-substitutions; X = F, Br; M = methyl). Indeed, five of the nine NxxCl crystal structures are isomorphous with their NxxBr analogues as the NpmCl/Br, NpoCl/Br, NmoCl/NmoBr, NopCl/Br, and NooCl/Br pairs. In the NxxCl series, the favored hydrogen bonding mode is aggregation by N–H···Npyridine interactions, though amide···amide intermolecular interactions are noted in NpoCl and NmoCl. For the NoxCl triad, intramolecular N–H···Npyridine interactions influence molecular planarity, whereas NppCl·H2O (as a monohydrate) exhibits O–H···O, N–H···O1W, and O1W-H···N interactions as the primary hydrogen bonding. Analysis of chlorine-containing compounds on the CSD is noted for comparisons. The interaction environments are probed using Hirshfeld surface analysis and contact enrichment studies. The melting temperatures (Tm) depend on both the lattice energy and molecular symmetry (Carnelley’s rule), and the melting points can be well predicted from a linear regression of the two variables. The relationships of the Tm values with the total energy, the electrostatic component, and the strongest hydrogen bond components have been analyzed.
Short abstract
Physicochemical properties, conformational analyses, and crystal structures of a 3 × 3 isomer grid of N-(chlorophenyl)pyridinecarboxamides (NxxCl) (x = para-/meta-/ortho-) are reported. Interaction environments are probed using Hirshfeld surface analysis and contact enrichment studies. Melting temperatures (Tm) depend on the lattice energy and molecular symmetry (Carnelley’s rule). Relationships of Tm with the total energy, electrostatic component, and strongest hydrogen bond components are analyzed.
Introduction
Organohalogens (as a class of organic chemicals) have seen a dramatic increase in research activity over the past 3 decades in a range of scientific fields such as atmospheric chemistry, pharmaceuticals, and agrochemicals.1−14 These research studies include both basic and applied research together with industrial applications.5,9 Ongoing structural chemistry research on organohalogens includes investigations on halogen bonding and intermolecular interactions;14 these studies have led to considerable developments and insights into our understanding of bonding and aggregation modes.14−32 Extensive structural studies have been undertaken on series of organohalogens. Examples include the investigation of fluorine in benzamides33−36 and potential uses of bromine and iodine in agrochemicals.37−39
Organochlorines have attracted considerable interest in the pharmaceutical sector40−46 and especially in agrochemicals (herbicides and pesticides) with uses as antihelminthic drugs such as niclosamide (an orally bioavailable chlorinated salicylanilide).41 Some of these have raised public concern mainly due to their disposal, waste treatment, and environmental problems.4,47,48 In tandem with drug development, there has been a surge in the study and use of halogens in new drugs and especially fluorine and chlorine in pharmaceuticals.33−36,40−46
The role and importance of the Cambridge Structural Database (CSD) as a tool for understanding structural systematics have been noted.49 As such, the development of structural systematics in pharmaceutical sciences is critical as one seeks to establish correlations in physicochemical relationships between series of molecules.14 In analyzing the electronic properties of series of compounds (such as n × m carboxamide isomer grids), the ability to observe trends in fundamental properties is essential. As advances in this area continue to be made, it is essential that our ability to assess tens, hundreds, or thousands of related structures is made easier.49 A key is to reduce the number of parameters and elucidate genuine relationships and correlations to aid in the development of new pharmaceutical drugs.7,8,41−43,49
We have previously reported several isomer grids of benzamides and carbamates including the mono-substituted, methyl-, fluoro-, and chlorobenzamides and the related methyl and methoxycarbamates.50−59 In expanding the isomer grid series, the increased numbers of compounds for analysis and for comparisons can be appreciated with what is already available for study on the CSD.49 In analyzing the electronic structure, one can ascertain the effects at the intramolecular and intermolecular level and derive trends and correlations in isoelectronic series of molecules such as the nine-member N-(chlorophenyl)pyridinecarboxamide NxxCl series (Scheme 1) described herein. This series is used for comparisons with related benzamide isomer grids.50−54 These benzamides are readily synthesized from the three p-/m-/o-pyridinoyl chlorides and three p-/m-/o-aminochlorobenzene isomers using standard synthetic and purification procedures.50,51 They are chemical analogues of the related N-(fluorophenyl)pyridinecarboxamides (NxxF).50 Nine NxxCl single crystal structures (Figures 1–6) and their conformational analyses and physicochemical properties are described (Figures 7–13). Together, these are analyzed and compared to highlight correlations with crystal properties and molecular charge densities and also to make notable comparisons with related series of isomers.50−53
Scheme 1. The NxxCl Series of Molecules with Nx Representing the C5H4NC=O (Pyridinoyl) and xCl the −HNC6H4Cl (Aminochlorobenzene) Moieties (x = Para-/Meta-/Ortho-substitutions).

The numbering scheme as used in the interplanar calculations (non-H atoms only) and Figures 1–6 is shown.
Figure 1.
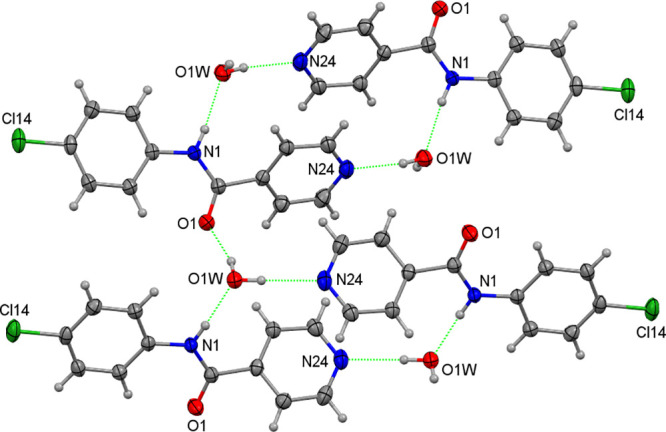
A view of [NppCl·H2O]2 linked by an O1W–H2W···O1 interaction.
Figure 6.

Molecular structure and intramolecular hydrogen bonding in NooCl.
Figure 7.

ATR-IR spectra of the NxpCl triad.
Figure 13.
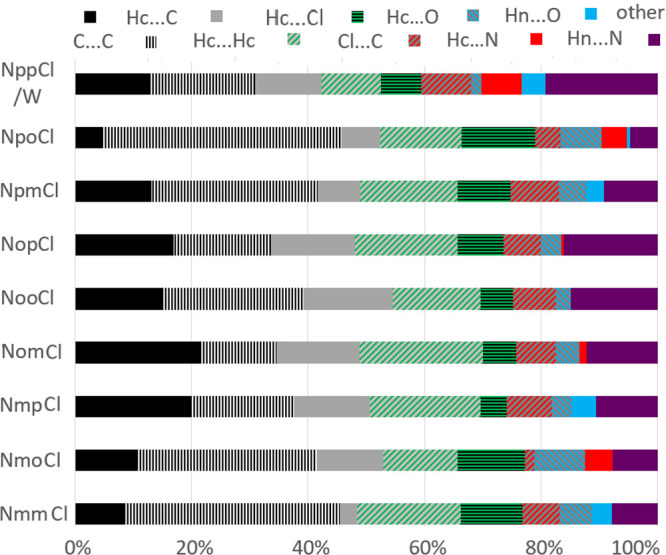
Contact proportions in the nine NxxCl isomer crystals.
Experimental Section
Materials, Methods, and Equipment
All chemicals, materials, vendors, and spectroscopic and crystallographic methods together with computational programs and equipment are as reported previously.50−53 Chemicals and silica (Davisil) were used as purchased from Sigma Aldrich; TLC alumina and silica plates were from Fluka. Melting points were measured using a Stuart Scientific SMP40 automated melting point apparatus. IR spectroscopy was recorded using a Perkin Elmer Spectrum GX FTIR spectrometer by the attenuated total reflection (ATR) method: spectral bands are quoted in cm–1. NMR spectroscopy was performed on a Bruker BioSpin UltraShield NMR spectrometer (293 ± 1 K) at 400 or 600 MHz for 1H and 100.62 MHz for 13C resonance. The 1H spectra were recorded in CDCl3 and DMSO-d6 with the 13C spectra analyzed in CDCl3. The NMR chemical shift values (δ) are in ppm referenced to TMS, and coupling constants (J) are quoted in Hz.
Single crystal X-ray data collections for the nine NxxCl crystal structures (Scheme 1) together with data reduction, structure solution, and refinements60−62 are as described for the previously reported 3 × 3 isomer grids56,59 and are fully detailed in the Supporting Information (Table S1). Selected crystallographic and structural information is analyzed and compiled with pertinent structural details provided in the main paper in Tables 1 and 2. Molecular structures and hydrogen bonding diagrams (Figures 1–6) are depicted with displacement ellipsoids drawn at the 30% probability level.63,64 The computational calculations65−67 were performed as described previously.56 Optimizations and conformational analyses in the gas phase were performed using the DFT method [B3LYP/6-311++G(d,p)].66,67 All calculations were performed using Gaussian0965 for Linux/Unix operating on an SGI Altix ICE 8200EX high-performance computing system at the ICHEC (Galway, Ireland). The gas phase data are presented in a diagram as a 3 × 3 grid to highlight trends in the position of the substituent and displayed from the pp to oo (Figure 12; in Supporting Information Section II as enlarged diagrams).
Table 1. Selected Crystallographic Data for NxxCl (Full Details Available; Table S1, Supporting Information).
| structure | crystal system; space group | Z′ | volume (Å3) | R, wR2R-factors,a GoF |
|---|---|---|---|---|
| NppCl·H2O | orthorhombic; Pbca | 1 | 2344.89(8) | 0.042, 0.108, 1.03 |
| NpmCl | monoclinic; P21/n | 1 | 1090.45(4) | 0.035, 0.102, 1.07 |
| NpoCl | monoclinic; Cc | 1 | 1108.06(11) | 0.026, 0.067, 1.10 |
| NmpCl | monoclinic; P21 | 1 | 531.27(3) | 0.043, 0.114, 1.03 |
| NmmCl | monoclinic; P21/n | 1 | 1068.45(4) | 0.038, 0.115, 1.11 |
| NmoCl | monoclinic; P21/c | 1 | 1078.89(9) | 0.066, 0.149, 1.08 |
| NopCl | triclinic; P1̅ | 1 | 543.73(3) | 0.038, 0.105, 1.05 |
| NomCl | triclinic; P1̅ | 1 | 532.00(5) | 0.038. 0.124, 1.08 |
| NooCl | orthorhombic; Pbca | 1 | 2226.2(11) | 0.051, 0.120, 0.89 |
R-factor definitions as R[F2 > 2σ(F2)], wR(F2).60
Table 2. Salient NxxCl Structural Features (Interplanar Angles, Distances, and Interactions in Å or °)a.
| structure | C6/C5N (°) | C6/amide (°) | C5N/amide (°) | N···N/Oc (Å) | primary H bonds |
|---|---|---|---|---|---|
| NppCl·H2O | 47.68(5) | 7.58(7) | 40.43(5) | 2.831(2)b | hydrate packing (2 × N···O/O···O) |
| 2.838(3)b | |||||
| 2.903(2)b | |||||
| NpmCl | 1.52(9) | 17.96(6) | 18.23(7) | 3.1373(17) | amide···pyridine |
| NpoCl | 83.24(7) | 69.59(8) | 27.43(11) | 2.797(2)c | amide···amide |
| NmpCl | 7.65(14) | 32.11(10) | 32.02(9) | 3.079(3) | amide···pyridine |
| NmmCl | 56.53(4) | 30.44(4) | 27.03(5) | 3.0842(13) | amide···pyridine |
| NmoCl | 16.30(17) | 38.87(17) | 25.54(15) | 2.884(3)c | amide···amide |
| NopCl | 2.86(7) | 1.26(7) | 1.75(7) | 2.6631(16) | intra as (N–H···N) |
| NomCl | 1.07(6) | 7.86(5) | 6.91(5) | 2.6536(13) | intra as (N–H···N) |
| NooCl | 8.6(2) | 9.7(2) | 1.2(2) | 2.624(4) | intra as (N–H···N) |
C6 is the (C11, ..., C16) benzene plane, C5N is the (C21, ..., C26) pyridine ring plane, and the amide is represented by the five atom C21–C1(=O1)N1–C11 plane (Scheme 1) and with reference to Figures 1–6.
NppCl monohydrate structure with N1···O1W, O1W···O1, and O1W···N1 hydrogen bonding.
Represents N···O (amide···amide) with the intramolecular N···N interactions underlined.
Figure 12.
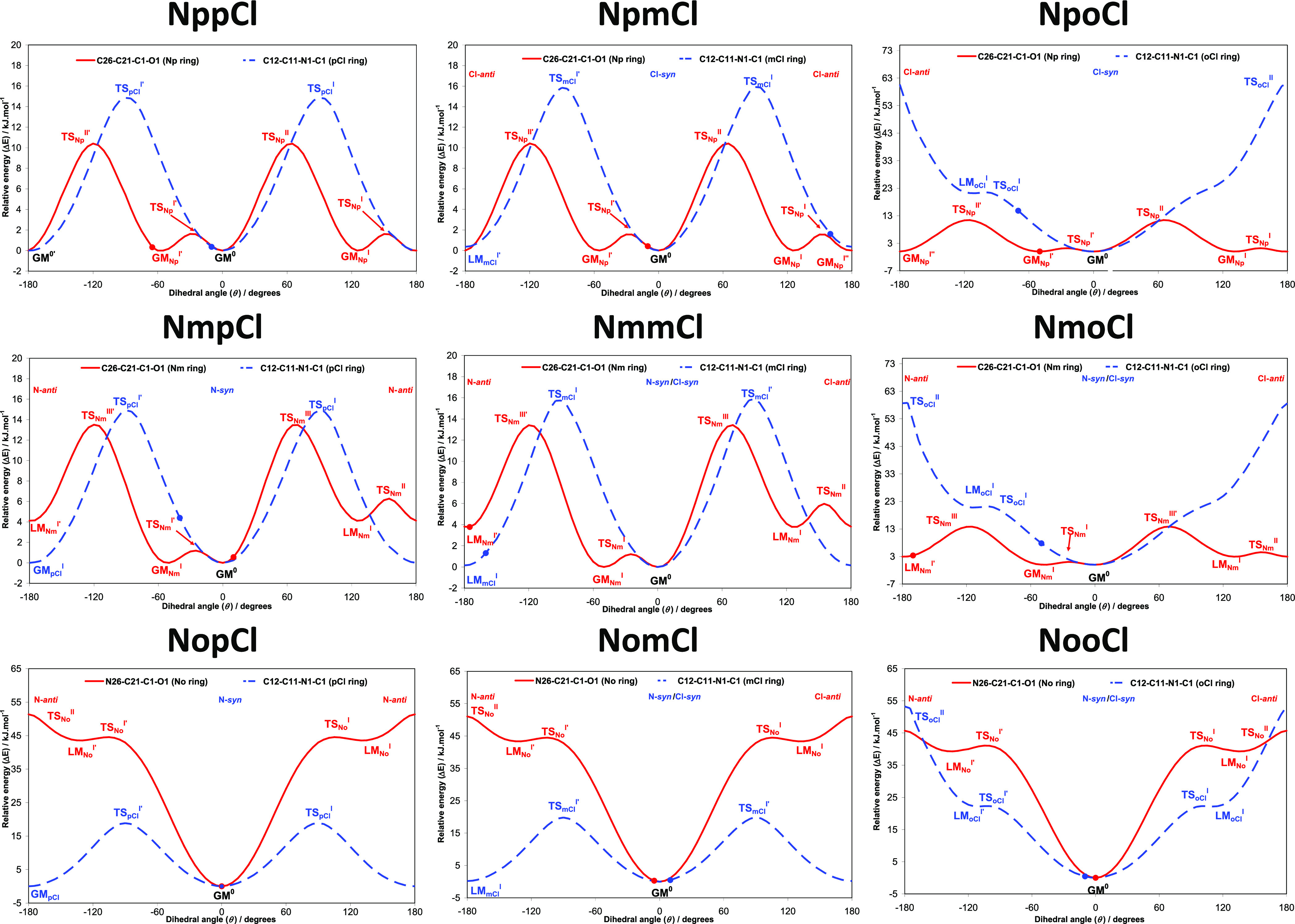
The potential energy surface (PES) conformational analysis for the NxxCl isomers optimized in the gas phase: the equivalent solid-state angle is depicted by (·). Transition states (TS) and global minima (GM) are indicated and labeled. Enlarged high-resolution figures are provided in the Supporting Information.
The average NxxCl molecular volume (i.e., cell volume (Å3)/Z) is 273 Å3, discounting the NppCl monohydrate. The largest molecular volumes are for NooCl (278 Å3) and NpoCl (277 Å3). The smallest volumes are for the NmpCl and NomCl structures (both 266 Å3). The calculation for NppCl·H2O is at ∼255 Å3 per NppCl, taking into account the volume of the tightly bound monohydrate molecule (as ∼38 Å3).63
Methods68−74
The electrostatic energy Eelec was computed from the charge density models transferred from the ELMAM2 database of multipolar atoms68 using the MoProSuite software.69 The structures as obtained from SHELX refinement were modified by elongation of the N–H and C–H bonds to standard distances retrieved from neutron diffraction studies.70 The molecules were rendered electrically neutral after charge density transfer by applying a uniform valence population shift to all atoms. The electrostatic energy between interacting molecules was obtained by the summation over pairs of multipolar charged atoms belonging to each entity. The lattice electrostatic energy was computed with the VMoPro module in real space. The energy was summed over successive parallelepiped shells surrounding the unit cell. The summations were carried over the [−9a,9a] × [−9b,9b] × [−9c,9c] space around the molecule containing 193 unit cells where convergence is largely achieved.
The total energy was computed with the CrystalExplorer19 software71 between the asymmetric unit and a cluster of surrounding molecules within a distance of 3.80 Å. The energy components calculated within this procedure are electrostatic, polarization, dispersion, exchange-repulsion, and finally the total interaction energy. These energy calculations were performed at the B3LYP/6-31G** level of theory.66,67 The structures used were the same as for the electrostatic energy calculation on the multipolar model. Diagrams are included in the main paper text as Figures 8–11 and in the Supporting Information (Section IV pp 56–68) as Figures S01–S06.
Figure 8.
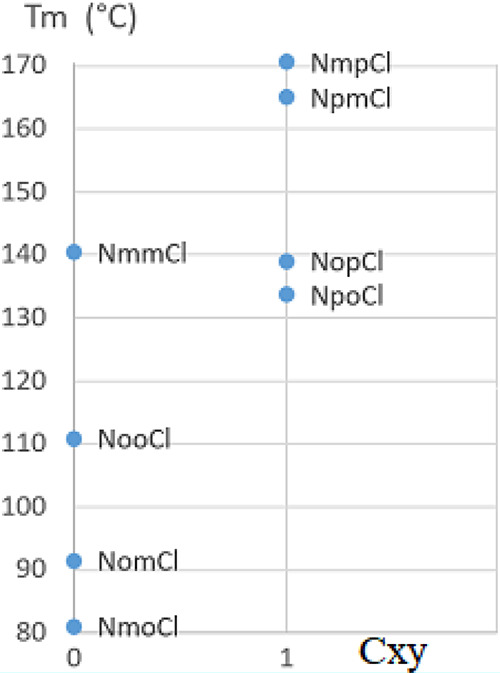
Melting point in the isomers classified according to descriptor CXY derived from Carnelley’s rule: CXY = 1 when x or y is p (para), CXY = 0 (remainder).
Figure 11.
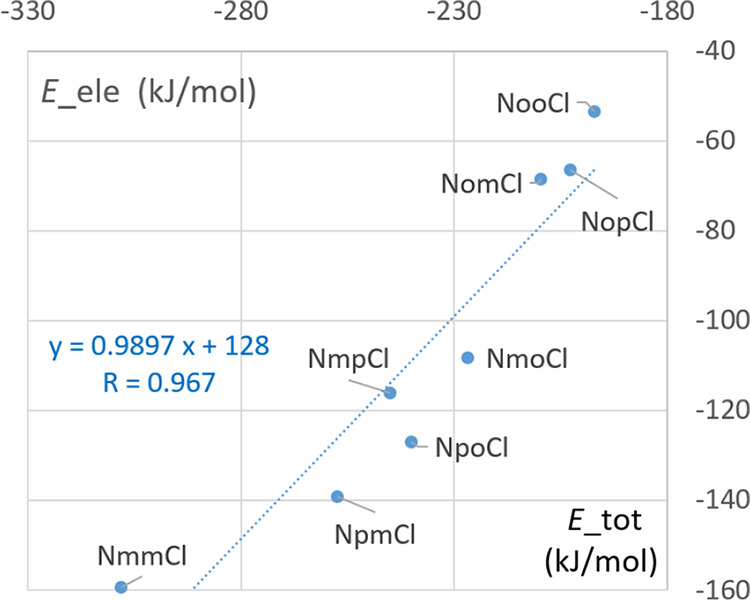
Scatterplot of total and electrostatic energy from CrystalExplorer.71
Results and Discussion
NxxCl Crystal Structures
The nine N-(chlorophenyl)pyridinecarboxamide crystal structures (NxxCl) are grouped in triads for structural comparisons with pertinent structural data presented in Tables 1 and 2. Comparisons are made with the Clxx series56 (as their amide-bridge reversed isomers) together with the related NxxF,50 (methyl) NxxM,51 and NxxBr analogues.54
The NpxCl Triad
NppCl crystallizes as a monohydrate with the amide N–H donor, O=C, and Npyridine acceptor groups engaged in hydrogen bonding interactions with the water molecule O1W. In the crystal structure, two NppCl·H2O aggregate through (amideN1–H1···O1W–H2W···N24pyridine) hydrogen bonds and form R44(18) hydrogen bonded rings about inversion centers (Figure 1). The (NppCl·H2O)2 units are linked by 2 × (O1W–H1W···O1=C1) and 2 × (C1=O1···H1W–O1W) hydrogen bonds per aggregate. These four strong intermolecular interactions form a 2D sheet that is effectively ∼21 Å wide. Overall, 2D sheets interlock into a 3D structure by using 2 × (C13–H13···O1=C1) and 2 × (C1=O1···H13–C13) weak H-bonds per aggregate. This hydrate aggregation is similar to related benzamide hydrates with all strong hydrogen bonding donors and acceptors used (e.g., in Clpm·2H2O75 and Clmm·H2O(56)). The closest contacts with the para-chlorine Cl14 atom involve three H atoms (H23, H25, and H26) on symmetry related molecules but with all of the H···Cl14 distances larger than 3.0 Å.
NpmCl and NpoCl Structures: Isomorphous Behavior49,76−83
NpmCl is isomorphous with NpmM(51) and NpmBr(54) in the monoclinic space group P21/n but is not isomorphous with NpmF(50) (see below). The hydrogen bonded N–H···N chains in NpmCl contrast with conventional N–H···O=C (amide···amide) interactions in NpoCl. Furthermore, two C–H···O=C contacts are noted in NpmCl in the absence of N–H···O=C interactions (Figure 2). The amide···pyridine N–H···Npyr hydrogen bonded chains are augmented by two weaker C–H···Npyr interactions. There are up to six Cl···H–C close contacts at dHCl < 3.6 Å with symmetry related molecules, though the shortest distance, H15···Cl13, is larger than 3.2 Å. In NpoCl, amide···amide hydrogen bonding as 1D chains along the c-axis direction is the primary interaction mode (Figure 3). Chains are weakly linked by C–H···Npyridine contacts. NpoCl is isomorphous with both NpoM(51) and NpoBr(54) in space group Cc but differs slightly from the NpoF and NpmF structures where N–H···N interactions dominate. However, both NpxF structures also crystallize in space group Cc and the series of structures can be considered as being on the continuum of isomorphic behavior.49,50,52 In NpoCl, the closest contacts between the chlorine Cl12 and H atoms involve H13 on a symmetry related molecule (though with H13···Cl12 > 3.1 Å). Therefore, in summary, both NpmCl and NpoCl exhibit an isomorphous behavior with their methylated (M)51 and brominated (Br)54 congeners but not with their fluorinated (F) analogues.50
Figure 2.
A view of the C–H···O1=C1 interactions in NpmCl.
Figure 3.
Crystallographic autostereogram of the 1D amide···amide chains along the c axis in NpoCl (atoms drawn as van der Waals spheres).
The NmxCl Triad
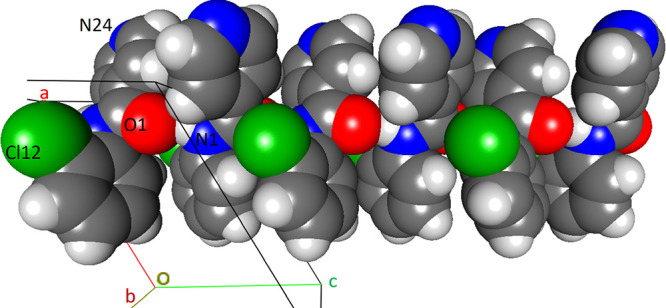
The NmxCl triad structures are not isomorphous with any of their NmxF(50) and NmxM(51) congeners, although there is an isostructural relationship between NmmCl and NmmF. NmpCl aggregates by zig-zagamideN–H···Npyridine chains of interactions along the b-axis direction in the monoclinic space group P21 and forms a 2D herringbone structure ∼16.5 Å wide (Figure 4). Chains are linked by C–H···O=C interactions and form a rumpled sheet. Short C22–H22···C2227,29 interactions form relays of contacts in tandem with amideN–H···Npyridine. The Cl14 atoms are not involved in any strong hydrogen or halogen bonding and are positioned in the lattice while involved in multiple aromatic H atom contacts. The closest contact involves the Cl14 and H25 atoms on symmetry related molecules (with H25···Cl14 at ∼3.0 Å).
Figure 4.
Crystallographic autostereogram of the amideN–H···Npyridinezig-zag chains in NmpCl.
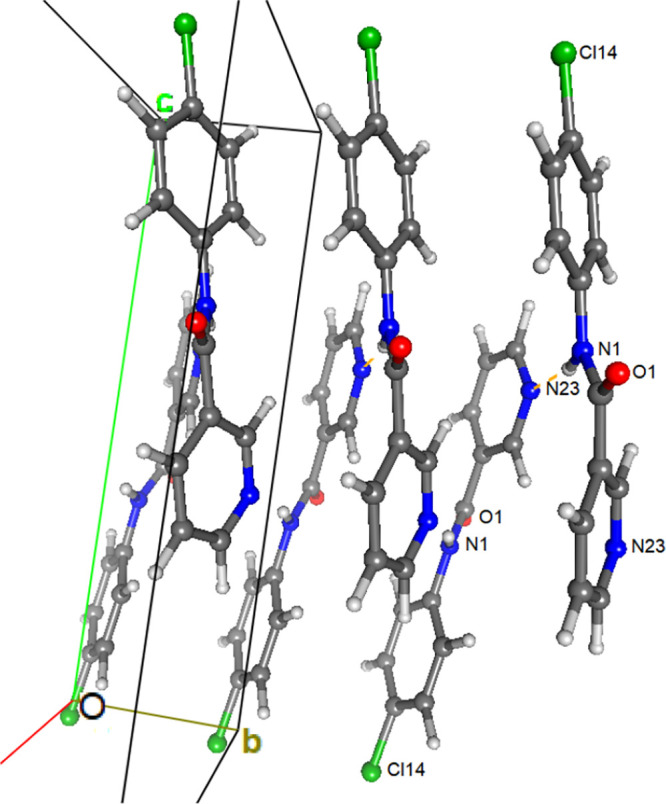
NmmCl with 1D zig-zag N–H···N chains is (at least) isostructural with NmmF in space group P21/n. Aggregation is assisted by the alignment of 1D chains via C14–H14···O1 interactions and formation of 2D sheets (Figure 5). In doing so, series of tetrameric units are generated in the NmmCl crystal structure and with C–H···π(arene) interactions generate ruffled sheets. In contrast, NmoCl is isomorphous with NmoBr(84) (TICDOZ01)49 with N–H···O=C intermolecular interactions along the b-axis direction and short intramolecular interactions between the ortho-Cl12 and the N–H group. In addition, there are Cl12···C14 contacts between the ortho-Cl12 and symmetry related chlorinated aromatic rings.
Figure 5.
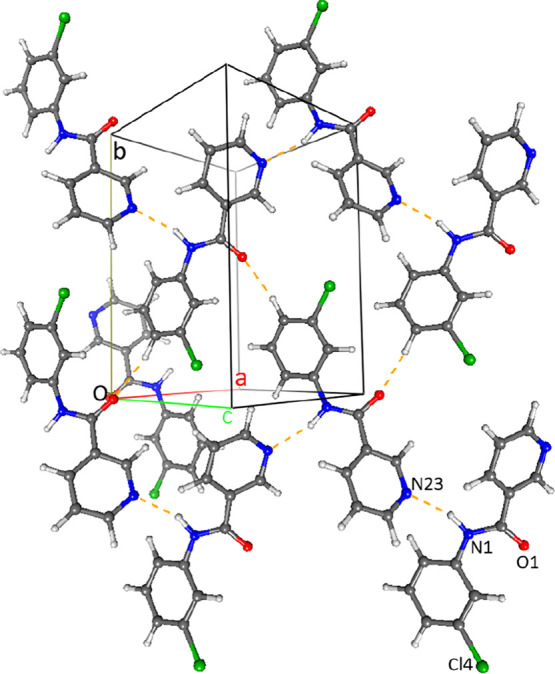
Crystallographic autostereogram showing the 1D zig-zag N–H···N chains as linked by C–H···O interactions in NmmCl.
The NoxCl Triad: Relatively Planar Molecules with Aromatic Stacking32
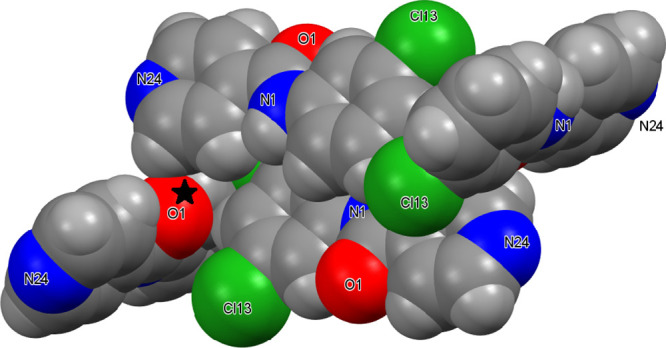
All three NoxCl have their benzene and pyridine rings aligned close to co-planarity (Table 2); this is largely influenced by two intramolecular N–H···N and C–H···O interactions. Both NopCl (reported previously as GEPQIC)85 and NooCl are isomorphous with the NopBr and NooBr congener structures,54 respectively.83 The NopCl crystal structure with an intramolecular N1–H1···N22pyridine contact has aromatic stacking and long-distance C–H···O/Cl interactions (dHCl = 2.88 Å) resulting in 2D sheet formation. Likewise, NomCl has two intramolecular H-bonds per molecule: the short N1–H1···N22 and a weaker C–H···O contact. Consequently, there are no strong intermolecular hydrogen bonds but only two C–H···O and one C–H···Cl (dHCl ∼2.90 Å) weak H-bonds; the closest C···C aromatic stacking distance is 3.4873(17) Å.32NooCl, isomorphous with NooBr,54 is relatively planar due to the intramolecular Cl22···H1(N1)···N22 bifurcated hydrogen bonding arrangement (Figure 6) and is similar in structure to NooF,50NooM,51 and Cl-NooM (a side-product from the NooM synthesis).51 The intermolecular interactions are typically weak and comprise C–H···O and C–H···π(arene) contacts (with C···C aromatic stacking distances ≥3.60 Å). Overall, the relatively planar NoxCl triad compares well with the NoxM, NoxF, and Cl-NoxM series, and in each of these series, it is usually the para-derivative that has its arene rings twisted most from co-planarity.50,51
Isomorphous Relationships: Summary and Analysis of NxxCl and NxxBr(18,49,54,83–87)
Isomorphous relationships between structures in the 3 × 3 isomer grids show an overlap between five NxxCl isomers and their NxxBr analogues (as the NpmCl/Br, NpoCl/Br, NmoCl/Br,84NopCl85/Br, and NooCl/Br pairs). This correlates well with what has been noted with five of the Clxx/Brxx amide-bridge reversed analogues (see Table 3).54,56 Furthermore, for the ’pm’ or ’po’ sets of crystal structures, the methylated analogues NpmM and NpoM are isomorphous with the Cl/Br pairs and further extend the structural series overlap.54
Table 3. Isomorphous Relationships between NxxCl (this work) and NxxBr:54,84 Comparisons with the Amide-Bridge Reversed Clxx(56) and Brxx(54,83)a.
|
NxxCl and NxxBr isomer grids |
Clxx and Brxx isomer grids |
||||
|---|---|---|---|---|---|
| NxxCl | space group | NxxBr(54) | Clxx(56) | space group | Brxx(54) |
| NppCl·H2O | Pbca ≠ P21 | NppBr | Clpp | P21/c | Brpp |
| NpmCl | P21/n | NpmBr | Clmp (Z′ = 4) | P1̅ ≠ P1̅ | Brmp (Z′ = 2) |
| NpoCl | Cc | NpoBr | Clop | Pbca | Brop |
| NmpCl | P21 ≠ C2/c | NmpBr | Clpm | P1̅ ≠ C2/c | Brpm |
| NmmCl | P21/n ≠ P1̅ | NmmBr | Clmm·H2O | P21/c | Brmm·H2O |
| NmoCl | P21/c –P21/a | NmoBr(84) | Clom | C2/c | Brom |
| NopCl(85) | P1̅ | NopBr | Clpo | C2/c ≠ P1̅ | Brpo |
| NomCl | P1̅ ≠ C2/c | NomBr | Clmo | P1̅ ≠ P21/c | Brmo |
| NooCl | Pbca | NooBr | Cloo | C2/c | Broo |
In Table 3, 10 of the 18 structural pairs from the Clxx/Brxx and NxxCl/NxxBr isomer grids are isomorphous.82,83 These results support an extensive Cambridge Structural Database (CSD) study by Mukherjee and Desiraju18,49 where they noted a significant degree of similarity between pairs of structures presenting C–X bonds (X = Cl or Br).18 Such pairs are observed to adopt the same space group, number of molecules in the unit cell, and reduced unit cell parameters (within 1 Å). Using this, our study aimed to compare the Clxx grid56 with the NxxCl series and make structural comparisons with the Brxx and NxxBr analogues.54
The extent of isomorphous behavior between pairs of structures in the Cl/Br series is much greater than that noted for the Me/F or F/Cl analogous pairs (NmmF and NmmCl are isostructural in P21/n).50,51,54 Moreover, there are examples where three structures exhibit an isomorphous behavior; e.g., the NpmM, NpmCl, and NpmBr triad is isomorphous in the monoclinic space group P21/n. Furthermore, NpoM, NpoCl, and NpoBr are isomorphous in space group Cc and are aggregating by amide···amide interactions.50,51,54 However, NpoF (and NpmF) differs in structure using amide···pyridine interactions, though also crystallizing in space group Cc.50 Of note are the isostructural Clmp (Z′ = 4) and Brmp (Z′ = 2) with two sets of similar unit cell axes in Clmp (a, b) and Brmp (b, c) and with the third axis (a) halved in Brmp (Supporting Information Table S4b). This represents the extent of overlap within these classes of functionalized benzamides.52−54 There is an extensive ’pp’ series with several closely related crystal structures.52,56 There are, however, no pairs of isomorphous NxxCl/Clxx structures involving amide-bridge swapped isomers,88,89e.g., NomCl/Clmo, as noted in the NmmM and Mmm crystal structures.51,53 Ojala and co-workers have commented on bridge-flipped isomers in an extensive series of benzylideneanilines and phenylhydrazones.88,89 In our series, the amide group N–H dominates as a pivot in the crystal structures and together with the Npyridine and halogen X reduces the possibility of bridge-flipping or amide-bridge swapping.50,51,54
The general trend for Cl/Br pairs of isomorphous structures is interesting,18,49 and in some cases, a methyl analogue (NxxM)51 is isomorphous with the Cl/Br pairs (NmoCl/NmoBr;84NmoM/NmoBr86 with NmoBr polymorphs84,86,87). However, it is also notable that the fluorinated Fxx(34,52)/NxxF50 do not tend to form isomorphous relationships with Me, F, or Cl to the same extent as the Clxx(56)/Brxx54 and NxxClthis work/NxxBr54 groups of structures.
Structural Aspects of Organic Chlorine
Fluorine has been extensively analyzed in terms of intermolecular interactions and contacts by using the CSD and other analytical methods.33−36,49,90,91 Chlorine contrasts with fluorine as it is often present as a chlorinated solvent such as a CH2Cl2 or CHCl3 solvate in crystal structures.
Analysis of C–H···Cl intermolecular interactions in molecular crystals as a function of the hybridization of the donor atom and acceptor atoms shows the C(sp2)–H···Cl–C(sp2) to be prevalent. Furthermore, upon cone correction, this type of C(sp2)-H···Cl intermolecular interaction exhibits a clear preference for angularity of ∼120° with the area approaching linearity also dominant. Analysis of N–H···Cl–C and O–H···Cl–C intermolecular interactions shows that they are less common than C–H···Cl–C interactions based on a statistical analysis as noted by the decrease in observed ’hits’.49 This was also noted in the analysis of several families of halogenated organic molecules by the contact enrichment ratio91 that confirmed that organic halogen atoms prefer to interact with the lowly charged HC hydrogen atoms (bound to a carbon atom) rather than with HO atoms (bound to O). On the other hand, O and N atoms that are stronger H-bond acceptors tend to form H-bonds with the more polar HN and HO hydrogen atoms.
The methodology described could be extended to investigate intermolecular interactions involving various other halogenated organic molecules containing bromine or iodine atoms. This is expected to provide a deeper understanding into the nature of such contacts and into the characteristics of interactions as a whole. Such analyses should be viewed in tandem with the structural similarity approach used by Mukherjee and Desiraju in their in-depth CSD study.18,49
Infrared Analysis
The ATR-IR spectra of all NxxCl derivatives can be correlated with their solid-state structures. For example, in comparison of the NxpCl spectra (Figure 7; Supporting Information p 55, ATR-IR diagram), three distinct spectra are observed as would be expected from calculated results. Indeed, NppCl·H2O forms N–H···O–H···O=C and O–H···Npyridine intermolecular hydrogen bonds involving NppCl and the water molecule (Scheme 2). Its spectrum contains a band at 3470 cm–1 indicating the water of crystallization (in the crystal structure as NppCl·H2O).75
Scheme 2. Intermolecular Hydrogen Bonding in NppCl·H2O and NmpCl and Intramolecular Hydrogen Bonding in NopCl.
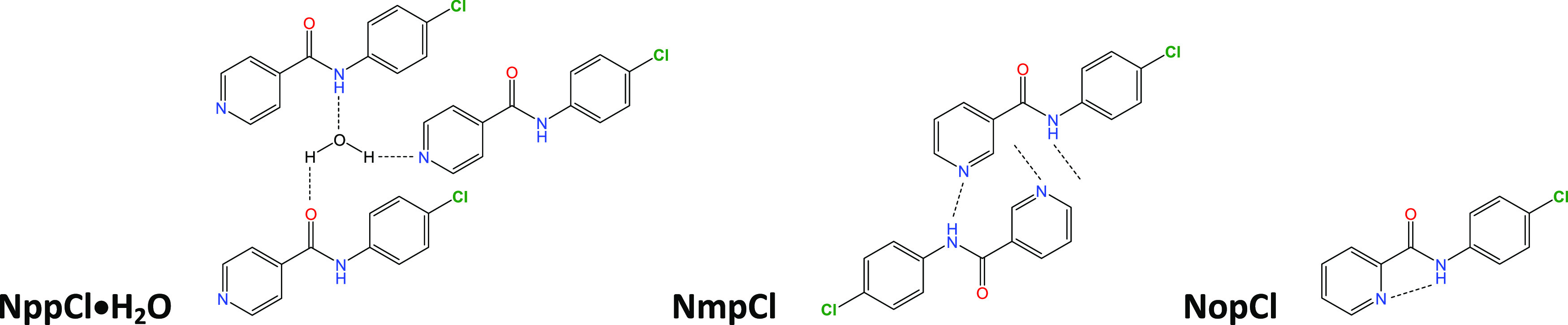
As expected, the NppCl molecule has two potentially strong acceptor groups, Npyridine and amide C=O, with one donor N–H group. It interacts with water having two potential O–H donors and either one or two acceptors as the O atom electron lone pairs. This effectively balances the total number of donors/acceptors in the crystal structure of NppCl·H2O.
Two distinct IR bands at 3231 and 3178 cm–1 in the NmpCl spectrum reveal an intermolecular hydrogen bond. This is as expected from the crystal structure results for the catemeric N–H···Npyridine chains arising in NmpCl (Figure 4). This is further highlighted for the NoxCl triad due to the presence of an intramolecular N–H···Npyridine interaction. In the NopCl spectrum, the very sharp strong band at 3330 cm–1 indicates that there is no strong intermolecular hydrogen bonding. For the NoxCl triad, the spectrum is indicative of an intramolecular hydrogen bond (Scheme 2) as noted in the NoxF(50) and NoxM(51) triads. Indeed, as shown in the Supporting Information (ATR-IR figure), there is a high degree of correlation between the interactions in the nine NxxCl crystal structures and their respective ATR-IR spectra. In structures with similar primary hydrogen bonding, e.g.,NoxCl, the ATR-IR spectra show similar features.
Melting Point Analysis92−99
Comparisons between the Clxx and NxxCl melting points are essential especially where there are structural relationships between the two series of isomers (Table 4). In previous work on related systems (NxxF, NxxM, Fxx, and Mxx),50−53 it has been shown that there is a general adherence to Carnelley’s rule that relates higher molecular symmetry and increased melting points.50−54,56
Table 4. Melting Point Ranges (°C) of the NxxCl (this work) and Clxx Isomers56,a.
| NxxCl | NpxCl | NmxCl | NoxCl |
|---|---|---|---|
| NxpCl | 139.0–140.0 (W) | 168.3–172.9 | 138.0–140.0 |
| NxmCl | 164.0–166.0 | 139.2–141.6 | 90.8–92.1 |
| NxoCl | 132.7–134.7 | 80.0–81.9 | 110.1–111.5 |
| Clxx | Clxp | Clxm | Clxo |
|---|---|---|---|
| Clpx | 206.2–208.4 | 150.1–151.3 | 131.5–134.5 |
| Clmx | 185.4–187.2 | 112.4–113.9 (W) | 95.0–105.0 |
| Clox | 167.9–169.9 | 134.5–137.8 | 134.4–138.0 |
Monohydrates are marked as (W).
The Clxx series provides an illustration of Carnelley’s rule93−95,98 (Table 4).56 An empirical function based on substituent positions and the lattice electrostatic energy was introduced and allowed a multilinear fit of the melting temperatures yielding a correlation coefficient with experimental values larger than 95%. The correlation coefficient between the melting points of Clxy(56) and NxyCl series is 51% (with x, y indicating o-/m-/p-substitution). Given the high degree of correlation, this model can be further refined in series of related benzamides and its possible predictive behavior evaluated.
The NxxCl melting points have been measured in a similar fashion to previous measurements and also independently compared using a blind test. Of interest in Table 4 is that the average NxxCl melting point is 130 °C, and this is ∼20°C less than the corresponding amide-bridged reversed Clxx isomers.56 How does this difference in melting points arise for isomers that differ by so little (as amide-bridge reversed structures)? The highest melting points are for NmpCl (170 °C) and NpmCl (165 °C), and the lowest are for NmoCl (68 °C) and NomCl (91 °C). The NppCl crystal as a monohydrate is kept separate and recorded for the sake of completion. The observed trends are what would be expected from molecular symmetry based on Carnelley’s rule93 and similar to our related series.50−54
As seen previously in the Clxx series56 (average melting point of 148 °C), the effect of chlorine substitution (compared to fluorine or methyl) is to bestow an average higher melting point of 17 °C compared to Fxx (131 °C), which is 15 °C greater than Mxx (116 °C) (in a trend of Br ≈ Cl > F > Me).93−98 Overall, the Clxx,56Fxx,52 and Mxx(53) series have higher average melting points than their corresponding amide-bridge reversed NxxCl (130 °C), NxxF (117 °C),50 and NxxM (113 °C)51 isomer grids. One partial answer must lie in the presence of intramolecular Npyr···H–Namide hydrogen bonds in NoxCl structures. The equivalent but weaker Clpyr···H–Namide hydrogen bonds are not formed in the Clox structures. The average Tm is 114 °C for NoxCl and 123 K for Clox.56 Globally, these subsets of structures with intramolecular H-bonds have lower melting points than their NmxCl and Clmx counterparts that have the same molecular symmetry level (Table 4). The presence of the intramolecular H-bond results in weaker intermolecular interactions and electrostatic energy, and consequently, Tm is decreased, as discussed in the next paragraph. The rest of the answer must lie in intramolecular interactions and how the molecules pack in their respective crystal structures.
Melting Points and Electrostatic Energy
To relate the melting point temperatures (Tm) to energies, additional analyses were conducted to identify correlations. The NppCl·H2O crystal structure, which has a different chemical content, was not included in the analysis.
The Gibbs free energy of a system depends on the temperature T and the enthalpy (ΔH) and entropy (ΔS) variations: ΔG = ΔH – TΔS. The free energy of melting vanishes at the temperature Tm, and therefore,
| 1 |
According to eq 1, the melting point temperature is expected to increase when the enthalpy change ΔHm is large. The crystal enthalpy is closely related to the computed lattice energy (the mechanical energy to separate the molecules to infinity while keeping their crystalline electron distributions and their nonrelaxed geometry). The electrostatic component (Eelec) can be estimated directly using the multipolar atom model transferred from the ELMAM2 electron density database.68 The relationship between Tm and the lattice or electrostatic energy is investigated here.
To see some trends, Table 5 shows the correlation between the melting points and several energetic and molecular symmetry descriptors of the nonhydrated NxxCl crystals. The quantities Tm and −Eelec indeed show a small correlation (R = 37%) in Figure S01 (Supporting Information). The three NoxCl compounds, with the intramolecular N–H···N hydrogen bond, have the weakest Eelec values. In our previous study of Clxx isomers,56 the two properties showed a higher correlation of R = 0.47, and the compounds with the strongest electrostatic lattice energy tended to have the highest melting points.
Table 5. Correlation Coefficients between the Experimental Tm Values and Computed Propertiesa.
| Cxy (Carnelley’s rule) | 0.76 |
|---|---|
| –Eelec | 0.37 |
| –(Eelec-EHB) | 0.44 |
| –E_tot | 0.46 |
| –(E_tot-EHB) | 0.64 |
| T_fit(Eelec, CXY) | 0.807 |
| T_fit(Eelec-EHB, CXY) | 0.870 |
| T_fit(E_tot, CXY) | 0.887 |
| T_fit(E_tot-Ehb, CXY) | 0.873 |
| T_fit(E_tot, Ehb, CXY) | 0.888 |
Correlations of Tm with T_fit melting points fitted by multiple regression are shown.
In the Clxx isomer series, it was observed that EHB, the electrostatic energy between acceptor and donor atoms of the strongest hydrogen bond in the crystal, has an influence on the melting point. The Tm values were more correlated (R = 0.63) with the −(Eelec-EHB) values than by considering −Eelec exclusively. This suggested that contributions to ΔHm are rather due to the weaker intermolecular interactions, as the strongest hydrogen bonds might subsist in the molten phases. The (Eelec-EHB) quantity refers to the total electrostatic energy corrected by removing the strongest hydrogen bond contribution. In the NxxCl series as presented herein, this correlation R = 0.44 is more moderate but still stronger than R(Tm,-Eelec) = 0.37 (Table 5).
Entropy is another key factor that plays a significant role in the melting point temperature in eq 1. Hence, according to Carnelley’s rule,92,93 a molecule with a higher rotational symmetry is expected to show a smaller increase in entropy /ΔSm when the crystal melts and, consequently, an increased Tm temperature.
The para-substituted NxxCl compounds have a higher symmetry than the unsymmetrical ortho- and meta-substituted isomers. In Figure 8, the compounds with a para-substitution clearly show, on average, higher Tm values than the other isomers (105.9 vs 152.1 °C). To model Carnelley’s rule by accounting for its dependence on the substituent positions, the CXY descriptor was defined for the NxyCl isomers: CXY = 1 when one of the substitution positions is located as para; CXY = 0 when there is no para-position. The resulting correlation between Tm and CXY is 0.76.
A double linear regression to fit Tm against the Carnelley-derived CXY function and the lattice energy was also undertaken. This model accounts simultaneously for the enthalpic and the entropic contributions to the melting point Tm. The scatterplot of the experimental Tm and of the ones fitted from (CXY,Eelec-EHB) data shows a correlation of 0.87 (Figure 9), which is lower than the high value of R = 0.961 observed for the Clxx benzamide series.56 The same double regression using (CXY,Eelec) properties leads to a fit of lower quality at R = 0.81 (Figure S02 Supporting Information). As observed also for the Clxx series,56 when the Eelec and CXY properties are combined, taking into account the EHB energies of the strongest H-bond as a third variable does not significantly improve the linear fitting (Table 5).
Figure 9.
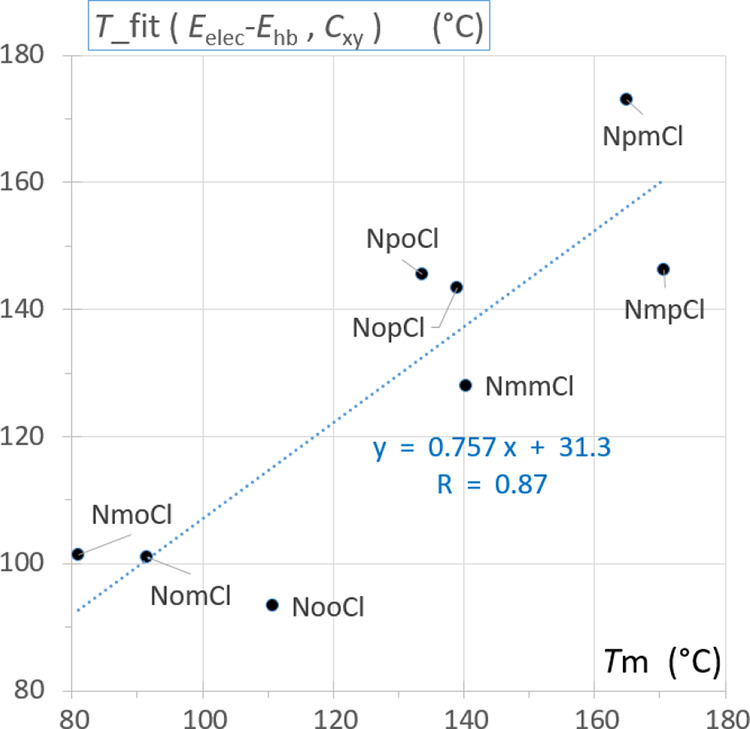
Double linear regression of the melting point Tm on the Carnelley molecule symmetry descriptor CXY and the Eelec-EHB value, the electrostatic lattice energy diminished by the strongest hydrogen-bond electrostatic energy.
The total lattice energy E_tot and its components have been computed with CrystalExplorer and are shown in Figure 10.71 The electrostatic energy E_ele as derived from CrystalExplorer71 and Eelec derived from the ELMAM268 electron density database show an excellent correlation (R = 0.967) (Figure 11), but the former values are on average 25% higher than the ELMAM2-derived ones (Figure S03).68 For the NxxCl series, the average electrostatic E_ele (from CrystalExplorer71) and dispersion E_disp values are −105 ± 38 and −257 ± 23 kJ/mol, which show that most of the lattice energy comes mostly from the dispersion component. This is related to the mostly hydrophobic character of the NxxCl molecules. The E_disp values show however low variations among the compounds, and as a result, the ranking of the E_tot values originates mostly from differences in E_ele values. This is confirmed by the scatterplot as depicted in Figure S03 (Supporting Information), which shows globally increasing E_tot values as E_ele is augmented. The E_tot values can be approximated from the E_ele ones by a well-defined linear equation that has a slope close to unity and has an intercept value of approximately 128 kJ/mol. The Tm melting points are much more correlated with the total energy −E_tot and −(E_tot-EHB) (R reaching 0.64) compared to the equivalent values issued from electrostatic energy −Eelec (Table 5; Figures S04 and S05; Supporting Information). The double linear fit of Tm on E_tot and CXY values yields a high R = 0.887 value (Figure S06; Supporting Information).
Figure 10.
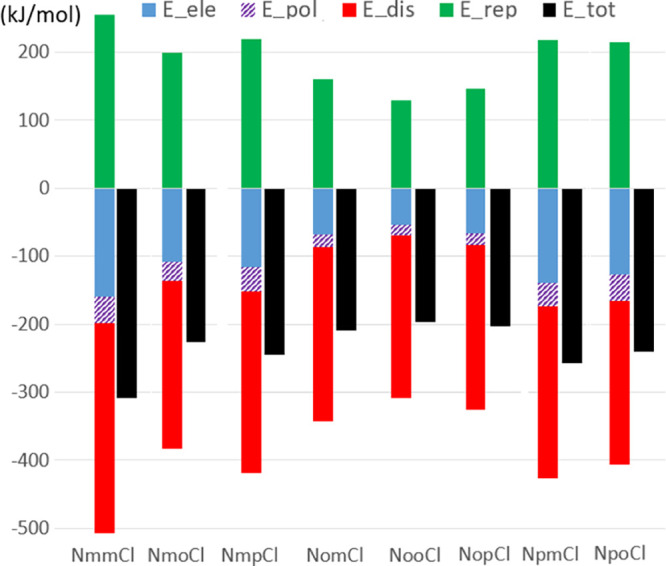
The components of the total lattice interaction energy of the NxxCl molecules computed on a cluster of surrounding molecules with CrystalExplorer using CE-B3LYP. B3LYP/6-31G(d,p) electron densities.71 In the summation of E_tot values, the electrostatic, polarization, dispersion, and repulsion components were scaled (coefficients 1.057, 0.74, 0.871, and 0.618) according to benchmarked energy models.71
Ab Initio Modeling Studies and Conformational Analysis of the NxxCl Isomer Grid
The molecular model geometries of the NxxCl isomers have been investigated and ab initio geometry optimizations undertaken using the DFT method (B3LYP/6-311++G(d,p)) with the Gaussian09 software.65 The three resulting optimized torsion angles α, β, and δ are tabulated in Table 6.
Table 6. Torsion Angles (°) of the Optimized NxxCl isomersa.
| α (°) | β (°) | δ (°) | |
|---|---|---|---|
| NppCl | 23.71 | 4.24 | 1.47 |
| NpmCl | 25.72 | 4.97 | 1.39 |
| NpoCl | 23.27 | 3.61 | 2.20 |
| NmpCl | 23.13 | 4.93 | 2.26 |
| NmmCl | 22.93 | 4.23 | 2.14 |
| NmoCl | 21.42 | 4.15 | 2.73 |
| NopCl | 0.00 | 0.00 | 0.00 |
| NomCl | 0.00 | 0.00 | 0.00 |
| NooCl | 0.00 | 0.00 | 0.00 |
The optimized geometries of the nine NxxCl isomers (Table 6) closely resemble the geometries of their equivalent isomer grids, i.e.,NxxF and NxxM.50,51 The NoxCl triad is completely planar with all torsion angles at 0.00°; the planarity of the NoxCl triad is assumed by the intramolecular N1–H1···N22 interaction.50,51 On the other hand, the NpxCl and NmxCl triads have torsion angles more or less deviating from planarity. On average, the α angle (para-/meta-pyridinyl ring, N-ring) is 23.36° (σ = 1.27°), whereas the β torsion angle (chlorophenyl ring, Cl-ring) is 4.36° (σ = 0.47°) and the δ torsion angle (amide linkage) is 2.03° (σ = 0.47°).
The conformational analysis was undertaken using the B3LYP/6–311++G(d,p) method and basis set.66,67 The PES conformational analysis diagrams (Figure 12) for the 3 × 3 NxxCl isomer grid reveal a significant similarity with their related NxxF and NxxM series.50,51 The N-ring and most of the Cl-ring PES profiles are similar with rotational barriers having comparable heights. However, the ortho-chlorophenyl ring (oCl-ring) shows a higher rotational barrier (53–60 kJ/mol) as compared to the oM-ring (35 kJ/mol) (NxoM triad)51 and the oF-ring (50 kJ/mol) (NxoF).50 This is rationalized by factoring in the larger atomic radius of chlorine compared to fluorine or the methyl group. Other differences are for NxoM/F/Cl triads and the effect of the ortho-methyl group on the shape and height of the β torsion angle C1–N1–C11–C12(Me) compared to both F and Cl that can be explained on both steric (size) and electronic grounds (intramolecular hydrogen bonding involving F and Cl).
Conformational analysis suggests that the N-syn conformation (Scheme 3) of the N-ring is more stable (by 3.9 kJ/mol) while the mCl-ring is just slightly stable (by 0.2 kJ/mol), making the Cl-anti conformation a possibility (Scheme 3). In addition, the N-anti conformation is plausible but is less probable in the gas phase. The orthooN-ring and oCl-ring can be stable only when they are positioned in the syn conformation. In summary, all modeling predictions are consistent with our previous studies on the NxxF and NxxM series.50,51
Scheme 3. Possible Conformations of NxxCl as Applied to the Ortho-/Meta-substitutions.

Comparisons of Calculated Models with Solid-State Structures
Differences between the modeled and solid-state torsion angles (N-ring and Cl-ring) are marked with a dot (·) on each of the NxxCl PES curves (Figure 12). The solid-state conformations of the NoxCl triad match those of the modeled structures with little or no torsion angle deviation. The NoxCl molecules are planar in both the solid state and as models, e.g.,NooCl (Figure 6). Therefore, the N1-H1···N22 intramolecular hydrogen bond and molecular planarity established in the optimized ab initio models is confirmed by the NoxCl solid-state structures. In NomCl, the Cl-syn conformation is preferred over Cl-anti; the Cl-anti would represent a disruption of the intramolecular hydrogen bonding, while the Cl-syn assists in the formation of C23–H23···Cl13 intermolecular interactions.
In the NpoCl crystal structure, the oCl-ring deviates by −70° from the optimized model. This deviation is necessary to allow for the formation of amide···amide (N1–H1···O1 = C1) intermolecular hydrogen bonds (Figure 3). The chlorine is positioned favorably, while the twisted oCl-ring allows for a closer aggregation of NpoCl molecules and hydrogen bond formation. A similar rotation of the oCl-ring arises in NmoCl where a less pronounced change in the Cl-ring torsion angle assists in N1–H1···O1 hydrogen bond formation. In tandem, the N-ring adopts the N-anti conformation that is essential for the formation of both C14–H14···Cl12 interactions and other important contacts that assist in structure aggregation.
Both aromatic rings in NmmCl are in the N-/Cl-anti conformation and opposite to the modeled gas phase structure (Figure 5). The flipping of the N-ring into an N-anti conformation was already noted in NmmF(50) and NmmM(51) as it is critical for the formation of N1–H1···N23 hydrogen bonds and zig-zag chains. It is unclear why the Cl-rings in both NmmCl and NpmCl isomers are in a slightly less stable geometry by adopting the plausible Cl-anti conformation (Figure 2); the chlorine atoms do not engage in any close contacts or halogen bonding but rather are situated in a relatively interaction-free position in the crystal structure. While the opposite Cl-syn conformation seems to be possible, there is no structural disorder observed (with a syn-/anti- swap), as noted for the analogous NomF isomer.50 There are no conformational differences between the optimized and solid-state structures for the NmpCl isomer, while for the NppCl isomer, this formalism is not applicable on symmetry grounds with both para-ring substitutions.
Contacts Analysis68−74,90,91
The intermolecular contact types on the Hirshfeld surface were analyzed in NxxCl using the MoProViewer software.72 The proportions of the main contacts in the nine NxxCl crystal structures are shown in Figure 13. Contacts between two chemical types (X,Y) are over-represented when their proportion Cxy is larger than that obtained by probability products of the chemical contents Sx and Sy on the Hirshfeld surface.72−74 Enrichment ratios are therefore obtained by dividing the actual proportion by the equiprobable reference value. The most enriched contacts are the strong N–H···N and N–H···O=C hydrogen bonding interactions with average enrichment ratios <E > larger than 4 (Table 7; Supporting Information Table S5). The standard deviations of EHnO and EHnN are large because for many crystals one of these two E values is zero, as only one of such hydrogen bond types occurs. The three NoxCl isomers have an intramolecular N–H···N hydrogen bond (not counted in the Hirshfeld statistics) but are devoid of an intermolecular one (Figure 6). The NmmCl, NmpCl, and NpmCl isomers display an intermolecular N–H···N hydrogen bond, whereas in both NmoCl and NpoCl isomers, an N–H···O=C hydrogen bond is observed (Figure 3). As the NxxCl molecules have two strong hydrogen bond acceptors with a deficit of strong donors (only one N–H group is available), weak hydrogen bonds are also favored as pyridineN···H–C and C=O···H–C. The enrichment ratios of ENHc and EOHc are −87% anti-correlated in the eight anhydrous crystal structure packings.
Table 7. Average X···Y Contact Enrichment Ratios between the Different Chemical Types in the Eight Nonhydrated NxxCl Crystal Structuresa.
| chem. | C | HC | Cl | N | HN | O |
|---|---|---|---|---|---|---|
| <surface> % | 35.5 | 37.6 | 13.8 | 5.2 | 2.9 | 5.1 |
| C | 1.2(5) | 1.0(4) | 0.9(3) | 1.0(6) | 0.8(7) | 0.6(3) |
| HC | 0.7(3) | 1.6(3) | 1.2(4) | 0.7(4) | 1.6(6) | |
| Cl | 0.6(7) | 0.1(2) | 0.0(1) | 0.4(4) | ||
| N | 0.9(11) | 4.3(55) | 0.1(2) | |||
| HN | 0.01(4) | 4.5(69) | ||||
| O | 0.1(3) |
The sample standard deviations are given between parentheses. The over-represented contacts are highlighted in bold characters. The second line shows the average chemical content on the Hirshfeld surface. The hydrophobic atoms C, HC, and Cl have been regrouped in the table. HC and HN refer to hydrogen atoms bound to carbon or nitrogen that are distinguished as they are chemically very different.
Among contacts between the C, HC, and Cl hydrophobic atoms, the weak Cl···HC hydrogen bonds are enriched. The C···C stacking contacts are also significantly enriched,32 as would be expected for heterocycles.69 All of the nine crystal structures have Cl···HC weak hydrogen bonds that are over-represented. This is easily understood as HC is the chemical type that has the largest representation at 37% on the Hirshfeld surface and as organic halogen atoms are favored contact partners for HC.90,91
The NxxCl isomers have mostly hydrophobic atoms (C, HC, and Cl) at their Hirshfeld surface, with a proportion reaching 86%. The amount of purely hydrophobic contacts within these atoms is remarkably stable at 77 ± 1.3% for the eight nonhydrated NxxCl isomers, and this corresponds to a global hydrophobic contacts enrichment of 1.03. In contrast, the polar···polar contacts only represent 3% of the Hirshfeld surface but are globally over-represented with E = 1.63. The cross polar/hydrophobic contacts make a total of 20% of the surface, are moderately under-represented at E = 0.84, and are mainly due to weak C–H···O and C–H···N hydrogen bonds.27,28
The NpoCl and NmmCl crystals are characterized by limited aromatic ring stacking as the two rings of the molecules have very different orientations (Table 2).32 Conversely, these two compounds have high amounts of weak C–H···π hydrogen bonds (EHcC = 1.56 and 1.42, respectively).29 In NpoCl, the two aromatic rings are nearly perpendicular (with C6/C5N = 83.24(7)° in Table 2), and this crystal packing consequently exhibits extensive C–H···π interactions.29 On the other hand, NmpCl and NomCl crystals show extensive aromatic ring stacking, and the two aromatic rings of each molecule are effectively parallel.32 In the NomCl packing, all the molecules are close to planarity and are essentially parallel [C6/C5N = 1.07(6)°], while in NmpCl, the aromatic rings have an orientation of C6/C5N = 7.65(14)°. Therefore, in summary, the C···C and C···HC enrichment values are −96.8% anti-correlated in the eight anhydrous NxxCl structures. Similarly, for contact proportions CXY, the anti-correlation of enrichments reaches −90.4%.
In broad terms, chlorine···chlorine contacts are generally avoided (with <E > = 0.6) but with the exception of NmpCl and NooCl. In these two crystal structures, the Cl···Cl contacts do not correspond to halogen bonds (where the σ-hole faces the electronegative crown) but are merely at the van der Waals contact level and result from the translation of molecules along a short unit cell axis.
To find some hints why the NxxF series50 shows poor isomorphism with the NxxCl series, the contact enrichments of F and Cl atoms were compared in Table S6 (Supporting Information). One major difference is that the NxxF series showed an average enrichment of only 1.3 for the F···HC weak hydrogen bonds when compared to 1.6 for the Cl···HC intermolecular interactions in NxxCl. Of further note is that Feng and co-workers have shown by rotational spectroscopy that in a competition between weak H-bonds in the CH2FCl·H2C=O adduct, the C–H···Cl intermolecular interaction is preferred to C–H···F.100
From a charge density topology point of view, the strengths of H···Cl hydrogen bonds appear to be also more important than that of the H···F type.101,102 Indeed, a starting degree of covalence appears at longer distances for Cl than for F.102 Accordingly, for a given internuclear distance H···halogen (halogen = F, Cl), the electron density at the bond critical point of H···Cl is larger than at that of H···F because the penetration of electron shells is more important in the case of Cl.101 Hence, due to the higher electronegativity of F compared to Cl, the H···F interaction tends to be more closed-shell in nature and a significant shared-shell character can be only present at very short H···F geometries. In addition, within the natural bond orbital theory (NBO),103 it has been established that the charge transfer from the acceptor (halogen) toward the X-H σ* molecular orbital can be considered as the signature of the X-H···(halogen) hydrogen bond strength. Again, due to the larger electronegativity of F, the charge transfer in hydrogen bonds will be less important with F than with Cl, leading to weaker interactions with the former acceptor. The interaction propensity of fluorine is different from that of chlorine and bromine, and this might explain the lower isomorphism of NxxF(50) with the NxxClthis work series compared to NxxBr.54
In conclusion, the nine NxxCl crystal structures fulfill the following contacts in order of priority: (i) one strong intra- or intermolecular hydrogen bond involving N–H with Npyridine or O=C; (ii) the remaining hydrogen acceptor atom interacts with HC atoms; (iii) weak C–Cl···HC hydrogen bonds are always formed; (iv) hydrophobic interactions between the HC and C atoms represent, on average, 50 ± 2% of the contact surface; and (v) aromatic ring stacking is favored when the two rings and their symmetry related partners have similar orientations,32 while weak C–H···π hydrogen bonding interactions occur mostly when the aromatic ring orientations differ significantly.27,29
In our previous studies with Clxx,56 we have noted the paucity of halogen bonding and notably Cl···Cl contacts in these amide-bridge reversed isomers (compared to NxxCl). This behavior is not too dissimilar to that observed for NxxCl. In related research, we have considered the competition between the F, O=C, N–H, and aromatic rings in terms of influencing interactions and aggregation.36 We have also speculated on the number of halogen atoms and type of halogen atom needed to tip the interactions from hydrogen bonding toward halogen bonding of the type C–Cl···O=C, C–Cl···Npyridine, and C–Cl···Cl–C. Indeed, research studies on the competition between interactions in crystal structure formation have been pursued with much interest recently in structural systematic studies of extensive series of molecules and in co-crystal formation.104−109 It has been noted that detailed studies are still rare.109 However, the ongoing structural systematic reports of series of closely related compounds together with both computational and database analyses should enable more in-depth analyses and predictive abilities in the near future.49,56,109,110
In research concerning the competition between hydrogen-bonding and halogen-bonding interactions in the crystal structures of pentachlorophenol (C6Cl5OH) and pentabromophenol (C6Br5OH), it has been pointed out that(C)O–H···O(H)–C is stronger than solitary C–Cl···Cl–C and C–Br···Br–C interactions, as observed from the topological properties of ρ(r) at the corresponding bond critical points (H···O > Br···Br > Cl···Cl).111 Similar conclusions were also raised with the electrophilic–nucleophilic interactions between the corresponding local charge concentration (CC) and charge depletion (CD) sites in the valence shell of atoms involved in the intermolecular interactions (H···O > Br···Br > Cl···Cl), here characterized by the topology of L(r) = −∇2ρ(r). In both crystal structures, neither O–H···Cl–C nor O–H···Br–C intermolecular hydrogen bonds are observed, indicating that O is a better acceptor in O–H···O(H)–C hydrogen bonds than Cl and Br in the former. On the other hand, halogen bonding of the C–Cl···O(H)–C and C–Br···O(H)–C type is not observed because involving O as an acceptor in (C)O–H···O(H)–C hydrogen bonds leads to stronger interactions. Consequently, if halogen bonding of the type C–Hal···O=C, C–Hal···Npyridine, or C–Hal···Hal-C should compete with H···O=C, H···Npyridine, and H···Hal-C hydrogen bonds, the best candidates should be found with the heavier halogens (Hal = Br, I); otherwise, the number of acceptors should be larger to permit Hal atoms to take the place of donors once the best donors have been used up.109,111 This is what recent structural research is beginning to show.49,109
Summary and Conclusions
The 3 × 3 isomer grid of NxxCl [N-chlorophenyl(pyridine)carboxamides] structures displays correlations with their NxxX (X = F, Br or M = Me) analogues. This is readily demonstrated with five isomorphous relationships between pairs of NxxCl and NxxBr structures.54,84,86 The NxxCl general behavior mimics the amide-bridge reversed Clxx series56 in its relationships with both methyl and bromo-substituted derivatives53,54 but not with the fluorine analogues.50,52 As such, there is a transition along the Me →F → Cl → Br series of structures where the increasing influence of the halogen atom is noted especially from F to Br. The impact on the structure and the increased structural overlap (isomorphous behavior) between the Cl and Br derivatives are noted here for NxxCl and in NxxBr.54 The matching of molecular crystal structures on the CSD readily demonstrates the value of systematic studies to the structural science community and the (bio)pharmaceutical sector in particular.49,110
N–H···N interactions dominate in comparison to N–H···O=C in the NxxCl series. This has been noted over several structural series between molecules where there is direct competition between O=C and Npyridine as acceptors of the N–H amide hydrogen bond donor group.50−54,56 The remaining O or N acceptor atom usually interacts with aromatic C–H groups. Weak C–H···Cl interactions are often present in the NxxCl structures but not in any predictable way. The planar NooCl structure is peculiar with its intramolecular Cl···HN···Npyridine synergistic combination. Aromatic ring interactions arise especially where symmetry favors stacking,32 and C–H···π interactions occur often where the aromatic plane orientations differ significantly.29 In models, the optimized geometries of the NxxCl isomers mostly resemble the geometries of related isomer grids, i.e.,NxxF and NxxM.50,51 They also mostly correspond with their crystal structures, and differences arise if there is a favorable interaction in the crystal structure that necessitates a change in NxxCl geometry. In doing so, the divergence between the models and solid-state geometry is more than compensated for in crystal packing forces and the resulting favorable lattice energy. At the solid/liquid boundary, the melting point of a member of the NxxCl series follows Carnelley’s rule on molecular symmetry but with distinct differences (typically lower average melting points) than noted for their Clxx analogues.56
The 18-member series of Brxx/NxxBr structures is in preparation for publication with additional contact analysis and for comparisons with NxxX (X = F,50 Clthis work or M = Me51) analogues together with their corresponding amide-bridge reversed isomers (Mxx,53Fxx,52Clxx56). The increasing role and influence of the heavier halogen in the crystal structures will be assessed in terms of the competition between hydrogen and halogen bonding interactions.109 Investigations on the physicochemical properties and trends of series of isomers of 72+ molecules (including polymorphs) will be available for future computational analysis.
Acknowledgments
This research was initially funded by the Programme for Research in Third Level Institutions (PRTLI) Cycle 4 (Ireland) and co-funded through the European Regional Development Fund (ERDF), part of the European Union Structural Funds Programme (ESF). J.F.G. thanks the School of Chemical Sciences, Dublin City University, for grants in aid of chemical and crystallographic research. The Irish Centre for High End Computing (ICHEC) is thanked for the support and assistance with the computational calculations (http://www.ichec.ie). P.M. thanks the T3 (PRTLI-IV) program for a studentship. We also thank Ms. Karoline Benedet (UNESC, Santa Catarina, Brazil) for technical assistance on the melting point measurements. J.F.G. together with E.A., E.E., B.G., and C.J. thanks the Université de Lorraine and Region Lorraine for a ″Chercheur d’Avenir″ grant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.cgd.2c00153.
Crystallography and melting point data (Section I); enlarged diagrams from Figure 12 (main paper) (Section II); ATR-IR and NMR data for the nine NxxCl isomers (NppCl·H2O to NooCl) (Section III); melting points and electrostatic energy diagrams (for ESI) (Section IV); and contact enrichment studies for NxxCl (Section V) (PDF)
Accession Codes
CCDC 2074347–2074355 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Gribble G. W. The diversity of naturally occurring organobromine compounds. Chem. Soc. Rev. 1999, 28, 335–346. 10.1039/a900201d. [DOI] [Google Scholar]
- Gribble G. W. The diversity of naturally produced organohalogens. Chemosphere 2003, 52, 289–297. 10.1016/S0045-6535(03)00207-8. [DOI] [PubMed] [Google Scholar]
- Gribble G. W. A recent survey of naturally occurring organohalogen compounds. Environ. Chem. 2015, 12, 396–405. 10.1071/EN15002. [DOI] [Google Scholar]
- Jin L. J.; Chen B. L. Natural Origins, Concentration Levels, and Formation Mechanisms of Organohalogens in the Environment. Prog. Chem. 2017, 29, 1093–1114. 10.7536/PC170563. [DOI] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Wang J.; Sánchez-Roselló M.; Aceña J. L.; del Pozo C.; Sorochinsky A. E.; Fustero S.; Soloshonok V. A.; Liu H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001-2011). Chem. Rev. 2014, 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Klencsàr B.; Sánchez C.; Balcaen L.; Todolì J.; Lynen F.; Vanhaecke F. Comparative evaluation of ICP sample introduction systems to be used in the metabolite profiling of chlorine-containing pharmaceuticals via HPLC-ICP-MS. J. Pharm. Biomed. Anal. 2018, 153, 133–144. 10.1016/j.jpba.2018.02.031. [DOI] [PubMed] [Google Scholar]
- Berger G.; Soubhye J.; Wintjens R.; Robeyns K.; Meyer F. Crystal packing and theoretical analysis of halogen- and hydrogen-bonded hydrazones from pharmaceuticals. evidence of type I and ii halogen bonds in extended chains of dichloromethane. Acta Crystallogr. 2018, B74, 618–627. 10.1107/S2052520618014221. [DOI] [Google Scholar]
- Banks R. E.; Smart B. E.; Tatlow J. C. Eds., Organofluorine chemistry: Principles and Commercial Applications, 1994, 11, Springer Science. [Google Scholar]
- Murphy C. D. New Frontiers in Biological halogenation. J. Appl. Microbiol. 2003, 94, 539–548. 10.1046/j.1365-2672.2003.01900.x. [DOI] [PubMed] [Google Scholar]
- Jeschke P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manage. Sci. 2010, 66, 10–27. 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]
- Jeschke P. Latest generation of halogen-containing pesticides. Pest Manage. Sci. 2017, 73, 1053–1066. 10.1002/ps.4540. [DOI] [PubMed] [Google Scholar]
- Latham J.; Brandenburger E.; Shepherd S. A.; Menon B. R. K.; Micklefield J. Development of halogenase enzymes for use in synthesis. Chem. Rev. 2018, 118, 232–269. 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]
- Mocilac P.; Gallagher J. F. Monohalogenated carbamates where hydrogen bonding rules without halogen bonding: is there a link between poor carbamate crystal growth and Z′ > 1?. CrystEngComm 2019, 21, 4048–4062. 10.1039/C9CE00318E. [DOI] [Google Scholar]
- Metrangolo P.; Resnati G. Eds., Halogen Bonding: Fundamentals and Applications; Structure and Bonding, 2008, 126, Springer: Berlin, [Google Scholar]
- Raatikainen K.; Rissanen K. Breathing molecular crystals: halogen- and hydrogen-bonded porous molecular crystals with solvent induced adaptation of the nanosized channels. Chem. Sci. 2012, 3, 1235–1239. 10.1039/c2sc00997h. [DOI] [Google Scholar]
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]
- Mukherjee A.; Desiraju G. R. Halogen bonds in some dihalogenated phenols: applications to crystal engineering. IUCrJ 2014, 1, 49–60. 10.1107/S2052252513025657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhothkina O.; Lieffrig J.; Jeannin O.; Fourmigué M.; Aubert E.; Espinosa E. Cocrystal or Salt: Solid State-Controlled Iodine Shift in Crystalline Halogen-Bonded Systems. Cryst. Growth Des. 2015, 15, 3464–3473. 10.1021/acs.cgd.5b00535. [DOI] [Google Scholar]
- Bauza A.; Mooibroek T. J.; Frontera A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. 10.1002/cphc.201500314. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116, 2476–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar M. H.; Hobza P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. 10.1021/acs.chemrev.5b00560. [DOI] [PubMed] [Google Scholar]
- Christopherson J. C.; Topić F.; Barrett C. J.; Friščić T. Halogen-Bonded Cocrystals as Optical Materials: Next-Generation Control over Light–Matter Interactions. Cryst. Growth Des. 2018, 18, 1245–1259. 10.1021/acs.cgd.7b01445. [DOI] [Google Scholar]
- Tepper R.; Schubert U. S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem., Int. Ed. 2018, 57, 6004–6016. 10.1002/anie.201707986. [DOI] [PubMed] [Google Scholar]
- Riel A. M. S.; Decato D. A.; Sun J.; Massena C. J.; Jessop M. J.; Berryman O. B. The intramolecular hydrogen bonded–halogen bond: a new strategy for preorganization and enhanced binding. Chem. Sci. 2018, 9, 5828–5836. 10.1039/C8SC01973H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R.; Claiser N.; Souhassou M.; Lecomte C.; Balkrishna S. J.; Kumar S.; Chopra D. Exploring the simultaneous σ-hole/π-hole bonding characteristics of a Br···π interaction in an ebselen derivative via experimental and theoretical electron-density analysis. IUCrJ 2018, 5, 647–653. 10.1107/S2052252518011041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiraju G. R.; Steiner T.. The weak hydrogen bond in structural chemistry and biology, 2001, Oxford University Press: UK., 10.1093/acprof:oso/9780198509707.001.0 [DOI] [Google Scholar]
- Domenicano A.; Hargittai I.. Strength from Weakness: Structural Consequences of Weak Interactions in Molecules, Supermolecules and Crystals, 2002, 68, NATO Science series II, Springer; Netherlands. [Google Scholar]
- Nishio M. CH/π hydrogen bonds in crystals. CrystEngComm 2004, 6, 130–158. 10.1039/b313104a. [DOI] [Google Scholar]
- Dance I.; Scudder M. Molecules embracing in Crystals. CrystEngComm 2009, 11, 2233–2247. 10.1039/b904479e. [DOI] [Google Scholar]
- Alvarez S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. 10.1039/c3dt50599e. [DOI] [PubMed] [Google Scholar]
- Martinez C. R.; Iverson B. L. Rethinking the term ″pi-stacking″. Chem. Sci. 2012, 3, 2191–2201. 10.1039/c2sc20045g. [DOI] [Google Scholar]
- Chopra D.; Guru Row T. N. Evaluation of the interchangeability of C–H and C–F groups: insights from crystal packing in a series of isomeric fluorinated benzanilides. CrystEngComm 2008, 10, 54–67. 10.1039/B709938J. [DOI] [Google Scholar]
- Donnelly K.; Gallagher J. F.; Lough A. J. Assembling an isomer grid: the isomorphous 4-, 3- and 2-fluoro-N’-(4-pyridyl)benzamides. Acta Crystallogr. 2008, C64, o335–o340. 10.1107/S0108270108012067. [DOI] [PubMed] [Google Scholar]
- Nayak S. N.; Reddy M. K.; Chopra D.; Guru Row T. N. Evaluation of the role of disordered organic fluorine in crystal packing: insights from halogen substituted benzanilides. CrystEngComm 2012, 14, 200–210. 10.1039/C1CE05441D. [DOI] [Google Scholar]
- Mocilac P.; Osman I. A.; Gallagher J. F. Short C–H···F interactions involving the 2,5-difluorobenzene group: understanding the role of fluorine in aggregation and complex C–F/C–H disorder in a 2 × 6 isomer grid. CrystEngComm 2016, 18, 5764–5776. 10.1039/C6CE00795C. [DOI] [Google Scholar]
- Luthe G.; Swenson D. C.; Robertson L. W. Influence of fluoro-substitution on the planarity of 4-chlorobiphenyl (PCB 3). Acta Crystallogr. 2007, B63, 319–327. 10.1107/S0108768106054255. [DOI] [PubMed] [Google Scholar]
- Klösener J.; Swenson D. C.; Robertson L. W.; Luthe G. Effects of fluoro substitution on 4-bromodiphenylether (PBDE 3). Acta Crystallogr. 2008, B64, 108–119. 10.1107/S0108768107067079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquesillo-Lazarte D.; Nemek V.; Cinčić D. Halogen bonded cocrystals of active pharmaceutical ingredients: pyrazinamide, lidocaine and pentoxifylline in combination with haloperfluorinated compounds. CrystEngComm 2017, 19, 5293–5299. 10.1039/C7CE01252G. [DOI] [Google Scholar]
- Naumann K. Influence of chlorine substituents on biological activity of chemicals: a review. Pest Manage. Sci. 2000, 56, 3–21. . [DOI] [Google Scholar]
- Chen W.; Mook R. A. Jr.; Premont R. T.; Wang J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. 10.1016/j.cellsig.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W. W.; Gopala L.; Bheemanaboina R. R. Y.; Zhang G. B.; Li S.; Zhou C. H. Discovery of 2-aminothiazolyl berberine derivatives as effectively antibacterial agents toward clinically drug-resistant Gram-negative Acinetobacter baumanii. Eur. J. Med. Chem. 2018, 146, 15–37. 10.1016/j.ejmech.2018.01.038. [DOI] [PubMed] [Google Scholar]
- Wang W.; Dong Z.; Zhang J.; Zhou X.; Wei X.; Cheng F.; Li B.; Zhang J. Acute and Subacute Toxicity Assessment of Oxyclozanide in Wistar Rats. Front. Vet. Sci. 2019, 6, 294. 10.3389/fvets.2019.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W. Y.; Ravindar L.; Rakesh K. P.; Manukumar H. M.; Shantharam C. S.; Alharbi N. S.; Qin H. L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandes M. Z.; Cavalcanti S. M. T.; Moreira D. R. M.; Filgueira de Azevedo W. Jr.; Leite A. C. L. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Kosjek T.; Heath E. Halogenated Heterocycles: Halogenated heterocycles as Pharmaceuticals 2011, 27, 219–246. [Google Scholar]
- Jayaraj R.; Megha P.; Sreedev P. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 2016, 9, 90–100. 10.1515/intox-2016-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani M.; Shankar U.; Jassal V. Recent strategies for removal and degradation of persistent & toxic organochlorine pesticides using nanoparticles: A review. J. Environ. Manage. 2017, 190, 208–222. 10.1016/j.jenvman.2016.12.068. [DOI] [PubMed] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocilac P.; Lough A. J.; Gallagher J. F. Structures and conformational analysis of a 3×3 isomer grid of nine N-(fluorophenyl)pyridinecarboxamides. CrystEngComm 2011, 13, 1899–1909. 10.1039/C0CE00326C. [DOI] [Google Scholar]
- Mocilac P.; Gallagher J. F. Structural systematics and conformational analyses of a 3×3 isomer grid of nine N-(tolyl)pyridinecarboxamides and three chlorinated relatives. CrystEngComm 2011, 13, 5354–5366. 10.1039/c1ce05169e. [DOI] [Google Scholar]
- Mocilac P.; Donnelly K.; Gallagher J. F. Structural systematics and conformational analyses of a 3×3 isomer grid of fluoro-N-(pyridyl)benzamides: physicochemical correlations, polymorphism and isomorphous relationships. Acta Crystallogr. 2012, B68, 189–203. [DOI] [PubMed] [Google Scholar]
- Mocilac P.; Tallon M.; Lough A. J.; Gallagher J. F. Synthesis, structural and conformational analysis of a 3×3 isomer grid based on nine methyl-N-(pyridyl)benzamides. CrystEngComm 2010, 12, 3080–3090. 10.1039/c002986f. [DOI] [Google Scholar]
- Hehir N.Structural systematics of halogenated benzamides. Dublin City University: Ireland. PhD thesis, 2017. [Google Scholar]
- Gallagher J. F.; Alley S.; Brosnan M.; Lough A. J. 1,1′-Fc(4-C6H4CO2Et)2 and its unusual salt derivative with Z′ = 5, catena-[Na+]2[1,1′-Fc(4-C6H4CO2–)2]·0.6H2O [1,1′-Fc = η5-(C5H4)2Fe]. Acta Crystallogr. 2010, B66, 196–205. [DOI] [PubMed] [Google Scholar]
- Gallagher J. F.; Farrell M.; Hehir N.; Mocilac P.; Aubert E.; Espinosa E.; Guillot B.; Jelsch C. At the Interface of Isomorphous Behavior in a 3 × 3 Isomer Grid of Monochlorobenzamides: Analyses of the Interaction Landscapes via Contact Enrichment Studies. Cryst. Growth Des. 2019, 19, 6141–6158. 10.1021/acs.cgd.9b00505. [DOI] [Google Scholar]
- Mocilac P.; Gallagher J. F. The First Phenyl-N-pyridinylcarbamate Structures: Structural and Conformational Analysis of Nine Methoxyphenyl-N-pyridinylcarbamates. Cryst. Growth Des. 2013, 13, 5295–5304. 10.1021/cg4010888. [DOI] [Google Scholar]
- Gallagher J. F.; Alley S.; Lough A. J. A structural systematic study of semi-rigid ferrocene derivatives as a 3 × 3 metallocene isomer grid: p–/m–/o-(FcC6H4)CONH(p–/m–/o-C6H4)CO2Et, [Fc = (η5-C5H5)Fe(η5-C5H4)]. Inorg. Chim. Acta 2016, 444, 113–125. 10.1016/j.ica.2016.01.028. [DOI] [Google Scholar]
- Mocilac P.; Gallagher J. F. Structural systematics and conformational analyses of an isomer grid of nine tolyl-N-pyridinylcarbamates. Struct. Chem. 2017, 28, 697–708. 10.1007/s11224-016-0851-5. [DOI] [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Oxford Diffraction Ltd ABSFAC and CrysAlisPro CCD/RED Version 1.171.33.55 Oxford Diffraction: Abingdon, Oxon, UK.
- Clark R. C.; Reid J. S. ABSFAC: a program for the calculation of the absorption during scattering in multifaceted crystals and similar samples. Comput. Phys. Commun. 1998, 111, 243–257. 10.1016/S0010-4655(98)00015-0. [DOI] [Google Scholar]
- Spek A. L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- Macrae C. F.; Edgington P. R.; McCabe P.; Pidcock E.; Shields G. R.; Taylor R.; Towler M.; van de Streek J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. 10.1107/S002188980600731X. [DOI] [Google Scholar]
- Frisch M. J.et al. , Gaussian 09 Revision B.01,2010, Gaussian Inc.Wallingford CT USA. [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. 3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-Consistent Molecular-Orbital Methods. 20. Basis Set For Correlated Wave-Functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Domagała S.; Fournier B.; Liebschner D.; Guillot B.; Jelsch C. An improved experimental databank of transferable multipolar atom models-ELMAM2. Construction details and applications. Acta Crystallogr. 2012, A68, 337–351. [DOI] [PubMed] [Google Scholar]
- Jelsch C.; Guillot B.; Lagoutte A.; Lecomte C. Advances in protein and small-molecule charge-density refinement methods using MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. 10.1107/S0021889804025518. [DOI] [Google Scholar]
- Allen F. H.; Bruno I. J. Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data. Acta Crystallogr. 2010, B66, 380–386. [DOI] [PubMed] [Google Scholar]
- Mackenzie C. F.; Spackman P. R.; Jayatilaka D.; Spackman M. A. CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. 10.1107/S205225251700848X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot B.; Espinosa E.; Huder L.; Jelsch C. MoProViewer: a tool to study proteins from a charge density science perspective. Acta Crystallogr. 2014, A70, C279. [Google Scholar]
- Hansen N. K.; Coppens P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. 1978, A34, 909–921. [Google Scholar]
- Volkov A.; Koritsanszky T.; Coppens P. Combination of the exact potential and multipole methods (EP/MM) for evaluation of intermolecular electrostatic interaction energies with pseudoatom representation of molecular electron densities. Chem. Phys. Lett. 2004, 391, 170–175. 10.1016/j.cplett.2004.04.097. [DOI] [Google Scholar]
- Mocilac P.; Gallagher J. F.; Jelsch C. Aggregation in Three Benzamide or Pyridylcarboxamide Hydrates: Formation of 1D Chains Comprising Water Molecules in a Chloro(pyridyl)benzamide Dihydrate. Croat. Chim. Acta 2018, 91, 281–288. [Google Scholar]
- Guru Row T. N. Hydrogen and fluorine in crystal engineering: systematics from crystallographic studies of hydrogen bonded tartrate–amine complexes and fluoro-substituted coumarins, styrylcoumarins and butadienes. Coord. Chem. Rev. 1999, 183, 81–100. 10.1016/S0010-8545(98)00184-2. [DOI] [Google Scholar]
- Tuchalski G.; Emmerling F.; Groger K.; Hansicke A.; Nagel T.; Reck G. X-ray investigations of nebivolol and its isomers. J. Mol. Struct. 2006, 800, 28–44. 10.1016/j.molstruc.2006.03.086. [DOI] [Google Scholar]
- Abad A.; Agullo C.; Cunat A. C.; Vilanova C.; de Arellano M. C. R. X-ray structure of fluorinated N-(2-chloropyridin-4-yl)-N’-phenylureas. Role of F substitution in the crystal packing. Cryst. Growth Des. 2006, 6, 46–57. 10.1021/cg049581k. [DOI] [Google Scholar]
- Cuffini S.; Glidewell C.; Low J. N.; de Oliveira A. G.; de Souza M. V. N.; Vasconcelos T. R. A.; Wardell S. M. S. V.; Wardell J. L. Nine N-aryl-2-chloronicotinamides: supramolecular structures in one, two and three dimensions. Acta Crystallogr. 2006, B62, 651–665. [DOI] [PubMed] [Google Scholar]
- Gelbrich T.; Hursthouse M. B.; Threlfall T. L. Structural systematics of 4,4′-disubstituted benzenesulfonamidobenzenes. 1. Overview and dimer based isostructures. Acta Crystallogr. 2007, B63, 621–632. [DOI] [PubMed] [Google Scholar]
- Wardell S. M. S. V.; de Souza M. V. N.; Vasconcelos T. R. A.; Ferreira M. L.; Wardell J. L.; Low J. N.; Glidewell C. C. Patterns of hydrogen bonding in mono- and di-substituted N-arylpyrazinecarboxamides. Acta Crystallogr. 2008, B64, 84–100. [DOI] [PubMed] [Google Scholar]
- Dumitru F.; Legrand Y.-M.; Barboiu M.; van der Lee A. Weak intermolecular hydrogen and halogen interactions in an isomorphous halogen series of pseudoterpyridine Zn-II complexes. Acta Crystallogr. 2013, B69, 43–54. [DOI] [PubMed] [Google Scholar]
- Bombicz P. The way from isostructurality to polymorphism. Where are the borders? The role of supramolecular interactions and crystal symmetries. Crystallogr. Rev. 2017, 23, 118–151. 10.1080/0889311X.2016.1251909. [DOI] [Google Scholar]
- Taylor R. G. D.; Yeo B. R.; Hallett A. J.; Kariuki B. M.; Pope S. J. A. An organometallic complex revealing an unexpected, reversible, temperature induced SC–SC transformation. CrystEngComm 2014, 16, 4641–4652. 10.1039/C4CE00070F. [DOI] [Google Scholar]
- Zhang Q.; Zhang S.-P.; Shao S.-C. N-(4-Chlorophenyl)picolinamide. Acta Crystallogr. 2006, 62, o4695–o4696. [Google Scholar]
- Percival D.; Storey J. D. A.; Harrison W. T. A. N-(2-Bromophenyl)pyridine-3-carboxamide. Acta Crystallogr. 2007, E63, o1851–o1852. [Google Scholar]
- Park Y.-T.; Jung C.-H.; Kim K.-W.; Kim H. S. Synthesis of 2-Pyridinylbenzoxazole: Mechanism for the Intramolecular Photosubstitution of the Haloarene with the Carbonyl Oxygen of the Amide Bond in Basic Medium. J. Org. Chem. 1999, 64, 8546–8556. 10.1021/jo9909498. [DOI] [Google Scholar]
- Ojala W. H.; Smieja J. M.; Spude J. M.; Arola T. M.; Kuspa M. K.; Herrera N.; Ojala C. R. Isostructuralism among bridge-flipped’ isomeric benzylideneanilines and phenylhydrazones. Acta Crystallogr. 2007, B63, 485–496. [DOI] [PubMed] [Google Scholar]
- Kassekert K. J.; Smith T. J.; Fermanich J. M.; Lystad K. M.; Gregoire T. A.; Ojala C. R.; Ojala W. H. Bridge-Flipped Isomers: Searching for Isomorphous Benzylideneanilines and Phenylhydrazones Reveals Unexpected Structures. Cryst. Growth Des. 2019, 19, 5896–5906. 10.1021/acs.cgd.9b00913. [DOI] [Google Scholar]
- Jelsch C.; Ejsmont C.; Huder L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. 10.1107/S2052252514003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelsch C.; Soudani S.; Ben Nasr C. Likelihood of atom-atom contacts in crystal structures of halogenated organic compounds. IUCrJ 2015, 2, 327–340. 10.1107/S2052252515003255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramowitz R.; Yalkowsky S. H. MELTING-POINT, BOILING-POINT, AND SYMMETRY. Pharm. Res. 1990, 07, 942–947. 10.1023/A:1015949907825. [DOI] [PubMed] [Google Scholar]
- Brown R. J. C.; Brown R. F. C. Melting point and molecular symmetry. J. Chem. Ed. 2000, 77, 724–731. 10.1021/ed077p724. [DOI] [Google Scholar]
- Vishweshwar P.; Nangia A.; Lynch V. M. Molecular complexes of homologous alkanedicarboxylic acids with isonicotinamide: X-ray crystal structures, hydrogen bond synthons, and melting point alternation. Cryst. Growth Des. 2003, 3, 783–790. 10.1021/cg034037h. [DOI] [Google Scholar]
- Slovokhotov Y. L.; Neretin I. S.; Howard J. A. K. Symmetry of van der Waals molecular shape and melting points of organic compounds. New J. Chem. 2004, 28, 967–979. 10.1039/b310787f. [DOI] [Google Scholar]
- Podsiadło M.; Bujak M.; Katrusiak A. Chemistry of density: extension and structural origin of Carnelley’s rule in chloroethanes. CrystEngComm 2012, 14, 4496–4500. 10.1039/c2ce25097g. [DOI] [Google Scholar]
- Dziubek K. F.; Katrusiak A. Structure-melting relations in isomeric dibromobenzenes. Acta Crystallogr. 2014, B70, 492–497. [DOI] [PubMed] [Google Scholar]
- Yalkowsky S. H. Carnelley’s Rule and the Prediction of Melting Point. J. Pharm. Sci. 2014, 103, 2629–2634. 10.1002/jps.24034. [DOI] [PubMed] [Google Scholar]
- Gamidi R. K.; Rasmuson A. Estimation of Melting Temperature of Molecular Cocrystals Using Artificial Neural Network Model. Cryst. Growth Des. 2017, 17, 175–182. 10.1021/acs.cgd.6b01403. [DOI] [Google Scholar]
- Feng G.; Gou Q.; Evangelisti L.; Vallejo-López M.; Lesarri A.; Cocinero E. J.; Caminati W. Competition between weak hydrogen bonds: C–H···Cl is preferred to C–H···F in CH2ClF–H2CO, as revealed by rotational spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 12261–12265. 10.1039/C4CP00919C. [DOI] [PubMed] [Google Scholar]
- Mata I.; Alkorta I.; Molins E.; Espinosa E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving H···X (X=H, C, N, O, F, S, Cl, π) Interactions. Chem. - Eur. J. 2010, 16, 2442–2452. 10.1002/chem.200901628. [DOI] [PubMed] [Google Scholar]
- Espinosa E.; Alkorta I.; Elguero J.; Molins E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. 10.1063/1.1501133. [DOI] [Google Scholar]
- Wheinhold F.; Landis C. R.. Valency and Bonding – A Natural Bond Orbital Donor-Acceptor Perspective, 2005, Cambridge University Press: Cambridge, UK. [Google Scholar]
- Corradi E.; Meille S. V.; Messina M. T.; Metrangolo P.; Resnati G. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly Processes. Angew. Chem., Int. Ed. 2000, 39, 1782–1786. . [DOI] [PubMed] [Google Scholar]
- Aakeröy C. B.; Fasulo M.; Schultheiss N.; Desper J.; Moore C. Structural Competition between Hydrogen Bonds and Halogen Bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. 10.1021/ja073201c. [DOI] [PubMed] [Google Scholar]
- An X.; Zhuo H.; Wang Y.; Li Q. Competition between hydrogen bonds and halogen bonds in complexes of formamidine and hypohalous acids. J. Mol. Model. 2013, 19, 4529–4535. 10.1007/s00894-013-1969-7. [DOI] [PubMed] [Google Scholar]
- Kowalska K.; Trzybiński D.; Sikorski A. Influence of the halogen substituent on the formation of halogen and hydrogen bonding in co-crystals formed from acridine and benzoic acids. CrystEngComm 2015, 17, 7199–7212. 10.1039/C5CE01321F. [DOI] [Google Scholar]
- Robertson C. C.; Wright J. S.; Carrington E. J.; Perutz R. N.; Hunter C. A.; Brammer L. Hydrogen bonding vs. halogen bonding: the solvent decides. Chem. Sci. 2017, 8, 5392–5398. 10.1039/C7SC01801K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeysekera A. M.; Day V. W.; Sinha A. S.; Aakeröy C. B. Mapping out the Relative Influence of Hydrogen and Halogen Bonds in Crystal Structures of a Family of Amide-Substituted Pyridines. Cryst. Growth Des. 2020, 20, 7399–7410. 10.1021/acs.cgd.0c01086. [DOI] [Google Scholar]
- Giangreco I.; Cole J. C.; Thomas E. Mining the Cambridge Structural Database for Matched Molecular Crystal Structures: A Systematic Exploration for Isostructurality. Cryst. Growth Des. 2017, 17, 3192–3203. 10.1021/acs.cgd.7b00155. [DOI] [Google Scholar]
- Brezgunova M. E.; Aubert E.; Dahaoui S.; Fertey P.; Lebègue S.; Jelsch C.; Angyan J. G.; Espinosa E. Charge Density Analysis and Topological Properties of Hal3-Synthons and Their Comparison with Competing Hydrogen Bonds. Cryst. Growth Des. 2012, 12, 5373–5386. 10.1021/cg300978x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.