

Abstract
The invasive leading edge represents a potential gateway for tumor metastasis. The role of fibroblasts from the tumor edge in promoting cancer invasion and metastasis has not been comprehensively elucidated. We hypothesize that crosstalk between tumor and stromal cells within the tumor microenvironment (TME) results in activation of key biological pathways depending on their position in the tumor (edge vs core). Here we highlight phenotypic differences between tumor-adjacent-fibroblasts (TAF) from the invasive edge and tumor core fibroblasts (TCF) from the tumor core, established from human lung adenocarcinomas. A multi-omics approach that includes genomics, proteomics, and O-glycoproteomics was used to characterize crosstalk between TAFs and cancer cells. These analyses showed that O-glycosylation, an essential post-translational modification resulting from sugar metabolism, alters key biological pathways including the cyclin-dependent kinase 4 and phosphorylated retinoblastoma protein (CDK4-pRB) axis in the stroma and indirectly modulates pro-invasive features of cancer cells. In summary, the O-glycoproteome represents a new consideration for important biological processes involved in tumor-stroma crosstalk and a potential avenue to improve the anti-cancer efficacy of CDK4 inhibitors.
Keywords: Tumor-Adjacent Fibroblasts (TAFs), O-glycosylation, Tumor microenvironment (TME), multi-omics, Lung Adenocarcinoma (LUAD)
Introduction
The leading edge of tumors represents one of the main sites of access for local invasion and metastasis (1); however, the role of fibroblasts from the tumor edge have been mainly overlooked. Much of our current understanding of cancer-associated fibroblasts (CAFs) is derived from fibroblasts harvested from the inner tumor, and the pro- and anti-tumor effects of CAFs are still being resolved (2–5). Single-cell studies have demonstrated the significant heterogeneity of CAFs in the tumor microenvironment (TME) (6,7), but the cell-cell interactions involving different tumor fibroblast subtypes, particularly those at the tumor edge, are not well known. Of the few studies that focus on fibroblasts at the invasive tumor edge, colorectal and hepatocellular carcinomas studies have found peritumor fibroblasts stimulate the proliferation and migration of cancer cells more strongly than CAFs (8,9). In order to better understand the role of these peritumor fibroblasts in tumor invasion and treatment response, fibroblast heterogeneity and cell-cell interactions at the invasive edge of the tumor need to be further explored.
While multi-omics analysis of fibroblasts has included epigenomic, genomic and transcriptomic data (10–12), post-translational modifications (PTM) of fibroblasts have not been well characterized. PTMs have gained significant interest in recent omics studies as they directly modulate numerous cancer hallmarks including cell cycle progression, migration and metabolism (12–15). Previous work showed that metabolic reprogramming toward the hexosamine biosynthesis pathway (HBP), an understudied glucose pathway leading to glycosylation, is associated with poor survival in the lung adenocarcinoma (LUAD) (16,17). The authors reported that this metabolic switch happens earliest in CAFs compared to the malignant cells across tumor compartments (fibroblasts, cancer, immune and endothelial cells). This finding suggests that tumor fibroblasts redirect their glucose toward HBP, leading to altered O-glycosylation, a PTM regarded to be as critical as phosphorylation and also initiated on serine and threonine residues (18,19). While previous results on CAFs assessed metabolic reprogramming toward HBP in the tumor stroma, direct measurements of the O-glycoproteome changes following tumor-stroma interactions using a multi-modal approach have yet to be reported.
O-linked glycosylation has been associated with several cancer hallmarks (20,21). However, very little is known about: 1) which proteins are O-glycosylated, 2) the effect(s) of O-glycosylation on protein function, and 3) how protein O-glycosylation impacts cell-cell crosstalk. To our knowledge, no published study has reported the O-glycoproteome of tumor fibroblasts nor undertaken a multi-omics approach that integrates the O-glycoproteome in the context of tumor-stroma crosstalk. The lack of data on the O-glycoproteome is largely due to the technical challenges associated with probing glycobiology (22) including the lack of automated techniques, small libraries of glycoproteins and limited availability of sugar-specific antibodies (18,23). Currently, mass spectrometry is the main method used to measure O-glycosylation, but the lability of O-glycans, the lack of universal enrichment methods due to O-glycan heterogeneity, and the limitations of bioinformatic search engines for O-glycopeptides (searching time and accuracy), as well as their quantification, add to the technical challenges. However, recent enrichment methods (24,25), employment of O-glycoproteases (26–28) and the new approach SimpleCell (29) have allowed us to improve the characterization and investigate the role of O-glycosylation in the TME.
This study focuses on the understudied peritumor fibroblasts from the invasive edge of primary LUAD tumors, referred hereon as Tumor-Adjacent-Fibroblasts (TAFs), and demonstrates the unconventional phenotype of TAFs compared to Tumor Core Fibroblasts (TCFs) harvested from the inner tumor which are more similar to conventional CAFs. We investigate the role of TAFs through multi-omics profiling of tumor-stroma cocultures by integrating transcriptomic, proteomic, and, for the first time, O-glycoproteomic data obtained by an optimized method of O-glycan enrichment (25) and precise spectra quantification of O-glycopeptides (30). We find that the O-glycoproteome provides additional biological insights on tumor-stroma crosstalk over genomic and proteomic data and highlights the role of O-glycosylation in modulating the CDK4-pRB axis of fibroblasts at the tumor invasive edge, among other important biological processes. Lastly, we show that targeting O-glycosylation in TAFs indirectly decreases pro-invasive features in cancer cells, establishing the O-glycoproteome as an important way to modulate the TME.
Materials and Methods
Cell Culture
HCC827 and HCC827-eGFP NSCLC adenocarcinoma cell lines were a generous gift from Dr Parag Mallick. The A549 were purchased from ATCC. All cells and primary cultures were grown in RPMI-1640 with L-Glutamine, supplemented with 10% fetal bovine serum (FBS) and 5% antibiotic solution (penicillin/streptomycin), at 5% CO2 and 37°C. These cells were not tested for mycoplasma nor authenticated by our laboratory.
Human Studies
Clinical aspects of this study were approved by the Stanford Institutional Review Board (IRB) in accordance with the Declaration of Helsinki guidelines for the ethical conduct of research. All patients involved provided a written informed consent. Collection and use of human tissues were approved and complied with data protection regulations regarding patient confidentiality (IRB protocol #15166). Following surgical resection of primary tumors from patients at Stanford Hospital, lung adenocarcinoma specimens were immediately processed in order to establish primary fibroblast cell cultures or embedded in Optimal Cutting Temperature Compound (OCT) for future use.
Tumor and Lung Specimen Dissociation
Immediately following surgery, fresh lung adenocarcinoma matched specimens were collected from different tumor regions, namely Normal Fibroblasts (NFs, >5 cm away from the tumor), Tumor-Adjacent Fibroblasts (TAFs, leading edge) and Tumor Core Fibroblasts (TCFs, tumor core). Samples were immersed and transferred from Stanford Hospital to the laboratory in MACS Tissue Storage Solution (Miltenyi Biotec). Then, tumors were cut into small pieces with dissecting scissors. The dissociation was performed utilizing the MACS Tumor Tissue Dissociation Kit (Miltenyi Biotec) for the tumor core samples or the Lung Dissociation Kit (Miltenyi) for the normal and adjacent tissues as per the manufacturer’s protocol. The single-cell suspensions were centrifuged at 500 × g for 5 minutes to pellet tumor cells, which were subsequently resuspended in RPMI-1640 and applied on a MACS SmartStrainer (70 μm, Miltenyi Biotec) for filtration Following centrifugation (500 × g, 5 minute), and red blood cells were removed using the Red Blood Cell Lysis Solution according to the manufacturer’s instructions (Miltenyi Biotec). Cells were then washed, resuspended in RPMI-1640 and plated in a 35 mm dish for fibroblast expansion. At confluency, all cells were trypsinized and transferred to a 25 cm2 flask until confluency, then all cells were split in two 75 cm2 flasks for further expansion. At confluency of the 75 cm2 flasks, primary fibroblasts were divided equally and frozen in 10 vials, designated here as “passage 1”. All cocultures experiments and resulting analyses including RNA-seq, mass spectrometry, flow cytometry, CyTOF, DON and palbociclib treatment, as well as immunofluorescences were realized using primary fibroblasts at passage 2. Briefly, one vial (passage 1) was thawed in a 75 cm2 flask, expanded until confluency, then split at a maximum dilution ratio of 1:10 or lower into subsequent flasks, designated here as passage 2. Passage 2 cells were collected at confluency of 80–95% to perform cocultures.
In Vitro Coculture Experiments and Conditioned Media Induction
HCC827 or A549 lung adenocarcinoma cells were cocultured at 1:1 ratio (8×105 cells each cell type) with primary fibroblasts for 24 hours. In parallel, monocultures (1.6×106 cells) were plated and, after 24 hours, conditioned media were collected and spun down at 500 × g for 5 minutes. The cells were rinsed PBS 1X and treated with conditioned media from the opposite cell type for 24 hours. Then, cocultures and monoculture controls or cultures induced with supernatant were rinsed with PBS 1X and lifted off tissue culture plates using TrypLE (Gibco), and cocultures were separated with the optimized amount of Anti-Fibroblast MicroBeads (40 μL, for up to three 100 mm Petri Dishes, Miltenyi Biotec) in accordance with the manufacturer’s protocol to reach minimal contamination in each compartment (Supplementary Fig.1A). Cells were counted and stored accordingly at −80 °C for further analysis including flow cytometry, mass cytometry, RNA-seq and proteomics.
Alternatively, HCC827-eGFP were cocultured at 1:1 ratio (4×105 cells each) with primary TAFs for up to 72 hours. After 24 hours, cells were treated with 10 μM 6-Diazo-5-oxo-L-norleucine (DON, Sigma Aldrich), 1 μM Palbociclib (Sigma Aldrich) or a combination of both for 48 hours. Then, cocultures and monoculture controls were rinsed with PBS 1X, lifted off tissue culture plates using TrypLE (Gibco), counted, pelleted, and stored accordingly in 500 μL of flow cytometry buffer (FCB) at −80 °C for further flow cytometry analysis. Drugs were dissolved in water and non-treated controls with RPMI medium only.
Flow Cytometry Analysis
After separation with Anti-Fibroblast MicroBeads (Miltenyi Biotec), the sorted cell compartments and monocultures were counted and aliquots of 1×106 cells were prepared per condition. Aliquots were incubated for 5 minutes with 1 μL of Zombie Aqua™ fixable viability dye in PBS 1X, then washed with FCB (0.5% BSA, 0.02% NaN3, and 2 mM EDTA in PBS 1X) and centrifuged (500 × g, 5 minutes) before adding paraformaldehyde (PFA, EMS) at a final concentration of 16% for 10 minutes at room temperature in FCB. Cells were then centrifuged at 500 × g for 5 minutes at 4 °C to pellet cells, PFA was removed, and cells were washed again with FCB. Cells were either stored long-term at −80 °C in 500 μL of FCB or permeabilized with 100 μL of eBioscience™ Permeabilization Buffer (Invitrogen) diluted at 1X concentration for 30 minutes on ice with a master mix of primary antibodies. After the incubation with primary antibodies, cells were washed with FCB and spun down at 500 × g for 5 minutes at 4°C (2X). These steps were repeated with the secondary antibody as needed. After the last wash with FCB, cells were resuspended in 500 μL of FCB, strained and analyzed using a BD LSRFortessa™ X-20 (BD Biosciences). Results were analyzed using Cytobank single-cell analysis software.
Mass Cytometry
After separation with Anti-Fibroblast MicroBeads (Miltenyi Biotec), the sorted cell compartments and monocultures were counted and aliquots of 5×105 cells were prepared per condition. To assess cell viability for mass cytometry, cell pellets were incubated for 5 minutes in 1 mL PBS containing cisplatin (Sigma-Aldrich) at a final concentration of 0.5 μM at room temperature. Cisplatin reaction was quenched by adding complete RPMI-1640 media (10% FBS) and subsequent centrifugation for 5 minutes at 500 × g. Cell pellets were resuspended in cell culture media or FCB and were fixed by adding PFA (EMS) at a final concentration of 16% for 10 minutes at room temperature. Cells were centrifuged at 500 × g for 5 minutes at 4 °C to pellet cells and remove PFA and washed once with FCB. Cell pellets were resuspended in FCB and stored at −80 °C until all conditions of the same clinical specimen were collected.
HCC827 cell lines monocultures and cocultures with NFs, TAFs and TCFs were analyzed using mass cytometry, as previously described (31).
Projections onto the EMT–MET PHENOSTAMP Phenotypic Map
PHENOSTAMP was downloaded from GitHub under https://githubcom/anchangben/ PHENOSTAMP. Briefly, FCS files previously gated in Cytobank according to vimentin, cytokeratins, CD104, slug and low signaling (Supplementary Fig.1B), or from flow cytometry were uploaded into R, and the PHENOSTAMP algorithm was used to project HCC827 and A549 monocultures and cocultures on the 2D EMT–MET state map, as previously described (31).
Immunohistochemistry and scoring of LUAD Tissue Microarray (TMA)
An existing lung adenocarcinoma tissue microarray (TMA), derived from anonymized pre-existing research samples in the Stanford pathology department, was analyzed by IHC; this TMA analysis did not involve collection of new tissues. IHC was performed on 4-μm Formalin-Fixed Paraffin-Embedded (FFPE) sections using the Ventana BenchMark XT automated immunostaining platform (Ventana Medical Systems, Roche) in accordance with the manufacturer protocol. IHC quantification was assessed for both cancer cells and fibroblasts, independently. Briefly, we analyzed a lung TMA of 260 LUAD cores (1 patient sample per core) that we qualitatively classified, based on visual assessment, into two distinct groups as “mixed” or “compartmentalized”. We defined cores as “mixed” when cancer cells were intermixed with stromal cells and it was not possible to distinguish between stromal and cancer cells based on morphology and with the help of a pathologist. In contrast, cores were classified as “compartmentalized” when the cancer cells areas were sharply delineated by at least one stream of spindly stromal cells crossing a diameter of the core. Out of the 260 LUAD cores analyzed, 30 samples were classified as compartmentalized and used as our surrogate leading-edge cohort, i.e., harboring TAFs-like fibroblasts. Fibroblasts from the “mixed” cores were classified as TCFs-like. Then, we quantified Ki67 positivity in both fibroblasts and cancer cells in all 260 samples. Samples were scored as follows; 0: no staining, 1: low proliferation (less than 10 positive cells), 2: medium proliferation (between 10–50 positive cells) and 3: high proliferation (more than 50 positive cells).
RNA Sequencing and Analysis
After in vitro coculture experiments, 1×105 cells were pelleted, stored in RNA later stabilization solution (Invitrogen) and sent to MedGenome Inc (Foster City, CA, USA) for RNA extraction, library preparation and analysis. Briefly, the RNA was extracted using a Macherey-Nagel Nucleospin analyzer (Fisher Scientific) in accordance with the manufacturer’s protocol. Sample and library quality control were performed with Qubit (ThermoFisher) and Tapesation (Agilent) bioanalyzers. The library was prepared using Illumina TruSeq stranded mRNA and the NovaSeq PE100 sequencing run type.
Expression levels of RNA sequencing data were quantified using a quasi-mapping two-phase inference algorithm implemented in Salmon (version 091) (32). Transcript sequences GRCh38 from Gencode were used for reference transcriptome indexing. Transcript-per-million (TPM) value was used as normalized expression level unit. Quantified gene expressions were then log transformed for downstream analysis.
Highly-variable genes with variances over 75th percentile were included for differentially-expressed gene (DEG) analysis. DEGs were identified between NFs, TAFs and TCFs, as well as before and after coculture with HCC827 from paired patient samples. DEG analysis was performed in R using paired Student’s t-test. False discovery rate was used to adjust the p values to ensure the control for multiple testing. We used two criteria for DEGs: (i) false discovery rate less than 0.05, and (ii) average fold change greater than 1.5. A permutation test on two class-paired samples from R package SAM (33) was also used to verify the differentially expressed genes.
O-glycopeptide Enrichment
After in vitro coculture experiments, 1×106 cells were pelleted and resuspended in 150 μL of lysis buffer (50 mM Tris, pH 8.0, 2% sodium dodecyl sulfate (SDS), 1X protease inhibitor cocktail, 25 mM NaF, 100 μM Na3VO4, 5 mM EGTA and 5 mM EDTA). Lysates were sonicated for a few seconds on ice with a Q500 sonicator at 20% intensity (Qsonica) to shear DNA/reduce viscosity, then centrifuged at 14000 rpm (4 °C, 10 min) for supernatant collection. Protein quantification was performed using a Pierce Bicinchoninic acid (BCA) protein assay kit (Thermo Scientific), and 300 μg of proteins were used for O-glycans enrichment, adapted as previously described (25).
Peptide identification, quantification, data processing and statistical analysis
The resulting MS raw data files were searched using Byonic software (Protein Metrics) (34). For total protein samples, data were searched using the entire human proteome downloaded from Uniprot (35) (reviewed, 20428 entries), with decoys appended within the Byonic environment as reversed sequences. Cleavage specificity was set as fully specific for C-terminal to R and K residues (fully tryptic), with two missed cleavages allowed, and fragmentation type was set to QTOF/HCD. Precursor mass tolerance was set to 10 ppm with fragment mass tolerance set to 0.4 Da. The total common max value for modifications was set to 3 and the total rare max was set to 1. Modifications used were carbamidomethyl at cysteine (+57,021644, fixed), oxidation at methionine (+15994915, common2) and acetylation at the protein N-temrinal (+42010565, rare1). The protein FDR cutoff was set to 1%. Unique databases of identified proteins were created for each total protein sample file using the “Create focused database” feature in Byonic. Following searching of all total protein runs, identified proteins in all focused database files were combined into one fasta file, with 4,495 non-redundant entries to make an appropriately-sized database for glycopeptide searching (36). Using this database, glycopeptide raw files were searched in Byonic using a glycan database of 9 common O-glycans provided by Byonic, with the glycan modifications being denoted as common2 in Byonic (meaning they could each occur twice in a sequence). The total common max value was set to 3 and the total rare max was set to 1. Other modifications were: carbamidomethyl at cysteine (+57,021644, fixed), oxidation at methionine (+15994915, common2), acetylation at the protein N-terminal (+42010565, rare1) and deamidated asparagine (+0984016, rare1). Cleavage specificity was set as fully specific for C-terminal to R and K residues (fully tryptic), with three missed cleavages allowed. Precursor mass tolerance was set to 10 ppm with fragment mass tolerance(s) set to 20 ppm for both HCD and EThcD, and protein FDR was set to 1% to create the “ focused database” feature in Byonic for standard shotgun proteomic experiments on unenriched tryptic peptides from HEK293 whole cell lysate.
Quantitative information was extracted from MS1 spectra of all identified peptides, using an in-house R script based on MSnbase package (30) as the AUC of the extracted ion current (XIC) of all remaining peptides after the alignment of the chromatographic runs. Then, protein abundance changes were analyzed using the Generic Integration Algorithm. All quantitative information is expressed in terms of Z-scores at protein or at peptide level, according to (37) in which the log2 ratios, were: calculated comparing the AUC of peptides in TAFs cocultures against the average of the controls; pondered using the corresponding statistical weight, calculated at spectrum level, according to WSPP model; and rescaled, and standardized to a normal distribution N(0,1). The validity of the null hypothesis at each level (spectrum, peptide, and protein) was carefully checked by plotting the cumulative distributions. The variances at the scan, peptide and protein levels, and protein expression changes were determined only with non-modified peptides. Afterwards, in the intact glycoproteomics analysis, the glycan-containing peptides were included in the analysis to determine those deviating more than expected from the rest of the (non-modified) peptides belonging to the same protein, applying the same method used in (37). The final statistical comparison was performed using Student’s-t test.
Immunofluorescence (IF)
IF images were generated on cell culture and primary tissue samples. TAFs:HCC827-eGFP cocultures and HCC827-eGFP monocultures were grown on coverslips using the same culture conditions previously described here, in the In Vitro Coculture methods section. Coverslips were rinsed 3 times with a cold solution of 5% BSA, washed in PBS and fixed for 10 minutes in PFA 4%. Then, the coverslips were washed again and incubated in 0.1% Triton X-100 for 10 minutes. Next, samples were blocked with 10% goat serum diluted in PBS for 30 minutes, and incubated with a combination of the primary antibodies in blocking solution in a humidified chamber overnight at 4 °C. After three washes in PBS, the coverslips were incubated with a combination of the secondary antibodies in blocking solution for 1 hour at RT. The samples were intensively washed with PBS and mounted on slides with a drop of mounting medium containing DAPI (Invitrogen). The specimens were observed under the BZ-X800 fluorescence microscope (Keyence).
Quantification and statistical analysis
Statistical testing for PHENOSTAMP results were performed with GraphPad Prism Software 8.4, using two-way ANOVA, followed by the Fisher least significant difference (LSD) multiple comparison test. Error bars in the figures represent the SD Statistical significance: *p <0.05; **p <0.01. Statistical testing for Ki67 quantification results were performed with GraphPad Prism Software version 9.1, using Dunn’s multiple comparison test following ANOVA. Boxes represent the mean: *p <0.05. Gene set enrichment analyses overlap, and activity z-scores p values have been calculated using the Ingenuity Pathway Analysis Software (Qiagen) and the previously described method (38). Metabolic pathway gene overlaps were calculated using a hypergeometric test. Statistical analysis of RNA-seq and mass spectrometry data are described in their respective method sections.
Data and materials availability:
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Sylvia Plevritis (sylvia.plevritis@stanford.edu). This study did not generate new unique reagents and all the reagents and antibodies used here are available in the Key Resource Table. The data generated in this study are available within the article and its supplementary data files. Gene expression data generated in this study were deposited on Gene Expression Omnibus (GEO) at GSE186235. The flow cytometry and CyTOF data, as well as the mass spectrometry raw files, will be shared upon individual request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| E-Cadherin PE/Cy7 (clone 67A4) | BioLegend | Cat#324116; RRID:AB_2563096 |
| Vimentin AF700 (clone RV202) | Novus Bio | Cat#NBP1-97672AF700 |
| α-SMA APC-Cy7 (clone SPM332) | Abcore | Cat#AC12-0261-05 |
| CD10 BV421 (Clone HI10a) | BioLegend | Cat#312217; RRID:AB_10899409 |
| GFAT2 FITC (clone aa175-201) | LSBio | Cat#LS-C259501 |
| FSP1 PerCP-Cy5 (clone NJ-4F3-D1) | BioLegend | Cat#370009; RRID:AB_2572075 |
| p21 AF594 (clone 195720) | R&D Systems | Cat#IC1047T |
| Ki67 AF647 (clone B56) | BD Biosciences | Cat#558615; RRID:AB_647130 |
| pRB (Thr826) | Invitrogen | Cat#44-576G; RRID: AB_2533683 |
| Capase-8 AF700 (clone 90A992) | Novus Bio | Cat#NB10-056527AF700 |
| IgG anti-rabbit PE (clone poly4064) | BioLegend | Cat#406421; RRID: AB_2563484 |
| CD44 (clone 156-3C11) | Cell Signaling Tecchnology | Cat#3570; RRID:AB_2076465 |
| LSP1 (clone EPR5997) | Abcam | Cat#ab133506 |
| pan-cytokeratin (clone AE1/AE3 + 5D3) | Abcam | Cat#ab86734; RRID:AB_10674321 |
| Vimentin (clone EPR3776) | Abcam | Cat#ab92547; RRID:AB_10562134 |
| Cy3-conjugated goat anti-rabbit IgG | Abcam | Cat#ab6939; RRID:AB_955021 |
| Cy5-conjugated goat anti-rabbit IgG | Abcam | Cat#ab97077; RRID:AB_10679461 |
| Cy3-conjugated goat anti-mouse IgG | Abcam | Cat#ab97035; RRID:AB_10680176 |
| Ki67 (clone MIB-1) | Agilent | Cat#GA62661-2 |
| CD44 AF488 (clone IM7) | BioLegend | Cat#103016; RRID:AB_493679 |
| CD24 APC/Cy7 (clone ML5) | BioLegend | Cat#311132; RRID:AB_2566347 |
| Twist AF647 (clone 927403) | R&D Systems | Cat#IC9160R-025; RRID:AB_2687921 |
| MUC1 BV421 (clone CD227) | BD Biosciences | Cat#743405; RRID:AB_2741478 |
| CyTOF Antibodies | ||
| CD44 (clone IM7, metal Ln, mass 115) | Biolegend | Cat#103051; RRID:AB_2562799 |
| cleaved Caspase 3 (clone C92-605, metal Nd, mass142) | BD Biosciences | Cat#559565; RRID:AB_397274 |
| phospho-Src pY418 (clone K98-37, metal Nd, mass 144) | BD Biosciences | Cat#560094; RRID:AB_1645532 |
| phospho-EGFR Tyr1068 (clone D7A5, metal Nd, mass 145) | Cell Signaling Tecchnology | Cat#3777; RRID:AB_2096270 |
| TROP2 (clone 77220, metal Nd, mass 148) | R&D Systems | Cat#MAB650; RRID:AB_2205665 |
| Oct 3/4 (clone O50-808, metal Nd, mass 150) | BD Biosciences | Cat#561555; RRID:AB_10715577 |
| Notch3 (clone MHN3-21, metal Eu, mass 151) | Biolegend | Cat#345402; RRID:AB_1953253 |
| Cytokeratin 8 (clone SP102, metal Sm, mass 152) | Abcam | Cat#ab227644 |
| PD-L1 (clone 29E.2A3, metal Eu, mass 153) | Biolegend | Cat#329702; RRID:AB_940372 |
| MUC1 (clone SPM492, metal Sm, mass 154) | Abcam | Cat#ab227740 |
| RUNX1 (clone 1C5B16, metal Gd, mass 155) | Biolegend | Cat#659302; RRID:AB_2563194 |
| Snail (clone C15D3, metal Gd, mass 156) | Cell Signaling Tecchnology | Cat#3879; RRID:AB_2255011 |
| E-Cadherin (clone 67A4, metal Gd, mass 158) | Biolegend | Cat#324102; RRID:AB_756064 |
| Nanog (polyclonal, metal Tb, mass159) | Cell Signaling Tecchnology | Cat#3580; RRID:AB_2150399 |
| phospho-H3 S28 (clone HTA28, metal Gd, mass 160) | Biolegend | Cat#641002; RRID:AB_1227659 |
| CD24 (clone ML5, metal Dy, mass 161) | Biolegend | Cat#311102; RRID:AB_314851 |
| phospho-SMAD2(S465/467)/3(S423/425) (clone D27F4, metal Dy, mass 162) | Cell Signaling Tecchnology | Cat#8828; RRID:AB_2631089 |
| phospho-NFKb p65 S529 (clone 10-895.12.50, metal Dy, mass 163) | BD Biosciences | Cat#558393; RRID:AB_647284 |
| phospho-S6 S235/pS236 (clone N7-548, metal Dy, mass 164) | BD Biosciences | Cat#624084 |
| phopsho-Rb S807/S811 (clone J112-906, metal Ho, mass 165) | BD Biosciences | Cat#558389; RRID:AB_647229 |
| Cytokeratin 7 (clone SP52, metal Er, mass 167) | Abcam | Cat#ab183344 |
| Twist1/2 (polyclonal, metal Er, mass 168) | Bioss | Cat#BS-2441R; RRID:AB_10856752 |
| Slug (clone C19G7, metal Yb, mass 172) | Cell Signaling Tecchnology | Cat#9585; RRID:AB_2239535 |
| CD104 (clone 58XB4, metal Yb, mass 173) | Fluidigm | Cat#3173008B |
| Vimentin (clone D21H3, metal Yb, mass 174) | Cell Signaling Tecchnology | Cat#5741; RRID:AB_10695459 |
| phospho-AMPK T172 (clone 40H9, metal Lu, mass 175) | Cell Signaling Tecchnology | Cat#2535; RRID:AB_331250 |
| Biological Samples | ||
| Fresh human lung adenocarcinomas | Dr. JB Shrager, chief of thoracic surgery, Stanford University | |
| Chemicals, Peptides and Recombinant Proteins | ||
| EDTA | Sigma Aldrich | Cat#E7889 |
| Sodium azide | Sigma Aldrich | Cat#S2002 |
| Paraformaldehyde 16% | Electron Microscopy Sciences (EMS) | Cat#15710 |
| Cisplatin | Sigma Aldrich | Cat#P4394 |
| 6-Diazo-5-oxo-L-norleucine (DON) | Sigma Aldrich | Cat#SD2141 |
| Trizma base | Sigma Aldrich | Cat#T1503 |
| Palbociclib | Sigma Aldrich | Cat#PZ0383 |
| Protease inhibitor cocktail | Roche | Cat#11697498001 |
| Sodium fluoride (NaF) | Sigma Aldrich | Cat#215309 |
| Sodium orthovanadate (Na3VO4) | Sigma Aldrich | Cat#S6508 |
| EGTA | Sigma Aldrich | Cat#E4378 |
| tris(2-carboxyethyl)phosphine (TCEP) | Sigma Aldrich | Cat#C4706 |
| Pierce™ Trypsin Protease, MS Grade | ThermoFisher Scientific | Cat#90058 |
| Iodoacetamide | Sigma Aldrich | Cat#I6125 |
| Ammonium bicarbonate | Sigma Aldrich | Cat#11213 |
| PNGase F | New England Biolabs | Cat#P0704S |
| Acetonitrile | ThermoFisher Scientific | Cat#A955-500 |
| Formic acid | ThermoFisher Scientific | Cat#A117-50 |
| Anhydrous DMSO | ThermoFisher Scientific | Cat#AA439985Y |
| Triton X-100 | Sigma Aldrich | Cat#T8787 |
| Reagents | ||
| RPMI-1640 | Gibco | Cat#11875093 |
| Fetal Bovine Serum | Gibco | Cat#26140079 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat#15140122 |
| Tissue storage solution | Miltenyi Biotec | Cat#130-095-929 |
| MACS SmartStrainer | Miltenyi Biotec | Cat#130-093-237 |
| Red Blood Cell Lysis Solution | Miltenyi Biotec | Cat#130-094-183 |
| Anti-Fibroblast MicroBeads | Miltenyi Biotec | Cat#130-050-601 |
| TrypLE | Gibco | Cat#12605-010 |
| eBioscience™ Permeabilization Buffer | Invitrogen | Cat#00-8333-56 |
| Cell strainer snap cap tubes | Corning | Cat#352235 |
| RNA later stabilization solution | Invitrogen | Cat#AM7020 |
| C18 columns | ThermoFisher Scientific | Cat#60109-001 |
| Phenylboronic Acid (PBA) columns | Agilent | Cat#12102018 |
| ProLong™ Gold Antifade Mountant with DAPI | Invitrogen | Cat#P36935 |
| Goat serum | ThermoFisher Scientific | Cat#88RNG001 |
| Critical Commercial Assays | ||
| MACS Tumor Tissue Dissociation Kit | Miltenyi Biotec | Cat#130-095-929 |
| MACS Lung Dissociation Kit | Miltenyi Biotec | Cat#130-095-927 |
| Zombie Aqua™ Fixable Viability Kit | BioLegend | Cat#423102 |
| Pierce BCA protein assay kit | ThermoFisher Scientific | Cat#23225 |
| Experimental Models: Cell Lines | ||
| Ex vivo derived human NFs, TAFs and CAFs | This study | |
| Human HCC827 adenocarcinoma cell line | Mallick lab, Stanford University | RRID:CVCL_2063 |
| Human HCC827 Luc2-eGFP adenocarcinoma cell line | Mallick lab, Stanford University | |
| Deposited data | ||
| RNAseq data | The list of DEGs is accessicible in the Supplementary Data. The raw data will be deposited on GEO. | |
| CyTOF data | Upon request. | |
| Flow Cytometry data | Upon request. | |
| Mass Spectrometry (O-glycoproteomics) data | The list of O-glycopeptides and putative O-glycosites is accessible in the Supplementary Data. The raw mass spectometry files will be shared upon request. | |
| Software and Algorithms | ||
| Cytobank | Cytobank Inc., Santa Clara, USA | |
| PHENOSTAMP | Karacosta et al., 2019, https://github.com/anchangben/ PHENOSTAMP | |
| R | https://www.R-project.org | |
| Ingenuity Pathway Analysis (IPA) Software | Quiagen, Redwood City, USA | |
| GraphPad Prism 8.4 | GraphPad Software, San Diego, USA | |
| MSnbase package | Bioconductor, Gatto and Lilley, 2012 | |
| Byonic | Protein Metrics, Cupertine, USA, Bern et al., 2012 | |
| Mass-tag cell barcoding (MCB) algorithm | Zunder et al., 2015, https://github.com/nolanlab/single-cell-debarcoder/releases/latest REAGENT | |
| X-Shift | Samusik et al., 2016, https://github.com/nolanlab/vortex | |
| Salmon version 0.9.1 | Patro et al., 2017, https://github.com/COMBINE-lab/salmon/releases | |
| Databases | ||
| Uniprot | https://www.uniprot.org/ | |
| OGP | Huang et al. bioRixv, http://www.oglyp.org/ | |
Results
The fibroblasts from the invasive tumor edge have a distinct phenotype compared to those from the tumor core.
Fibroblasts from the leading edge of tumors have been shown to facilitate invasion of cancer cells (9) but have not been fully characterized. To better understand fibroblasts from the invasive tumor edge, we established matched primary fibroblast cell cultures from different tumor regions, namely Normal Fibroblasts (NFs, >5 cm away from the tumor), Tumor-Adjacent Fibroblasts (TAFs, invasive tumor edge) and Tumor Core Fibroblasts (TCFs, tumor core) (Fig. 1A) from fresh human LUAD clinical specimens (Table 1). We find that the three subtypes of fibroblasts present striking differences (Fig. 1). The fibroblast typical morphology is defined as elongated cells with extended cell features that show a fusiform or spindle-like shape. In addition, fibroblasts are often identified by their ability to adhere to plastic and their lack of markers that indicate other cell lineages (2,31). NFs demonstrate the typical spindle-like morphology commonly associated with fibroblasts and TCFs show enlarged cellular bodies, also described as cruciform or stellate shaped, which agrees with the commonly described “activated” fibroblast morphology, also known as CAF phenotype (2,39,40) (Supplementary Fig. S1C). In contrast, TAFs are filiform with a striated organization, a distinct morphology for tumor fibroblasts. We then profiled the NFs, TAFs and TCFs using flow cytometry and RNA sequencing (RNA-seq). To visualize the flow cytometry data, we applied the high-dimensional embedding tSNE algorithm to project α-SMA, FSP1, GFAT2, CD10, p21 and vimentin markers for each cell onto a two-dimensional space. NFs, TAFs and TCFs clustered into distinct regions on the tSNE plot with some overlap between NFs and TAFs. For each clinical specimen, α-SMA and FSP1 were consistently high in TCFs, intermediate in the NFs and low in TAFs; the other markers were more variable across specimens (Fig. 1B, Supplementary Fig. S2A and S2B). From the RNA-seq data, we found that none of the canonical CAFs markers were significantly differentially expressed among the different fibroblast subtypes (Supplementary Fig. S2C and Supplementary Data S1), but the functional gene set enrichment analysis revealed that NFs were uniquely enriched for glycogen degradation, TAFs, for the thioredoxin redox metabolism, as well as lipid signaling mediated by Tubby pathways, and TCFs, for inflammatory and pro-fibrotic pathways including IL-10 and NF-κB signaling (Fig. 1C–1E, Supplementary Fig. S2D and Supplementary Data S2). Of note, all fibroblast subtypes, including NFs, were enriched for pathways associated with a stress response (p38 MAPK), suggesting that our NFs may also be partially activated (Fig. 1E).
Figure 1. Analysis of LUAD fibroblasts derived from different tumor regions.
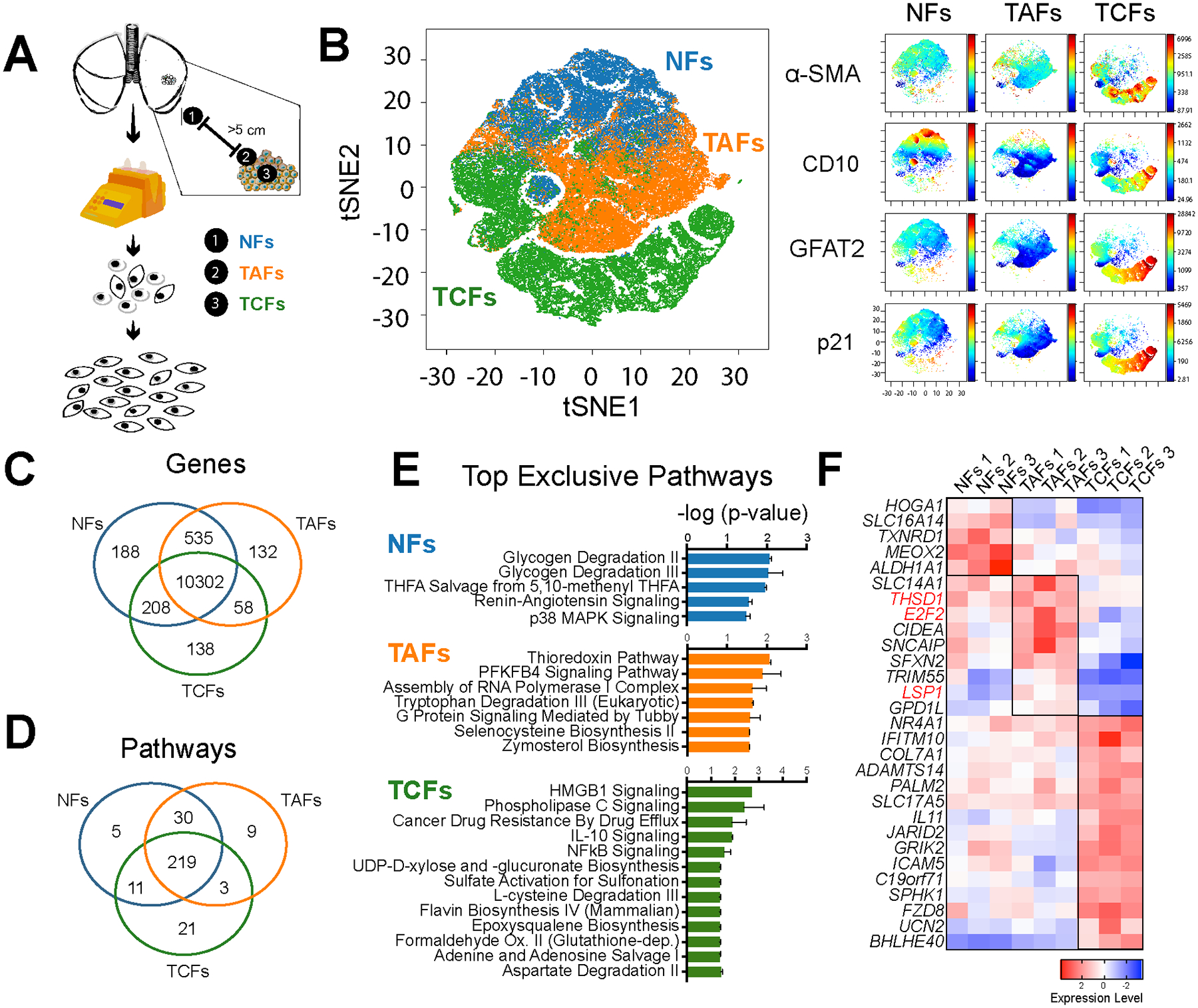
A, Fibroblast expansion workflow of LUAD fresh specimens. B, Phase contrast microscopy images of NFs, TAFs and TCFs. Specimen 2 is displayed here. C, Representative viSNE analysis of NFs, TAFs and TCFs, using α-SMA, FSP1, GFAT2, CD10, p21 and vimentin as clustering markers. Other biological replicates are available in Supplementary Fig. S1C. Venn diagram showing the gene overlap measured by RNAseq (TPM >1 in all biological replicates of each subtype). D, Venn diagram showing pathway overlap (E) and bar graph (F), showing the top exclusive pathways in NFs (n=3), TAFs (n=3) and TCFs (n=3), analyzed with Ingenuity Pathway Analysis software. G, Heatmap showing relative normalized gene expression of top DEGs in NFs, TAFs and TCFs.
Table 1.
Clinical annotations and histopathological information of LUAD specimens analyzed in this study
| Specimen | NFs | TAFs | TCFs | Tumor size (cm) | Differentiation status | Prior Treatment | Comorbordities | Smoker status | Common mutations | Classification | Node status | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | X | X | X | 3.5 | Well | None | None | Never | EGFR exon 19 deletion | Acinar 85%, lepidic 10%, pipillary 5% | N0 | ||||||||||||||||||
| 2 | X | X | X | 2.6 | Moderate | None | Hypertension | Former | None | Papillary 50%, acinary 30%, lepidic 20% | N1 | ||||||||||||||||||
| 3 | X | X | X | 3.5 | Moderate | None | Hypertension | Never | None | Acinar 70%, papillary 25%, lepidic 5% | N1 | ||||||||||||||||||
| Specimen | Sex | Stage | Tumor size (cm) | Differentiation status | Prior Treatment | Comorbordities | Smoker status | Common mutations | Classification | Node status | |||||||||||||||||||
| 59 | F | 2 | 2.1 | Unknown | None | Hypercholesterolemia, hypertension, disc rupture L4-L5 | Never | Unknown | Invasive adenocarcinoma, papillary predominant | N0 | |||||||||||||||||||
| Number of patients | Grade | Histology | Stage | Cancer cell/Stroma compartmentalization | |||||||||||||||||||||||||
| Poor | Moderate | Well | Unknown | Adeno | Broncho-alveolar | Mixed adeno/broncho-alveolar | 1 | 2 | 3 | 4 | Y | N | |||||||||||||||||
| 160 | 54 | 138 | 53 | 15 | 217 | 32 | 11 | 146 | 70 | 43 | 1 | 30 | 230 | ||||||||||||||||
Several interesting markers were identified among the genes most uniquely expressed in TAFs, as compared to NFs and TCFs, including the E2F2 transcription factor (Fig. 1F), known for its role in proliferation of, and association with, a poor prognosis in LUAD (41,42). TAFs also overexpressed Thrombospondin Type 1 Domain Containing 1 (THSD1), a marker of hematopoietic stem cell (43), and the fibrocyte marker leukocyte-specific protein 1 (LSP1). In addition, TAFs show a moderate expression of α-SMA (Fig. 1B, Supplementary Fig. S2B and S2C) as well as a significant increase in antigen presentation compared to TCFs (Supplementary Fig. S2E), which are also known fibrocyte properties (44). This fibrocyte signature is intriguing because fibrocytes are antigen-presenting leukocytes derived from peripheral blood mononuclear cells implicated in wound healing and displaying fibroblast-like properties. Fibrocytes are present in injured organs and have both inflammatory features of macrophages and tissue remodeling properties of fibroblasts (44). Taken together, these results show a distinct subtype of tumor fibroblasts, namely TAFs, which display some fibrocyte-like properties and suggest a hematopoietic origin.
TAFs, but not TCFs, induce a mesenchymal phenotype and proliferation in LUAD cancer cells.
To understand how the fibroblasts isolated from different tumor regions affect cancer cells, we analyzed lung cancer adenocarcinoma cells, using the HCC827 cell line, after cell-cell contact cocultures for 24 hours with NFs, TAFs and TCFs, and performed RNAseq analysis on the sorted compartments (Fig. 2A). HCC827 cells were chosen for their well characterized epithelial-to-mesenchymal (EMT) transition under TGF-β stimulation at the single-cell resolution, using mass cytometry (31). Strikingly, the images of HCC827 cocultured with TAFs and TCFs greatly differ: HCC227 intermixed with TAFs, but HCC827 formed tightly packed islands of epithelial cells when cocultured with TCFs (Fig. 2B and Supplementary Fig. S3A). Then, we performed gene set enrichment analysis on the differentially expressed genes (DEGs) obtained from the sorted cancer cells cocultures relative to their matched monoculture, and highlighted the differences in functional enrichment between HCC827 cocultures with TAFs versus TCFs (Fig. 2C–2E and Supplementary Data S3). Of note, cancer cells cocultured with the TAFs showed a significant increase for pathways associated with migration, invasion and micropinocytosis, among others; in contrast, TCFs promoted functional enrichment of differentiation pathways and glycosaminoglycan production in the cancer cells. These transcriptional differences are consistent with the morphological differences observed in the HCC827s in the cocultures with TAFs vs TCFs.
Figure 2. RNAseq analysis of HCC827 after coculture with NFs, TAFs and TCFs.
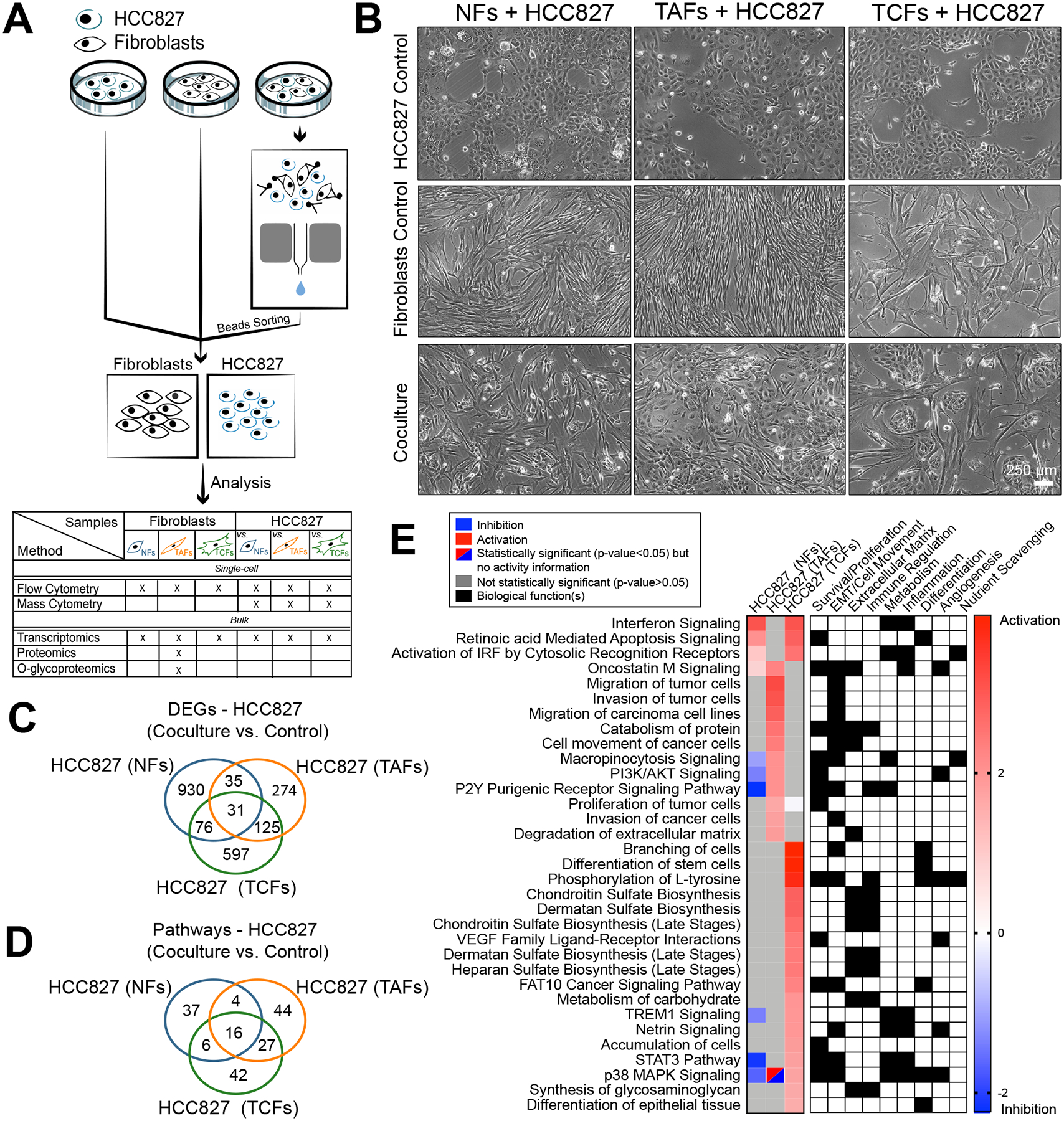
A, Coculture, cell separation and downstream analysis workflow. B, Phase contrast microscopy images of NFs, TAFs and TCFs cocultures from specimen 2 with HCC827 lung adenocarcinoma cell line. Biological replicates are available at Supplementary Fig. S2A. C, Venn diagram showing the gene overlap between HCC827 cocultures with NFs (n=3), TAFs (n=3) and TCFs (n=3) compared to their monoculture control measured by RNAseq (p < 0.05, fold-change > 0.5). Venn diagram showing the pathways overlap and (D), heatmap showing the main pathways/biological functions differences between HCC827 cocultures with NFs (n=3), TAFs (n=3) and TCFs (n=3) compared to their monoculture control (E) analyzed with the IPA software.
Next, HCC827 cocultures were analyzed with mass cytometry (CyTOF) using an EMT panel of markers and projected on PHENOSTAMP, a publicly available lung cancer reference map of EMT phenotypic states (31). Only the TAFs significantly induced EMT in the HCC827, as illustrated on the PHENOSTAMP projections (Fig. 3A and B). Similar results were also observed with the A549, another LUAD cell line (Supplementary Fig. S3B). In addition, we used the forced-directed layout and X-shift clustering algorithm to visualize the mesenchymal cell populations after coculture (Fig. 3C and Supplementary Fig. S3C–S3E). Again, only the HCC827 after direct cell-cell contact coculture with TAFs showed a mesenchymal phenotype, as confirmed by the position of the HCC827 cocultured cells (dark green cluster) with the other mesenchymal cells (Fig. 3C). HCC827 showed a significant decrease of epithelial markers, transcription factors and signaling, as well as an increase of mesenchymal markers after direct contact with TAFs. Interestingly, supernatant from TAFs did not promote a significant EMT response in HCC827, suggesting that direct contact between cancer cells and TAFs is necessary to induce the mesenchymal phenotype in the cancer cells (Supplementary Fig. S4A). We corroborated these findings in situ by IF on LUAD tumor serial sections using LSP1 as a TAF-specific marker. When compared to the other subtypes of fibroblasts analyzed in our study, we find LSP1 to be expressed exclusively in TAFs (Fig. 1F). To identify TAFs-like, TCFs-like and cancer cells in situ, we used a combination of LSP1, panCK and vimentin expression, spindle-shaped morphology and H&E staining (Supplementary Fig. S4B). These preliminary results demonstrate the colocalization of vimentin with panCK in cancer cells close to TAFs-like cells, suggesting a partial EMT phenotype in cancer cells proximal to TAF-like, but not TCF-like cells. These results need to be further validated using additional TAFs, TCFs and EMT markers at larger scale, ideally using highly multiplexed IF imaging instead of serial sections.
Figure 3. CyTOF analysis of HCC827 after coculture with NFs, TAFs and TCFs.
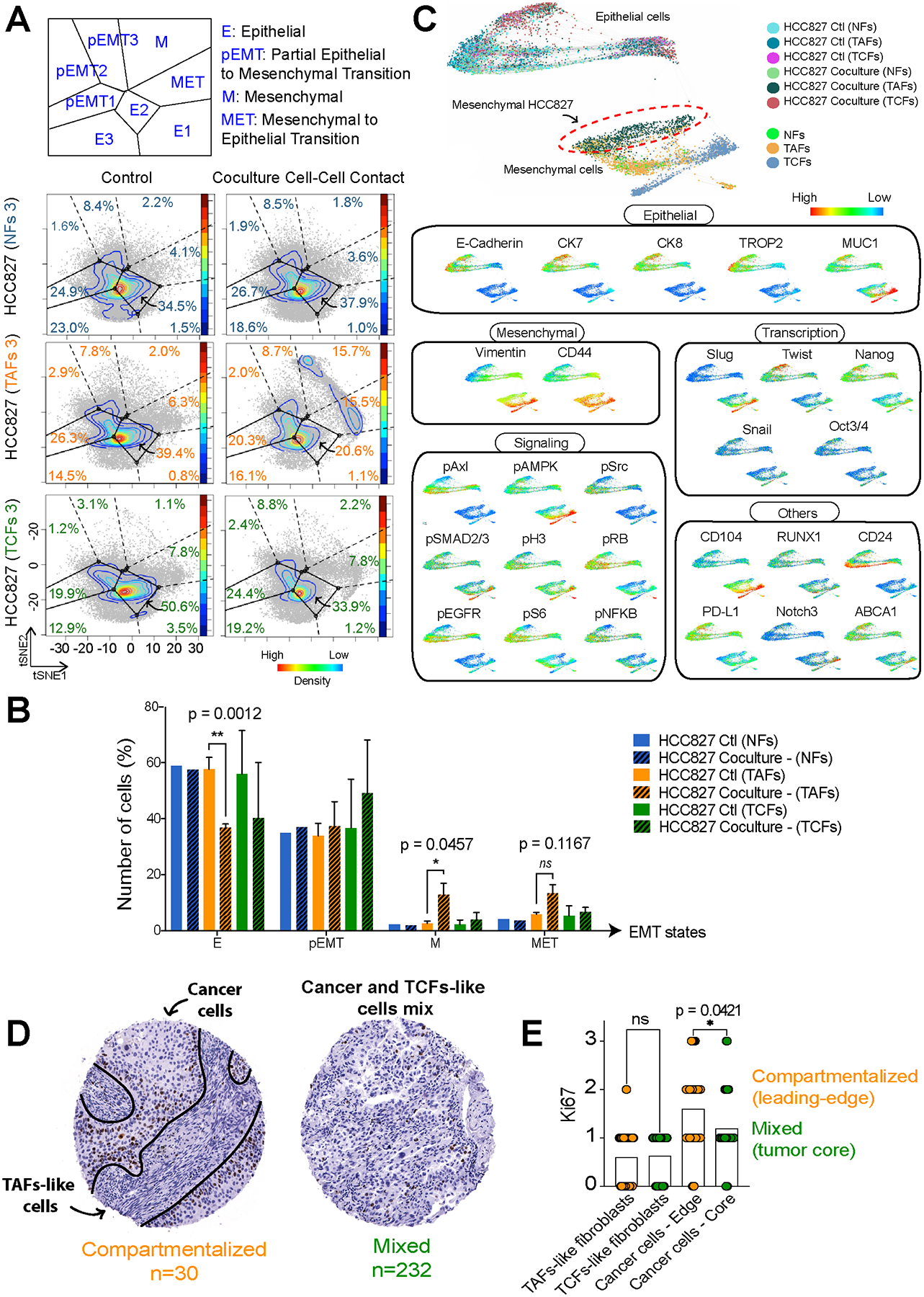
Projections of the HCC827 monoculture controls and matched cocultures after cell-cell contact with NFs, TAFs and TCFs onto the EMT–MET PHENOSTAMP phenotypic map (A) and bar graph quantification (B) of the number of cells in each region of the map. C, Single-cell force-directed layout colored by protein expression of indicated markers of HCC827 cocultures with NFs, TAFs and TCFs using X-shift clustering (arcsinh transformed data of specimen 3). See Supplementary Fig. S3C for expression scale bars and S3D and S3E for biological replicates. D, IHC representative examples of LUAD specimens from the lung TMA showing compartmentalized and mixed samples and (E) Ki67 quantification of fibroblasts and cancer cells.
Next, we quantified the effect of TAFs and TCFs on cancer cell proliferation using a tumor microarray (TMA) cohort of primary lung adenocarcinoma of 260 patients (Table 1). Because this TMA does not have cores derived from the invasive edge of the tumor, we classified the cores into two distinct groups described as “mixed” or “compartmentalized”. Briefly, we defined cores as “mixed” when cancer cells were intermixed with stromal cells without being able to easily distinguish between stromal and cancer cells based on morphology. In contrast, cores were classified as “compartmentalized” when the cancer cells areas were sharply delineated by streams of spindly stromal cells (Fig. 3D). Out of the 260 LUAD specimens included in our analysis, 30 samples were classified as compartmentalized. This group was used as our surrogate leading-edge cohort. We then analyzed Ki67 positivity in both fibroblasts and cancer cells in all 260 samples. Interestingly, we observed an increase in cancer cell proliferation at the leading edge of the cancer cell regions in the compartmentalized group, but not in the mixed specimens (Fig. 3E). These results suggest that cancer cells are likely to be more proliferative at the leading edge of the tumor compared to the tumor core, and warrant further investigation.
HCC827 cells induce differential metabolic reprogramming in TAFs vs TCFs.
To measure the effect of cancer cells on primary fibroblasts, we then analyzed the transcriptome of NFs, TAFs and TCFs 24 hours after coculture with HCC827 cells (Fig. 4A, 4B, Supplementary Fig. S4C–S4E and Supplementary Data S2). Gene set enrichment analysis shows that TAFs experience a significant decrease in cell movement and signaling, particularly lipid signaling (inositol compounds). Conversely, TCFs experience an increase in most of these pathways, as well as pathways associated with metabolism and immune regulation (Fig. 4C). Overall, TAFs activity is drastically downregulated without significantly affecting cell death (Supplementary Fig. S4F), as opposed to almost all of the same pathways significantly upregulated in NFs and TCFs cocultures (Supplementary Fig. S4C).
Figure 4. Gene expression analysis of NFs, TAFs and TCFs after coculture with HCC827 lung adenocarcinoma cancer cells.
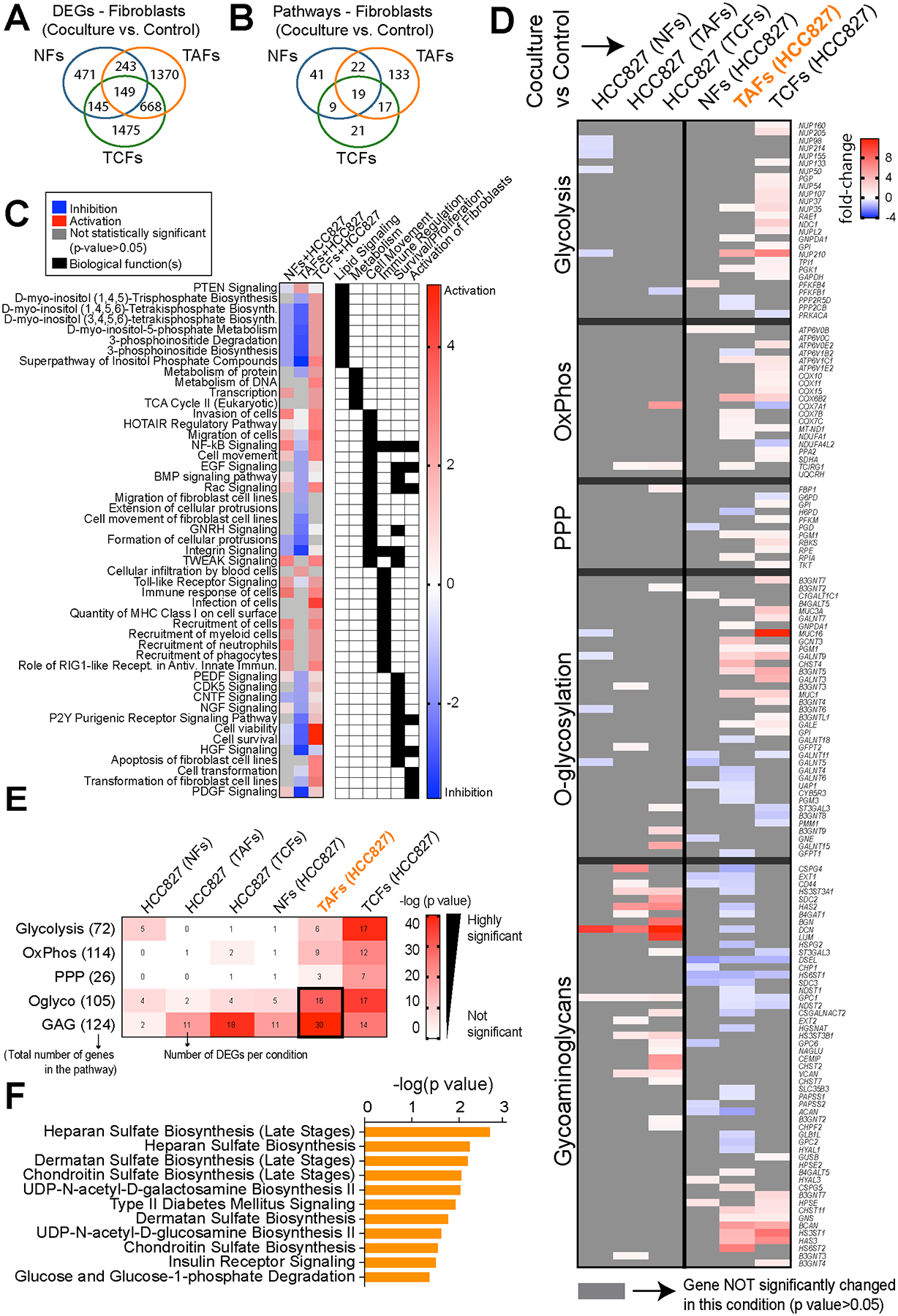
A, Venn diagram showing the gene overlap between NFs (n=3), TAFs (n=3) and TCFs (n=3) cocultures with HCC827 compared to their monoculture control measured by RNAseq analysis (p < 0.05, fold-change > 0.5). Venn diagram showing pathway overlap (B), heatmap showing the main pathways/biological functions differences analyzed with the IPA software (C) and heatmap showing the fold-change of DEGs in the main glucose metabolic pathways between NFs (n=3), TAFs (n=3) and TCFs (n=3) cocultures with HCC827, (D) and heatmap showcasing the number of overlap genes between DEGs in each condition and within each metabolic pathway (E). Color of heatmap represents -log(p-value) of the gene overlap calculated using a hypergeometric test. The numbers in parentheses show the total number of genes from each MSigDB gene set and tile numbers show the number of DEGs for each condition. See p values at Supplementary Fig. S4G. F, Bar graph showing exclusive pathways related to glycans biosynthesis enriched in TAFs cocultures compared to their monoculture control and analyzed with the IPA software.
Prior work from our group reported transcriptional alterations in CAFs and metabolic reprogramming toward HBP in primary LUAD tumors (16). Here, we explored the significant DEGs involved in metabolic reprogramming for all cocultures in the main glucose pathways (glycolysis, oxidative phosphorylation, pentose phosphate pathway, O-glycosylation and glycosaminoglycans biosynthesis) using the Molecular Signatures Database (MSigDB) gene sets. Strikingly, transcriptomic changes in metabolic pathways occur primarily in tumor fibroblasts as opposed to cancer cells (Fig. 4D, 4E and Supplementary Fig. S4G), as illustrated by the low number of genes significantly changed in cancer cell cocultures (relative to monoculture control). As expected from previously published work on CAFs, several metabolic pathways were found to be dysregulated in our TCFs, but, interestingly, we show that the main pathways modulated in TAFs were related to glycan biosynthesis (Fig.4E). We also highlight several other exclusive pathways related to glucose metabolism with unknown activity in the TAFs cocultures, including glucose degradation, nucleotide sugars and glycoproteins biosynthesis, all of which were identified through the Ingenuity Pathway Analysis (IPA) software (Qiagen, Fig. 4F, Supplementary Data S2 and S3). These transcriptomic results suggest that fibroblasts from the invasive edge of the tumor undergo differential metabolic reprogramming compared to TCFs. In particular, TAFs upregulate glycoproteins biosynthesis when in direct cell-cell contact with cancer cells, suggesting an important role of glycans at the tumor invasive edge.
The TAFs O-glycoproteome reveals previously unidentified O-glycoproteins.
Our transcriptomic analysis suggests that the hexosamine (aminosugars biosynthesis leading to O-glycosylation) and glycan biosynthesis pathways in TAFs are modulated by tumor-stroma crosstalk. Because these phenomena are largely uncharacterized, we assessed the effects attributed to cancer cells-TAFs crosstalk through mono- and coculture analysis on the proteome and O-glycoproteome, using mass spectrometry. Briefly, TAFs were sorted after coculture with HCC827; then, their protein extracts were digested with trypsin and enriched for O-glycopeptides, using an optimized method based on previously published work (25). For a control, a matched shotgun proteomic sample not subjected to O-glycopeptide enrichment was prepared and analyzed for each sample. Across all conditions, the proteomic analysis revealed 4495 unique proteins in which 337 O-glycoproteins were found to be O-glycosylated on 780 putative O-glycosites (Fig. 5A and Supplementary Data S4). Of the 337 O-glycoproteins identified in TAFs, 43 proteins were found to be differentially O-glycosylated (adjusted for protein level differences) after coculture with HCC827, most of them unknown O-glycoproteins according to the most comprehensive repository, the OGP database (23). The majority of these proteins were enzymes, transcription regulators, transporters or cytoskeleton proteins (Fig. 5B). O-glycans are classified into common core structures depending on their composition and sugar configuration (Tn antigen and core 1 to 8). Of the 43 differentially O-glycosylated proteins measured here, core 1 O-glycans, the most common core structure (45), were the most abundant (Fig. 5C). In addition, approximatively 50% of the O-glycans were sialylated, a common glycan modification in cancer that blocks further linear extension of O-glycan chains (45). Overall, we generated the first stromal O-glycoproteome in the context of tumor-stroma crosstalk with their putative O-glycosites, providing a new resource for developing targeted O-glycosylation strategies and tools.
Figure 5. Crosstalk with HCC827 alters the TAFs O-glycoproteome.
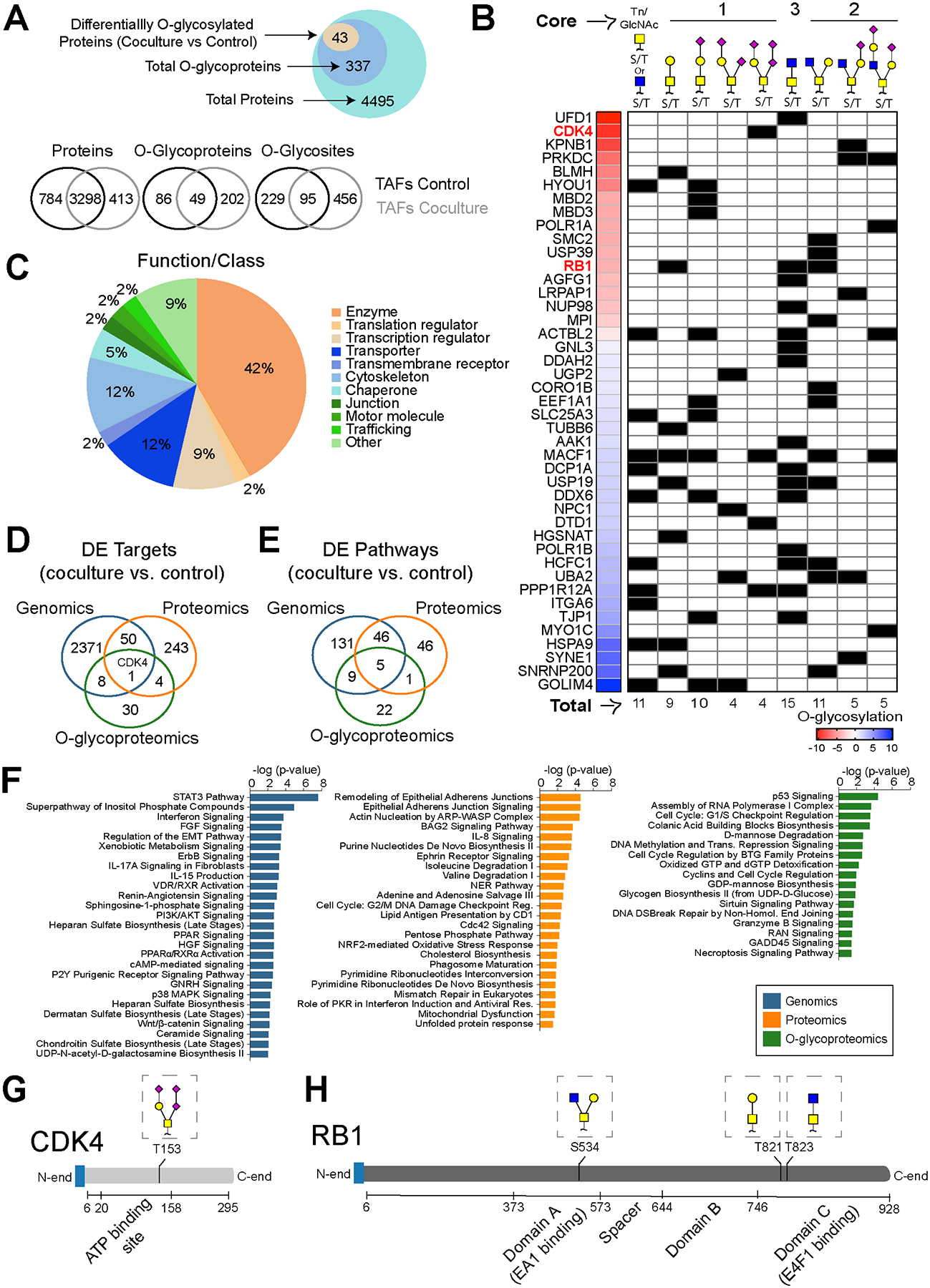
A, Venn diagram showing proteins, O-glycoproteins and O-glycosites found in TAFs monocultures and cocultures analyzed by mass spectrometry. B, Pie charts showing the main classes of the differentially O-glycosylated proteins after coculture identified by mass spectrometry. C, Heat map and summary table of the differentially O-glycosylated proteins and glycan heterogeneity after coculture with HCC827. Venn Diagram showing the overlap of targets (D) and the DE pathways identified in TAFs cocultures (relative to control monoculture) (E) using different methods of analysis. F, Bar graph highlighting a sample of the top enriched pathways in TAFs cocultures at the genomic, proteomic and O-glycoproteomic levels. CDK4 (G) and RB1 (H) protein domains and O-glycan positions.
Next, the differentially expressed proteins and O-glycoproteins were analyzed for pathway enrichment and compared with the transcriptomic data (Fig. 5D–F). The pathways measured with different assays show a modest overlap suggesting that each data dimension highlights unique molecular functional characterization in TAFs cocultures. Interestingly, the activity measured for shared pathways did not necessarily increase or decrease conjointly at the gene and protein levels, highlighting the relevance of a multi-omics approach to more accurately interpret biological activity (Supplementary Fig. S4H). Then, we compared the top unique pathways (Fig. 5F and Supplementary Data S5) and summarized the biological functions for each method of analysis (Supplementary Data S6). Briefly, cell proliferation, immune response and cell movement are regulated primarily at the gene expression level, energy, amino acids and nucleotide metabolism as well as cytoskeleton remodeling are regulated at the protein level, and O-glycosylation regulates cell cycle, cell death and glucose, mannose and guanosine metabolism. Taken together, these results support the use of a multi-omics approach that includes the transcriptomics, proteomics and O-glycoproteomics to more fully characterize biological functions.
O-glycosylation regulates the CDK4-pRB axis in TAFs and modulates the anti-cancer properties of CDK4/6 inhibitors.
To validate the relevance of the stromal O-glycoproteome in tumor-stroma crosstalk, we explored the role of O-glycosylation in the CDK4-pRB axis as an example, since this signaling pathway stood out from our analysis. Briefly, cyclin dependent kinase 4 (CDK4) phosphorylates the tumor suppressor retinoblastoma protein 1 (RB1), leading to its inactivation and uncontrolled cell proliferation. In our multi-omics analysis, only one target, CDK4, was found to be differentially upregulated at the gene, protein and O-glycosylation levels in the TAFs after coculture with HCC827 cells (Fig. 5D). In addition, both CDK4 and RB1 were found to be O-glycosylated at a higher level in TAFs after direct cell-cell contact with cancer cells. Interestingly, CDK4 is O-glycosylated at T153 in TAFs cocultures (Fig. 5G and Supplementary Fig. S4I), which is expected to be in the active site of the enzyme (46). Moreover, upon coculture, RB1 is O-glycosylated at S534, T821 and T823 glycosites (Fig. 5H), which are predicted to be located within transcription factor interaction domains of the protein. Given the role of EMT in resistance to CDK4 inhibitors (47), the pro-EMT effect of TAFs on cancer cells observed in Fig. 3, and the location of the O-glycans on CDK4 and RB1, we hypothesized that O-glycosylation of the stroma could affect the efficacy of CDK4/6 inhibitors, a recent family of anti-cancer agents targeting the CDK4-pRB axis (48).
Unfortunately, no specific O-glycosylation inhibitors to these sites are available, nor specific antibodies for O-glycans, due to the limited knowledge and tools available for O-glycobiology. In addition, primary fibroblast mutants from human clinical specimens are notoriously difficult to generate, due to their limited lifespan. To test this hypothesis, we treated TAFs:HCC827-eGFP cocultures with palbociclib, a clinically-approved CDK4/6 inhibitor, alone and in combination with 6-diazo-5-oxo-L-norleucine (DON), a glutamine analog that inhibits HBP and reduces O-glycosylation, using drug concentrations in accordance with previous studies performed in non-small cell lung cancer cell lines (49,50). The main goal of this experiment was to validate the link between O-glycosylation and the efficacy of CDK4 inhibitors in the tumor stroma by measuring the phosphorylation of RB by CDK4 with and without O-glycosylation. To quantify the effect of palbociclib alone and in combination with DON, we measured the proliferation marker Ki67 and phosphothreonine 826, the CDK4-preferred phosphosite on RB1 (uniport.org) in the cocultures at 48 hours post-treatment at single-cell resolution. We also used the marker p21 to differentiate proliferative from non-proliferative cells, since p21 stabilizes the Cyclin D/CDK4 complex leading to RB phosphorylation but is also one of many markers characterizing cell senescence (51,52). Briefly, we focused our analysis on two main cell populations, referred to as: (i) “proliferative” cells, defined as {Ki67high, pRBhigh, p21high}-cells; and (ii) “non-proliferative” cells, defined as {Ki67low, pRBhigh, p21high}-cells that were identified on a forced-directed layout projection of two independent experiments (biological replicates) using TAFs clinical specimens from different patients. The cocultures treated with palbociclib in combination with DON exhibited a decrease of proliferative TAFs and an increase of non-proliferative TAFs (Fig. 6A–E) compared to those treated with palbociclib or DON alone. This effect was not observed in the monocultures (Supplementary Fig. S5). Moreover, the combination treatment of DON and palbociclib led to a TCF-like morphology change in the TAFs, consistent with a decreased proliferation rate and increased size of the cells. These results indicate that O-glycosylation of the CDK4-pRB axis modulates the effect of palbociclib in TAFs cocultured with cancer cells; this effect is significantly improved when inhibiting O-glycosylation with DON.
Figure 6. O-glycosylation of the CDK4-Rb axis in TAFs modulates Palbociclib efficacy.
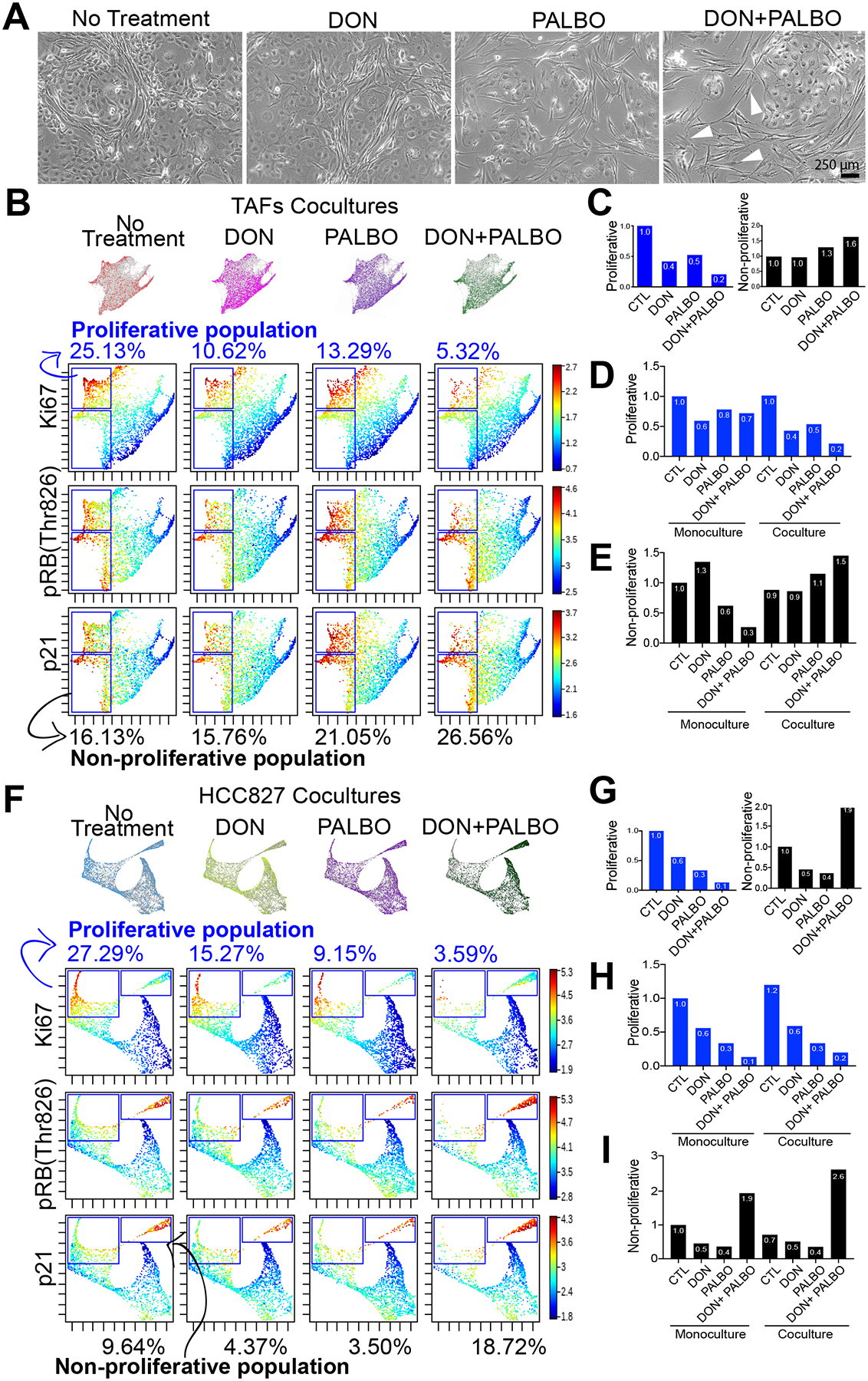
A, Phase-contrast microcopy of TAFs:HCC827 cocultures treated with 10 μM DON, 1 μM palbociclib or a combination of both. White arrowheads show examples of TAFs that have gained an activated TCFs-like morphology. B, Force-directed layout of TAFs cocultures showing proliferative (upper gate) and non-proliferative (lower gate) TAFs and (C) bar graph quantification of percentage ratios relative to non-treated cocultures. Bar graph quantification showing all conditions of TAFs mono- and cocultures relative to untreated monoculture for (D) proliferative and (E) non-proliferative TAFs. Force-directed layout of HCC827 cocultures showing proliferative (left gate) and non-proliferative (right gate) cancer cells (F) and bar graph quantification of percentage ratios relative to non-treated cocultures. G, Bar graph quantification showing all conditions of HCC827 mono- and cocultures relative to non-treated monoculture for (H) proliferative and (I) non-proliferative cancer cells. Results are from two independent experiments with biological replicates combined (TAFs from specimens 1 and 3).
Modulating O-glycosylation of the CDK4-pRB axis in TAFs indirectly decreases the mesenchymal features of cancer cells.
It is crucial to understand the effect of cancer treatment on the stroma, but the cancer cells remain the ultimate target in cancer. Although we did not measure the HCC827 O-glycoproteome because it was not significantly altered at the transcriptomic level, we did measure the proliferation and mesenchymal features in the cancer cells following the combination treatment. Briefly, our analysis suggests that the combination of palbociclib and DON is more efficacious than palbociclib alone in reducing cancer cell proliferation. However, O-glycosylation of the CDK4-pRB axis in the cancer cells does not seem to be significantly modulated by cell-cell contact with TAFs, since no significant difference was observed between mono- and cocultures treated with DON (Fig. 6F–I and Supplementary Fig. S6).
Lastly, to assess whether the TAFs morphological transformation into TCF-like cells observed in the cocultures treated with DON and palbociclib affects the cancer cells, we analyzed the emergence of EMT (or partial EMT) by measuring the expression of CD44 in the cocultures by IF. Our rationale for analyzing the mesenchymal marker CD44 was based on our CyTOF analysis presented in Fig. 3, which shows a significantly lower expression of CD44 in the HCC827 cocultured with TCFs compared to TAFs. The IF staining on cocultures supports our hypothesis that TAFs may display a TCF-like phenotype with the combination treatment by showing a strong decrease of CD44 in the HCC827 when cocultured with TAFs, and by displaying a pattern resembling TCFs:HCC827 coculture morphology (Fig. 7 and Supplementary Fig. S7). However, more validations and quantifications are needed to confirm the effect of the treatment alone versus the indirect effect of the treated TAFs on the cancer cells. Overall, these results suggest not only that inhibiting O-glycosylation in combination with palbociclib reduces pro-tumor properties in TAFs and HCC827, but also that these changes in the stroma are likely to decrease the induction of mesenchymal features in cancer cells.
Figure 7. Decreased O-glycosylation with DON treatment indirectly prevents EMT in HCC827.
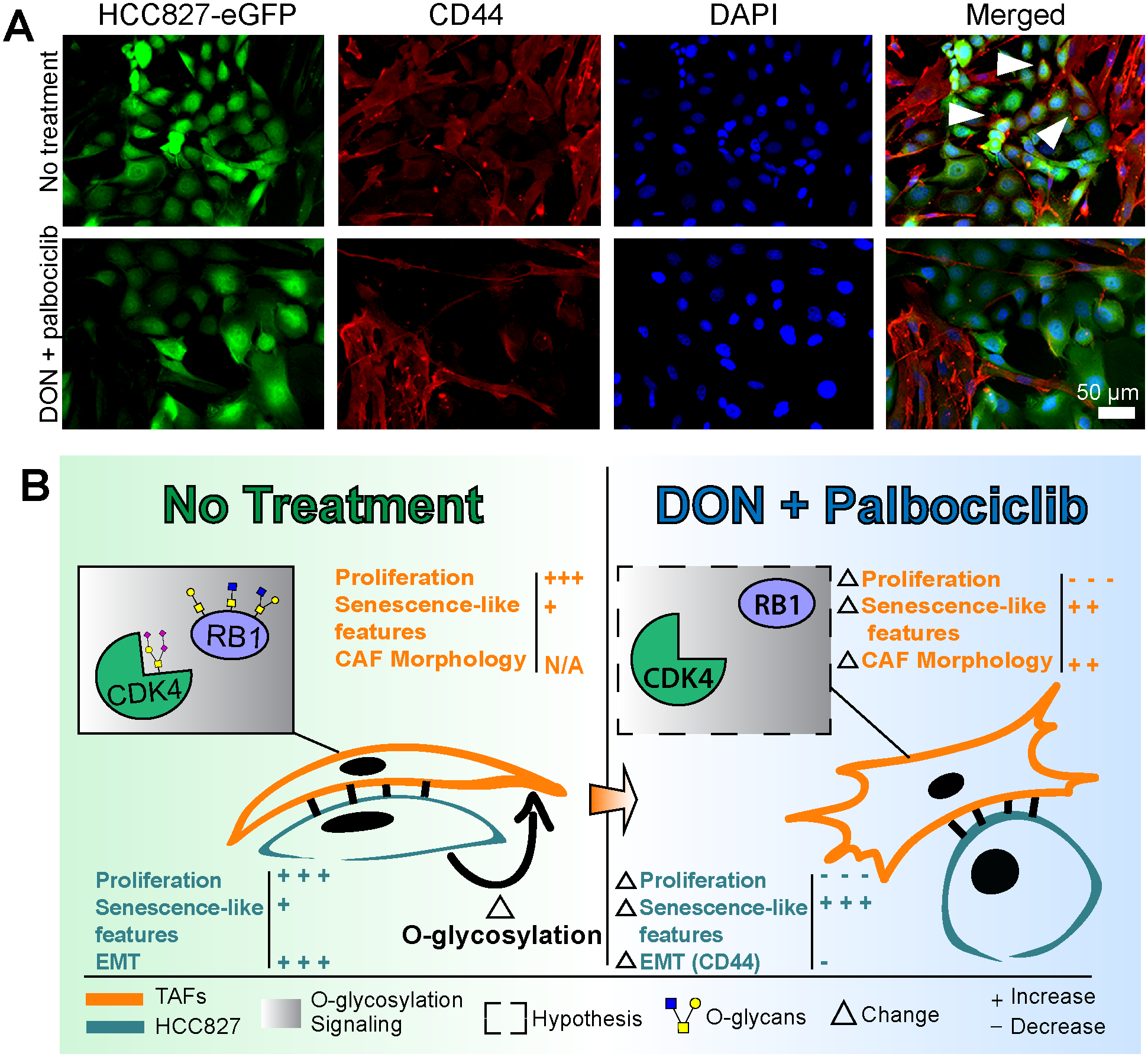
A, Immunofluorescence staining of HCC827-eGFP:TAFs coculture showing a decrease of the mesenchymal marker CD44 in cancer cells after treatment with DON and palbociclib. Cancer cells are represented by green pseudo color. TAFs are represented by red pseudo color only. CD44+ cancer cells are represented by co-staining with green and red pseudo colors. White arrowheads represent examples of CD44+ cancer cells. B, Summary figure of the main findings of the study showing that EMT in HCC827 and O-glycosylation of the CDK4-pRB axis in the stroma are induced in TAFs:HCC827 cocultures; this is prevented with the combination treatment of DON and palbociclib.
Discussion
The role of tumor fibroblasts at the tumor leading edge have been understudied. Moreover, studying fibroblasts in general is challenging for several reasons, including their phenotypic plasticity and lack of adequate markers. Here, we have characterized tumor-adjacent fibroblasts, designated here as TAFs, with a multi-omics approach that includes PTMs through O-glycoproteomics. We use single-cell mass cytometry to demonstrate the increased ability of TAFs to induce EMT in cancer cells compared to TCFs, as well as the pro-migratory and proliferative role of the stroma at the tumor leading edge. In addition, our transcriptomic analysis highlights that metabolic reprogramming at the leading edge is happening mainly in the stroma, as opposed to cancer cells, and can be measured in the context of tumor-stroma crosstalk through the O-glycoproteome. Our multi-omics analysis of TAFs highlights O-glycosylation of the CDK4-pRB axis as a consequence of tumor-stroma crosstalk, and suggests a new avenue for improving the efficacy of CDK4/6 inhibitors on cancer cells by modulating TAFs. Lastly, our study generated the first stromal O-glycoproteome of more than 300 O-glycoproteins in the context of tumor-stroma crosstalk with their putative O-glycosites, providing a new resource for developing targeted O-glycosylation strategies and tools.
Compared to TAFs, CAFs have been extensively studied. Here, we highlight key differences between CAFs-like fibroblasts from the tumor core, referred here as TCFs and TAFs, derived from primary LUAD. Our transcriptomic analysis showed an increase of invasion and cell movement signatures in HCC827 when cocultured with TAFs vs TCFs. Our CyTOF experiment confirmed the ability of TAFs (and not TCFs) to induce EMT more robustly in cancer cells in coculture experiments, which was only observed with direct cell-cell contact between TAFs and cancer cells. More precisely, our CyTOF results show a downregulation of cytokeratins and molecular signaling including pAXL, pSRC, pRB, pEGFR, pS6 and pNFKB in cancer cells when cocultured with TAFs, but not TCFs (in addition to the changes in E-cadherin, vimentin, CD44, CD24, Twist and MUC1 measured with PHENOSTAMP). These findings suggest that contact of TAFs with cancer cells decrease the cancer cells’ expression of cytokeratins, an important constituent of cellular junctions, which in turn disrupts cell-cell signaling. This general phenomenon has been associated with EMT (53,54) but the specific molecules mediating these interactions between cancer cells and TAFs warrants further investigation.
Another reason why these results are compelling is that TAFs express low to undetectable levels of the canonical CAFs markers. These observations are consistent with a recent murine study. Zhao et al. showed that TAFs, which they refer to as “peri-tumor fibroblasts,” isolated from human hepatocellular carcinoma specimens promoted proliferation and migration of cancer cells at a greater level compared to TCFs in a cancer mouse model (8). This effect was driven by STAT3 signaling, which we also find to be particularly enriched in human TAFs in our transcriptomic analysis (Supplementary Fig. S4D). Moreover, we found that TAFs are morphologically distinct from TCFs--consistent with a study of TAFs, which again referred to them as “peri-tumor fibroblasts”--isolated from tongue squamous cell carcinoma and shown to have different cell organization in coculture compared to CAFs (55). Ba et al. observed that CAFs grow in a more disorganized manner compared to peri-tumor fibroblasts. These observations are consistent with our results: we show that primary TAFs are organized in striated patterns compared to primary TCFs, which present as randomly dispersed during culture.
To date, the most promising anti-CAF therapies target the markers α-SMA and FAP but have had disappointing results, suggesting complex pro- and anti-tumor properties for CAFs (5,40). Alexeyenko et al. characterized the transcriptional landscape of normal skin and prostate fibroblasts after cell-cell contact cocultures with PC-3 tumor cells, and showed that normal fibroblasts have an inhibitory effect on cancer cells. Interestingly, the authors identified BHLHE40 and NR4A1 as top differentially expressed genes in the cancer-inhibitory fibroblasts after coculture with cancer cells. Of note, both of these markers were exclusively upregulated in our TCFs (Fig. 1F). In our cocultures of TCFs and cancer cells, TCFs surrounded the islands of cancer cells, which established striking epithelial morphology as opposed to the mesenchymal phenotype generally described when cancer cells are cocultured with CAFs (2,56,57). In addition, the cancer cells showed increased cell differentiation and glycosaminoglycan production. Differentiation of cancer cells has been associated with a less proliferative and invasive behavior in the histopathological classification of LUAD (58). However, physical proximity with CAFs and cancer cells has also been reported to reduce drug accumulation in carcinoma cells and increase treatment resistance (59). In contrast, in our cocultures of TAFs and cancer cells, the cancer cells showed an increased mesenchymal morphology, which is associated with increased migration features and proliferation. Interestingly, cancer cells are often described as either proliferative or migrative, also designated as the “Go or Grow” dichotomy (60), but our single-cell results support both phenomena to be happening at the same time in different cell populations. Overall, our results suggest greater pro-tumor roles for TAFs compared to TCFs, but future work exploring the meaning of these morphological and behavioral differences is needed to better understand the heterogeneous roles of cancer fibroblasts in cancer progression and drug response.
It is unclear whether TAFs and TCFs originate from the same cell lineage. Our results highlight LSP1 and THSD1 as exclusively overexpressed in TAFs compared to NFs and TCFs. THSD1 has been previously described as a marker of hematopoietic stem cell (43) and LSP1 is a common fibrocyte marker (44). Fibrocytes are considered to be mesenchymal cells that arise from monocyte precursors with both the inflammatory features of macrophages and the tissue remodeling properties of fibroblasts (44). Our transcriptomic results also show that TAFs express significantly higher expression of MHC class I and II molecules compared to TCFs MHC class II markers, which are normally expressed only by professional antigen-presenting cells including macrophages (61). These results propose that TAFs may be hematopoietic-derived cells recruited at the invasive edge of the tumor as they display fibrocyte-like properties. However, lineage-tracing analysis should be performed to confirm this hypothesis.
Previous results from our group highlighted the role of activated fibroblasts in metabolic reprogramming toward the HBP (16). Here, our work confirms that when either TCFs or TAFs are cocultured with cancer cells, they are both metabolically reprogrammed, but differently with TAFs that predominantly involve pathways associated with glycans biosynthesis. These results motivated us to expand our multi-omics approach to cover the O-glycoproteome in addition of the transcriptome and the proteome of TAFs in the context of tumor-stroma crosstalk. Interestingly, these different methods showed a modest overlap of biological functions and unique pathways enriched in each molecular dimension. Of note, O-glycosylation implicated protein associated with the regulation of the cell cycle, cell death and metabolism. This is not surprising as several factors involved in proliferation, apoptosis and cellular energetics have been shown to be stabilized by O-glycosylation (20). Interestingly, CDK4 was the only target differentially upregulated in TAFs, after coculture with cancer cells, across all -omics platforms used in this study. In-depth analysis of our O-glycoproteome data revealed the importance of the CDK4-pRB axis in the stroma as both of these proteins were differentially O-glycosylated after TAFs:HCC827 coculture. CDK4 is known to phosphorylate the tumor suppressor RB, leading to its inactivation. The inactive RB releases the transcription factor E2F2, which leads to cell cycle progression and proliferation (48); notably, the transcription factor E2F2 was uniquely upregulated in TAFs, relative to the TCFs and NFs, at the gene expression level. Our study is the first to identify specific O-glycosites with their associated O-glycoforms in the CDK4-pRB axis. Wells et al. have previously reported RB to be O-glycosylated in HeLa S3 cells at an unknown site but, to our knowledge, we are the first to report CDK4 to be O-glycosylated at T153 and to identify RB O-glycosites at S534, T821 and T823. Interestingly, T153 is predicted to be located in the ATP pocket of CDK4 (46,62), which is also known as the binding site for most CDK4 inhibitors (51). Glycans can physically restrict access to a binding site or chemically modify the affinity for a target (63). The tools to validate if O-glycosylation of CDK4 at T153 affects the binding affinity of palbociclib are not readily accessible, due to the limited availability of glycan antibodies and the challenges associated with generating primary fibroblast mutants. Instead, we inhibited O-glycosylation with DON and showed that the combination treatment reduces proliferation in both TAFs and HCC827 cocultures at a greater level than palbociclib alone. In addition, the combination treatment of DON and palbociclib promoted a TCFs-like morphology in cocultured TAFs, which, in turn, appears to reduce EMT in the HCC827. This aligns with previous studies where altering protein glycosylation mediated many cellular changes associated with EMT and cell–cell adhesion (21,64). However, we cannot exclude the possibility that some changes in the fibroblast morphology may be attributed to the reduced confluency caused by the treatment in this experiment, another limitation of our study.
The tumor suppressor role of RB has been extensively reviewed (65,66) but, to our knowledge, only one group previously reported RB to be regulated by O-glycosylation. We find RB to be O-glycosylated at T821 and T823 among the three O-glycosites detected. Lentine et al. showed that dephosphorylation of T821 is required for the cell to undergo apoptosis (67). In addition, these O-glycosites are located in the binding domain of the E4F1 protein, which binds to RB and indirectly stabilizes p21 (68). Although we did not observe an increase of apoptosis in the cocultured TAFs after inhibiting O-glycosylation with DON, we did observe an overall increase of p21 in the cocultured TAFs, which has been associated with senescence (52). These results also implicate O-glycosylation as a new avenue to regulate senescence, specifically by modulating RB interactions with E4F1, which impact p21 stabilization. However, we acknowledge that DON can also affect other metabolic enzymes using glutamine, and that a site-directed mutagenesis approach to prevent O-glycosylation of CDK4 or RB would be preferable to confirm the role of O-glycosylation in reducing palbociclib affinity in the CDK4-pRB signaling axis.
In summary, we characterized the crosstalk between cancer cells and fibroblasts derived from different tumor regions including normal, adjacent and tumor core. This work placed a greater focus on an understudied subtype of fibroblasts, namely those derived from the invasive tumor front, designated here as TAFs. We found that O-glycosylation regulates the CDK4-pRB axis in TAFs cocultured with cancer cells, highlighting the relevance of the O-glycoproteome as an important dimension in modulating protein signaling through metabolic reprogramming. Lastly, modulating O-glycosylation of the CDK4-pRB axis in TAFs may indirectly decrease the mesenchymal features of cancer cells by inducing a TCF-like phenotype in TAFs, which primarily showed anti-tumor roles in our study. In conclusion, the addition of O-glycoproteome data in combination with transcriptomic and proteomic data provides new insight into the biological control of the tumor leading edge by the stroma and indicates new avenues to improve cancer treatment. We hope that these results will motivate the development of targeted preclinical models to establish the significance of specific O-glycosylation events in the lung TME and promote the importance of glycobiology in cancer.
Supplementary Material
Significance.
A multi-omics analysis of spatially-distinct fibroblasts establishes the importance of the stromal O-glycoproteome in tumor-stroma interactions at the leading edge and provides potential strategies to improve cancer treatment.
Acknowledgments
GB was supported by the National Cancer Institute (R25CA180993) and Les Fonds de Recherche du Québec – Santé (GB #35603 and #267646). NMR was supported by the US National Institutes of Health (K00CA212454). LCM was supported by PHS Grant Number CA09302, awarded by the National Cancer Institute. The authors thank Dr Parag Mallick for providing the HCC827 cell lines used in this study. We also thank Dr Sean Bendall for CyTOF reagents, Winston L Trope for providing clinical specimen information, as well as Dr Marc Driessen, Dr Yuanyuan Li, and Dr Dhanya K Nambiar for their technical support.
Financial Support
National Cancer Institute grant R25CA180993 (SKP)
Les Fonds de Recherche du Québec – Santé grant #35603 and #267646 (GB)
US National Institutes of Health grant K00CA212454 (NMR)
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Deryugina EI, Kiosses WB, Jolla L, Facility MC, Jolla L. Intratumoral Cancer Cell Intravasation can occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017;19:601–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalluri R The biology and function of fibroblasts in cancer. Nat Publ Gr. 2016;16. [DOI] [PubMed] [Google Scholar]
- 3.Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. Elsevier B.V; 2016;99:186–96. [DOI] [PubMed] [Google Scholar]
- 4.Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. Springer US; 2019; [DOI] [PubMed] [Google Scholar]
- 5.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype Moulding of Stromal Cells in the Lung Tumour Microenvironment. Nature. 2018;1–23. [DOI] [PubMed] [Google Scholar]
- 7.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Z, Xiong S, Wang R, Li Y, Wang X, Wang Y, et al. Peri-tumor fibroblasts promote tumorigenesis and metastasis of hepatocellular carcinoma via Interleukin6/STAT3 signaling pathway. Cancer Manag Res. 2019;11:2889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivridis E, Giatromanolaki A, Koukourakis MI. Proliferating fibroblasts at the invading tumour edge of colorectal adenocarcinomas are associated with endogenous markers of hypoxia, acidity, and oxidative stress. J Clin Pathol. 2005;58:1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. Elsevier; 2018;33:463–479.e10. [DOI] [PubMed] [Google Scholar]
- 11.Manousopoulou A, Hayden A, Mellone M, Baquero DJG, White CH, Noble F, et al. Quantitative proteomic profiling of primary cancer-associated fibroblasts in oesophageal adenocarcinoma: CAF proteomic profiling in OAC. Br J Cancer. 2018;118:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. SSRN Electron J. Elsevier; 2018;178:160–175.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell. 2020;182:200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu JY, Zhang C, Wang X, Zhai L, Ma Y, Mao Y, et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell. Elsevier; 2020;182:245–261.e17. [DOI] [PubMed] [Google Scholar]
- 15.Li C, Di Sun Y, Yu GY, Cui JR, Lou Z, Zhang H, et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell. Elsevier Inc; 2020;38:734–747.e9. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Bouchard G, Yu A, Shafiq M, Jamali M, Shrager JBJB, et al. GFPT2-expressing cancer-associated fibroblasts mediate metabolic reprogramming in human lung adenocarcinoma. Cancer Res. 2018;78:3445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Lee HM, Cai F, Ko B, Yang C, Lieu EL, et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat Metab. Springer US; 2020;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.National Research Council. Transforming glycoscience: A roadmap for the future. 2012. [PubMed]
- 19.Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT-BG, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32:1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peixoto A, Relvas-Santos M, Azevedo R, Lara Santos L, Ferreira JA. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. 2019;14:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips RM, Lam C, Wang H, Tran PT. Bittersweet tumor development and progression: Emerging roles of epithelial plasticity glycosylations. 1st ed. Adv. Cancer Res Elsevier Inc.; 2019. [DOI] [PubMed] [Google Scholar]
- 22.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009;5:1087–104. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Wu M, Yang Z, Siyuan K, Mingqi L, Biyun J, et al. OGP: A Repository of Experimentally Characterized O-Glycoproteins to Facilitate Studies on O-Glycosylation. Genomics Proteomics Bioinformatics. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods. 2015;12:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Yuan Z-F, Fan J, Karch KR, Ball LE, Denu JM, et al. A Novel Quantitative Mass Spectrometry Platform for Determining Protein O-GlcNAcylation Dynamics. Mol Cell Proteomics. 2016;15:2462–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shon DJ, Malaker SA, Pedram K, Yang E, Krishnan V, Dorigo O, et al. An enzymatic toolkit for selective proteolysis, detection, and visualization of mucin-domain glycoproteins. Proc Natl Acad Sci U S A. 2020;117:21299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang W, Ao M, Hu Y, Li QK, Zhang H. Mapping the O-glycoproteome using site-specific extraction of O-linked glycopeptides (EXoO). Mol Syst Biol. 2018;14:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci U S A. 2019;116:7278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KTBG, Kong Y, Bennett EP, et al. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8:977–82. [DOI] [PubMed] [Google Scholar]
- 30.Gatto L, Lilley KS. Msnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics. 2012;28:288–9. [DOI] [PubMed] [Google Scholar]
- 31.Karacosta LG, Anchang B, Ignatiadis N, Kimmey SC, Benson JA, Shrager JB, et al. Mapping Lung Cancer Epithelial-Mesenchymal Transition States and Trajectories with Single-Cell Resolution. Nat Commun. 2019;10:5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. Nature Publishing Group; 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bern M, Kil YJ, Becker C. Byonic: Advanced peptide and protein identification software. Curr Protoc Bioinforma. 2012;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bateman A UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. Oxford University Press; 2019;47:D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khatri K, Klein JA, Zaia J. Use of an informed search space maximizes confidence of site-specific assignment of glycoprotein glycosylation. Anal Bioanal Chem. 2017;409:607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navarro P, Trevisan-Herraz M, Bonzon-Kulichenko E, Núñez E, Martínez-Acedo P, Pérez-Hernández D, et al. General statistical framework for quantitative proteomics by stable isotope labeling. J Proteome Res. 2014;13:1234–47. [DOI] [PubMed] [Google Scholar]
- 38.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 40.Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer. 2020;146:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao Z, Shi R, Yuan K, Wang Y. Expression and prognostic value of E2F activators in NSCLC and subtypes: a research based on bioinformatics analysis. Tumor Biol. Tumor Biology; 2016;37:14979–87. [DOI] [PubMed] [Google Scholar]
- 42.Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer. Springer US; 2019;19:326–38. [DOI] [PubMed] [Google Scholar]
- 43.Takayanagi SI, Hiroyama T, Yamazaki S, Nakajima T, Morita Y, Usui J, et al. Gene Expression Profiles of Non-Small Cell Lung Cancer: Survival Prediction and New Biomarkers. Blood. 2006;107:4317–25.16455951 [Google Scholar]
- 44.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brockhausen I, Stanley P. Chapter 10 O-GalNAc Glycans. Essentials Glycobiol. 2017;1–9. [Google Scholar]
- 46.Echalier A, Hole AJ, Lolli G, Endicott JA, Noble MEM. An inhibitor’s-eye view of the atp-binding site of CDKs in different regulatory states. ACS Chem Biol. 2014;9:1251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandey K, An HJ, Kim SK, Lee SA, Kim S, Lim SM, et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int J Cancer. 2019;145:1179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell. Elsevier; 2018;34:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W, Do KC, Saxton B, Leng S, Filipczak P, Tessema M, et al. Inhibition of the hexosamine biosynthesis pathway potentiates cisplatin cytotoxicity by decreasing BiP expression in non-small-cell lung cancer cells. Mol Carcinog. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naz S, Sowers A, Choudhuri R, Wissler M, Gamson J, Mathias A, et al. Abemaciclib, a selective CDK4/6 inhibitor, enhances the radiosensitivity of non–small cell lung cancer in vitro and in vivo. Clin Cancer Res. 2018;24:3994–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. Elsevier Ltd; 2018;28:911–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashraf HM, Moser J, Spencer SL. Senescence Evasion in Chemotherapy: A Sweet Spot for p21. Cell. Elsevier Inc; 2019;178:267–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garcia MA, Nelson WJ, Chavez N. Cell – Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb Perspect Biol. 2017;10:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ba P, Zhang X, Yu M, Li L, Duan X, Wang M, et al. Cancer associated fibroblasts are distinguishable from peri-tumor fibroblasts by biological characteristics in TSCC. Oncol Lett. 2019;18:2484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, Xiao C-H, Tan L-D, Wang Q-S, Li X-Q, Feng Y-M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo M, Luo Y, Mao N, Huang G, Teng C, Wang H, et al. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell Physiol Biochem. 2018;51:315–36. [DOI] [PubMed] [Google Scholar]
- 58.Barletta JA, Yeap BY, Chirieac LR. Prognostic significance of grading in lung adenocarcinoma. Cancer. 2010;116:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh A, Rozhok AI, et al. Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Res. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hatzikirou H, Basanta D, Simon M, Schaller K, Deutsch A. “Go or grow”: the key to the emergence of invasion in tumour progression? Math Med Biol. 2012;29:49–65. [DOI] [PubMed] [Google Scholar]
- 61.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. Nature Publishing Group; 2015;15:203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shafiq MI, Steinbrecher T, Schmid R. Fascaplysin as a specific inhibitor for cdk4: Insights from molecular modelling. PLoS One. 2012;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pinho SS, Reis CA. Glycosylation in cancer: Mechanisms and clinical implications. Nat Rev Cancer. Nature Publishing Group; 2015;15:540–55. [DOI] [PubMed] [Google Scholar]
- 64.Taparra K, Tran PT, Zachara NE. Hijacking the Hexosamine Biosynthetic Pathway to Promote EMT-Mediated Neoplastic Phenotypes. Front Oncol. Frontiers Media SA; 2016;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. [DOI] [PubMed] [Google Scholar]
- 67.Lentine B, Antonucci L, Hunce R, Edwards J, Marallano V, Krucher NA. Dephosphorylation of threonine-821 of the retinoblastoma tumor suppressor protein (Rb) is required for apoptosis induced by UV and Cdk inhibition. Cell Cycle. 2012;11:3324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fajas L, Paul C, Zugasti O, Le Cam L, Polanowska J, Fabbrizio E, et al. pRB binds to and modulates the transrepressing activity of the E1A-regulated transcription factor p120E4F. Proc Natl Acad Sci U S A. 2000;97:7738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.