

Abstract
We herein report the functionalization of α-C–H in alcohols through cross-dehydrogenative coupling reactions. Selectfluor was used as a mild oxidant. In situ-generated HF participated in the reaction and no external strong acid was necessary. A variety of heteroaryl-substituted alcohols were achieved with good yields and with good functional group tolerance.
We herein report the functionalization of α-C–H in alcohols through cross-dehydrogenative coupling reactions.
Alcohols are one of the most important types of compounds, because they exist universally in bioactive molecules and are also found in nature in the form of sugars, steroids, etc. Furthermore, alcohols can be used as versatile building blocks, and be transformed into other functional groups. The synthesis of alcohols is a classic topic in organic chemistry. Late-stage functionalization has attracted the interest of chemists because in this way complex molecules could be modified. In particular, functionalization of C–H is a very efficient way to build various structures, as pre-functionalization here is not needed. Thus, transformation of C–H in alcohols is a direct method to prepare diverse alcohols from simple molecules.
During the past several decades, activations of the α sp3 C–H groups of alcohols have been achieved through radical-involved processes. In the early research, functionalizations of α C–H groups in alcohols were always initiated by light or di-tert-butyl peroxide, and the generated radicals could add to alkenes, affording alkylated alcohols.1 This reaction was developed by Duan to construct hydroxyl-containing oxindoles through a tandem addition/cyclization reaction.2 Also, amino alcohols were prepared through addition of radicals generated from alcohols to unactivated alkenes.3 Reactions catalyzed by metals such as Rh,4 Fe,5 Cu and Co6 have also been applied. In 2009, addition of such radicals to alkynes was reported. Homoallylic alcohols were prepared using simple alcohol molecules.7 The reactions of alcohols and cinnamic acids were also reported and allylic alcohols were obtained.8 However, arylations of the α C–H in alcohols have attracted much interest because such structures are present in many bioactive molecules (Fig. 1). Great efforts have been expended to the synthesis of arylated alcohols. In 2014, Liu reported the reaction of alcohol molecules and isocyanide, and various alkyl-substituted phenanthridines were achieved.9
Fig. 1. Bioactive molecules containing alcoholic hydroxyl groups.

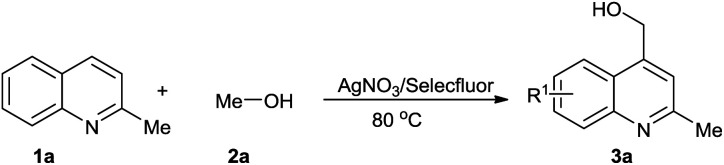
Cross-dehydrogenative coupling reactions have been the focus of many researchers and have been shown to be powerful and convenient tools in organic synthesis.10 The activation of the C–H groups in two molecules allows application of starting materials without further functionalization. This simple process with high atomic economy provides an ideal transformation for the preparation of various target compounds. The Minisci reaction has been widely explored since the 1960s. Palmer and McIntyre first reported the hydroxymethylation of quinolone in the presence of hydroxylamine-O-sulphonic acid (HSA).11 Minisci further developed an Fe-catalyzed process that resulted in good yields.12 PdCl2 (ref. 13) and TiCl3 (ref. 14) were also shown to be efficient catalysts for the Minisci reaction. Metal-free reactions initiated by TBHP15 and M2S2O8 (M = Na, K, NH4)16 have also been developed. Very recently, as visible light-promoted processes find applications in organic synthesis, visible light-induced Minisci reactions have been reported.17 However, a strong oxidant, external acid, and complex system are always needed for these reactions. In our research, we found that Selectfluor could be used as a mild oxidant for the functionalizations of C–H groups in ethers, and the HF generated in situ could be made to participate in the reaction process.18 Herein, we report an Ag/Selectfluor-catalyzed Minisci reaction, in which Selectfluor was used as a mild oxidant and no external acid was necessary (Scheme 1).
Scheme 1. Arylations of α sp3 C–H groups in alcohols.

We chose quinaldine as the model substrate and carried out the reactions (Table 1). When 0.25 eq. Selectfluor was used, only a trace amount of product was observed (Table 1, entry 1). Increasing the number of equivalents of Selectfluor led to better results (Table 1, entries 1–4), and a high yield was realized when 4 eq. Selectfluor were added (Table 1, entry 4). The yield was improved to 88% when 1 eq. AgNO3 was used (Table 1, entry 5). Further increasing of the amount of AgNO3 used showed no obvious improvement of the yields (Table 1, entry 6). Then we examined the ratio of the solvent components (Table 1, entries 6–8). An excellent yield was achieved when the volume ratio of MeOH to H2O was 9 : 1. The presence of a little water may have promoted the dissolution of Selectfluor and AgNO3. However, when more water was included, lower yields were observed, which may have been the result of its influence on the reactivity of the intermediate. Increasing and decreasing the reaction time from 4 h both showed a slight decrease of the yield (Table 1, entries 9 and 10). Increasing and decreasing the temperature from 80 °C also both led to a worse result (Table 1, entries 11 and 12).
Screening of the reaction conditionsa.
| |||||
|---|---|---|---|---|---|
| Entry | AgNO3 (eq.) | Selectfluor (eq.) | Time (h) | MeOH : H2Ob | Yieldc (%) |
| 1 | 0.5 | 0.25 | 4 | 3 : 1 | Trace |
| 2 | 0.5 | 0.5 | 4 | 3 : 1 | 7 |
| 3 | 0.5 | 1 | 4 | 3 : 1 | 24 |
| 4 | 0.5 | 4 | 4 | 3 : 1 | 76 |
| 5 | 1 | 4 | 4 | 3 : 1 | 88 |
| 6 | 2 | 4 | 4 | 3 : 1 | 89 |
| 7 | 1 | 4 | 4 | 1 : 1 | 88 |
| 8 | 1 | 4 | 4 | 9 : 1 | 96 |
| 9 | 1 | 4 | 2 | 9 : 1 | 93 |
| 10 | 1 | 4 | 6 | 9 : 1 | 93 |
| 11d | 1 | 4 | 4 | 9 : 1 | 90 |
| 12e | 1 | 4 | 4 | 9 : 1 | 85 |
Conditions: 0.2 mmol of 1a, AgNO3, Selectfluor, and 2 mL of solvent were added in a tube that was then sealed. The system was heated at the indicated temperature.
Volume ratio.
Isolated yields.
Reaction temperature was 60 °C.
Reaction temperature was 100 °C.
With the optimal conditions in hand, we explored a series of quinaldines substituted with various groups or atoms. Hydroxymethylation of substituted substrates went smoothly with high yields. 6-Substituted quinaldines were shown to be proper starting materials. Good yields were observed for a variety of introduced substituents, such as F (3b), Br (3c), I (3d), Me (3e) and CF3 (3f). Electron-withdrawing atoms and groups were favourable for high yields. However, molecule with electron-donating group gave a low yield (3e). This result may be caused by the lower stability of the in situ-generated intermediate containing an electron-withdrawing group. 8-Substituted quinaldines could also undergo this process. However, the yields were lower than those of 6-substituted substrates. We supposed that this was the result of steric effects. The substituents present on the 8-position may have protected the heterocycles from protonation, an effect harmful for the hydroxymethylation. Here again, electron-withdrawing substituents were more efficient than electron-donating groups. 7-Chloroquinaldine reacted under the standard conditions, giving the product (3l) in 94% yield. Multifluorinated heterocycles were used, and good results were obtained (3m, 3n). Lepidine was also hydroxymethylated on the 2-position in 85% yield (3o) (Scheme 2).
Scheme 2. Quinaldine substrate scope. Standard conditions: 1 (1 mmol), AgNO3 (1 eq.), Selectfluor (4 eq.), and 10 mL of solvent (MeOH/H2O = 9 : 1) were added into a tube that was then sealed. The system was heated at 80 °C for 4 h.

Next, we examined the scope of alcohols. To our delight, ethanol reacted with substituted quinaldines smoothly. Arylation of the α sp3 C–H in ethanol occurred with good yields. Various substituents, such as F (4a, 4e), Cl (4b, 4f), Br (4c) and I (4d), were tolerated and the position of the substituent showed little influence on the yield. 1-Propanol was also tested as a starting material and arylations using this starting material proceeded with good yields, albeit slightly lower than those for ethanol (4g, 4h, 4i). This result may be caused by a steric effect, leading to lower reactivity of the generated radicals (Scheme 3).
Scheme 3. Alcohol substrate scope. Standard conditions: 1 (1 mmol), AgNO3 (1 eq.), Selectfluor (4 eq.), and 10 mL of solvent (RCH2OH/H2O = 9 : 1) were added into a tube that was then sealed. The system was heated at 80 °C for 4 h.

To explore the reaction mechanism, some control experiments were carried out. When base was added together with starting materials, no product was obtained. When TEMPO was added, also no product was observed according to liquid chromatography-mass spectrometry (LC-MS) results. However, several new species were present besides the original heterocycle and alcohol. A peak at a position corresponding to a molecular weight of 161.0548 was assigned to salt 5, which was generated by the reaction of Selectfluor and AgNO3. The peak characterized by a molecular weight of 158.1548 was attributed to compound 7. We did not, however, observe a peak corresponding to compound 6, which had been expected to result from the combination of TEMPO and hydroxymethyl radical. We supposed that 6 was a hemiacetal, which due to its instability was hydrolysed rapidly to give 7. Furthermore, a kinetic isotopic effect was observed using [2H4]methanol (KIE = 2.8), indicating that the hydrogen atom abstraction from methanol was the rate-limiting step. Based on these data, a mechanism was derived (Scheme 4). First, the Ag(i) species was oxidized to Ag(ii) by Selectfluor, generating a fluorine radical and 5. Then the α-H in the alcohol molecule was abstracted by the fluorine radical, and an alkyl radical (Int I) was generated. Meanwhile, HF was released and combined with the heterocycles, giving an intermediate cation (Int II). This cation was attacked by Int I, resulting in the radical cation Int III. Then a proton was released and the radical Int IV was oxidized by Ag(ii) to give a cation (Int V). Finally, a proton was abstracted by external base, leading to the desired product. If the base was added at the beginning of the reaction, the released HF could be trapped, and the intermediate Int II was not generated, resulting in a disturbance of the process. Our results together with the derived mechanism indicated that the in situ-generated HF played a significant role in this reaction.
Scheme 4. Proposed mechanism.

In summary, we developed an efficient method for the arylation of α sp3 C–H groups in alcohols using heterocycles through CDC catalyzed by Ag. Selectfluor was used as a mild oxidant and no external acid was needed. Instead, in situ-generated HF played an important role. Various substituents were tolerated, such as halogens, alkyl groups, CF3, etc. Multi-substituted quinaldines also underwent this reaction smoothly. Common alcohols, such as methanol, ethanol and 1-propanol were explored as materials and all the alcohols could be used without further purification. Mechanism studies showed that a radical process occurred and the hydrogen abstraction step was determined to be the rate-limiting step.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support from the Natural Science Foundation (31972850), the Shandong Provincial Natural Science Foundation (ZR2016HM44, ZR2016BQ31, ZR2016JL009 and ZR2017ZC0529), the Shandong Key Research Program (2017GSF218061) and the University of Jinan are gratefully acknowledged.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09954a
Notes and references
- For selected examples for photoinitiated and di-tert-butyl peroxide-initiated addition of alcohols to aliphatic olefins, see: ; (a) Urry W. H. Stacey F. W. Huyser E. S. Juveland O. O. J. Am. Chem. Soc. 1954;76:450–455. doi: 10.1021/ja01631a037. [DOI] [Google Scholar]; (b) LaZerte J. D. Koshar R. J. J. Am. Chem. Soc. 1955;77:910–914. doi: 10.1021/ja01609a033. [DOI] [Google Scholar]; (c) Walling C. Huyser E. S. Org. React. 1963;13:91–149. [Google Scholar]; (d) Curran D. P. Synthesis. 1988:489–513. doi: 10.1055/s-1988-27620. [DOI] [Google Scholar]
- Meng Y. Guo L.-N. Wang H. Duan X.-H. Chem. Commun. 2013;49:7540–7542. doi: 10.1039/C3CC44055A. [DOI] [PubMed] [Google Scholar]
- Zhou W. Qian P. Zhao J. Fang H. Han J. Pan Y. Org. Lett. 2015;17:1160–1163. doi: 10.1021/acs.orglett.5b00088. [DOI] [PubMed] [Google Scholar]
- Shi L. Tu Y.-Q. Wang M. Zhang F.-M. Fan C.-A. Zhao Y.-M. Xia W.-J. J. Am. Chem. Soc. 2005;127:10836–10837. doi: 10.1021/ja0528331. [DOI] [PubMed] [Google Scholar]
- Zhang S.-Y. Tu Y.-Q. Fan C.-A. Zhang F.-M. Shi S. Angew. Chem., Int. Ed. 2009;48:8761–8765. doi: 10.1002/anie.200903960. [DOI] [PubMed] [Google Scholar]
- Cheng J.-K. Loh T.-P. J. Am. Chem. Soc. 2015;137:42–45. doi: 10.1021/ja510635k. [DOI] [PubMed] [Google Scholar]
- Obora Y. Hatanaka S. Ishii Y. Org. Lett. 2009;11:3510–3513. doi: 10.1021/ol901366q. [DOI] [PubMed] [Google Scholar]
- Cui Z. Shang X. Shao X.-F. Liu Z.-Q. Chem. Sci. 2012;3:2853–2858. doi: 10.1039/C2SC20712E. [DOI] [Google Scholar]
- (a) Li Z. Fan F. Yang J. Liu Z.-Q. Org. Lett. 2014;16:3396–3399. doi: 10.1021/ol501461u. [DOI] [PubMed] [Google Scholar]; (b) Xu Z. Hang Z. Liu Z.-Q. Org. Lett. 2016;18:4470–4473. doi: 10.1021/acs.orglett.6b01946. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see: ; (a) Guo S.-R. Kumar P. S. Yang M. Adv. Synth. Catal. 2017;359:2–25. doi: 10.1002/adsc.201600467. [DOI] [Google Scholar]; (b) Zhang S.-Y. Zhang F.-M. Tu Y.-Q. Chem. Soc. Rev. 2011;40:1937–1949. doi: 10.1039/C0CS00063A. [DOI] [PubMed] [Google Scholar]
- Palmer M. H. McIntyre P. S. Tetrahedron Lett. 1968;9:2147–2150. doi: 10.1016/S0040-4039(00)89763-4. [DOI] [Google Scholar]
- Citterio A. Gentile A. Minisci F. Serravalle M. Ventura S. Tetrahedron. 1985;41:617–620. doi: 10.1016/S0040-4020(01)96508-1. [DOI] [Google Scholar]
- Correia C. A. Yang L. Li C.-J. Org. Lett. 2011;13:4581–4583. doi: 10.1021/ol201774b. [DOI] [PubMed] [Google Scholar]
- Neubert T. D. Schmidt Y. Conroy E. Stamos D. Org. Lett. 2015;17:2362–2365. doi: 10.1021/acs.orglett.5b00861. [DOI] [PubMed] [Google Scholar]
- He T. Yu L. Zhang L. Wang L. Wang M. Org. Lett. 2011;13:5016–5019. doi: 10.1021/ol201779n. [DOI] [PubMed] [Google Scholar]
- (a) Zhou L. Okugawa N. Togo H. Eur. J. Org. Chem. 2017:6239–6245. doi: 10.1002/ejoc.201701321. [DOI] [PubMed] [Google Scholar]; (b) Liu Y. Jiang B. Zhang W. Xu Z. J. Org. Chem. 2013;78:966–980. doi: 10.1021/jo302450f. [DOI] [PubMed] [Google Scholar]; (c) Bohman B. Berntsson B. Dixon R. C. M. Stewart C. D. Barrow R. A. Org. Lett. 2014;16:2787–2789. doi: 10.1021/ol500776j. [DOI] [PubMed] [Google Scholar]
- (a) Huff C. A. Cohen R. D. Dykstra K. D. Streckfuss E. DiRocco D. A. J. Org. Chem. 2016;81:6980–6987. doi: 10.1021/acs.joc.6b00811. [DOI] [PubMed] [Google Scholar]; (b) Twilton J. Christensen M. DiRocco D. A. Ruck R. T. Davies I. W. MacMillan D. W. C. Angew. Chem., Int. Ed. 2018;57:5369–5373. doi: 10.1002/anie.201800749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Niu L. Liu J. Liang X.-A. Wang S. Lei A. Nat. Commun. 2019;10:1–7. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. Fan Y. Zhao H. Wang J. Zhang S. Wang W. Synlett. 2019;30:2096–2100. doi: 10.1055/s-0039-1690697. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.