
Effect of inhibitors on the affinity of NDM-1 by molecule dockinga.
| Inhibitors | Structures | Docking energy (kcal mol−1) | Inhibitors | Structures | Docking energy (kcal mol−1) |
|---|---|---|---|---|---|
| AMA |
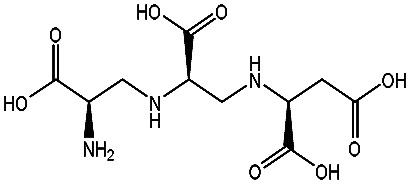
|
−98.41 ± 1.23 | d-Captopril |
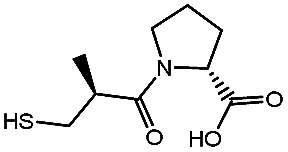
|
−61.76 ± 0.69 |
| Ebselen |
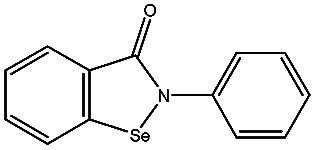
|
−78.03 ± 0.92 | EDTA |
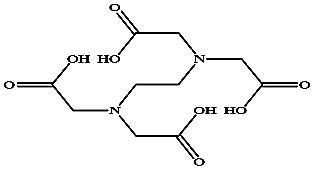
|
−109.19 ± 1.58 |
a
Molecular docking was carried out using the Auto-dock 4.0 software. Only ligand molecules were considered to be flexible during the docking, and only the free energy of their best orientation was used to compute the docking free energy.